
Lattice Distortions in the FeCoNiCrMn High Entropy Alloy Studied by Theory and Experiment


 ,
, {kind=link}
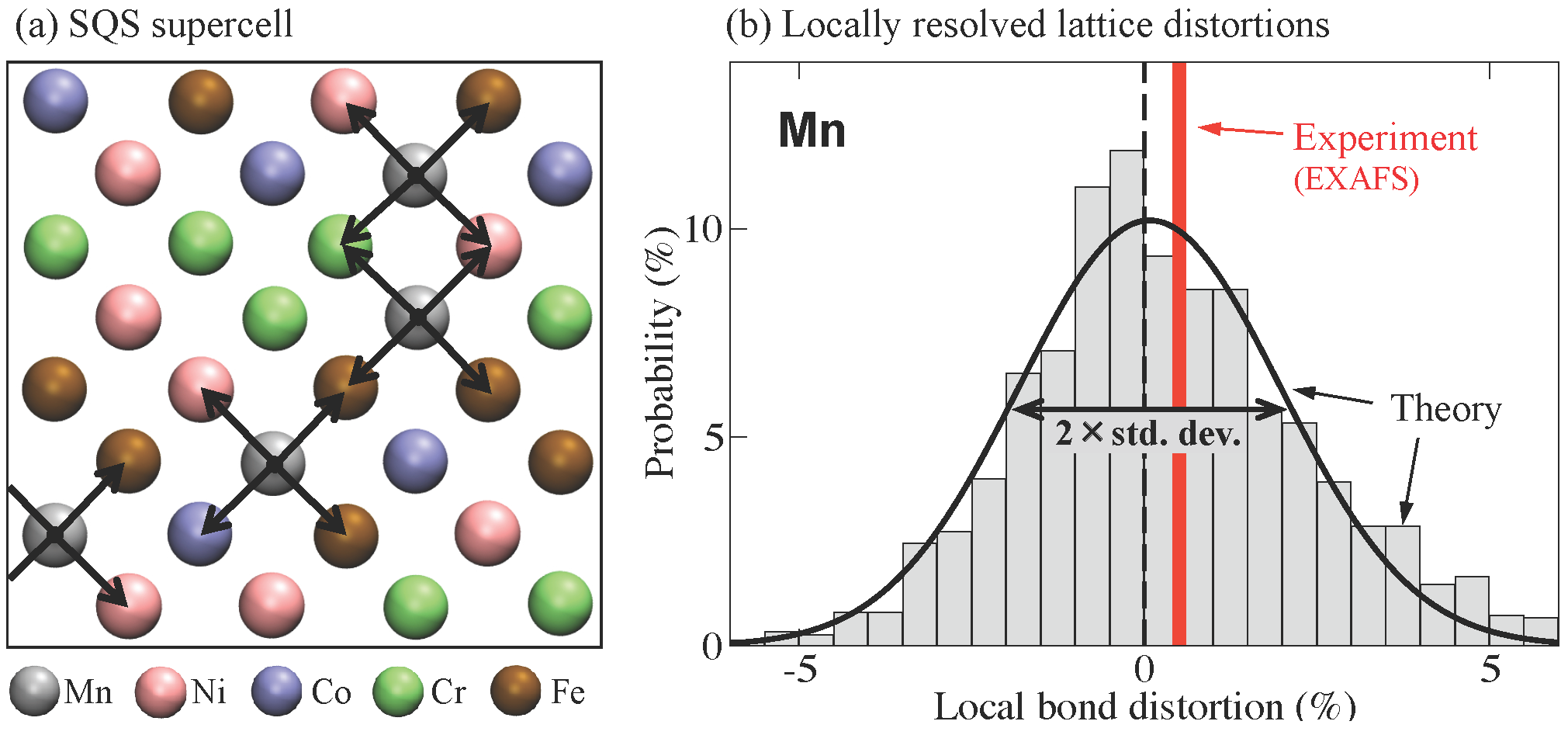{kind=link}
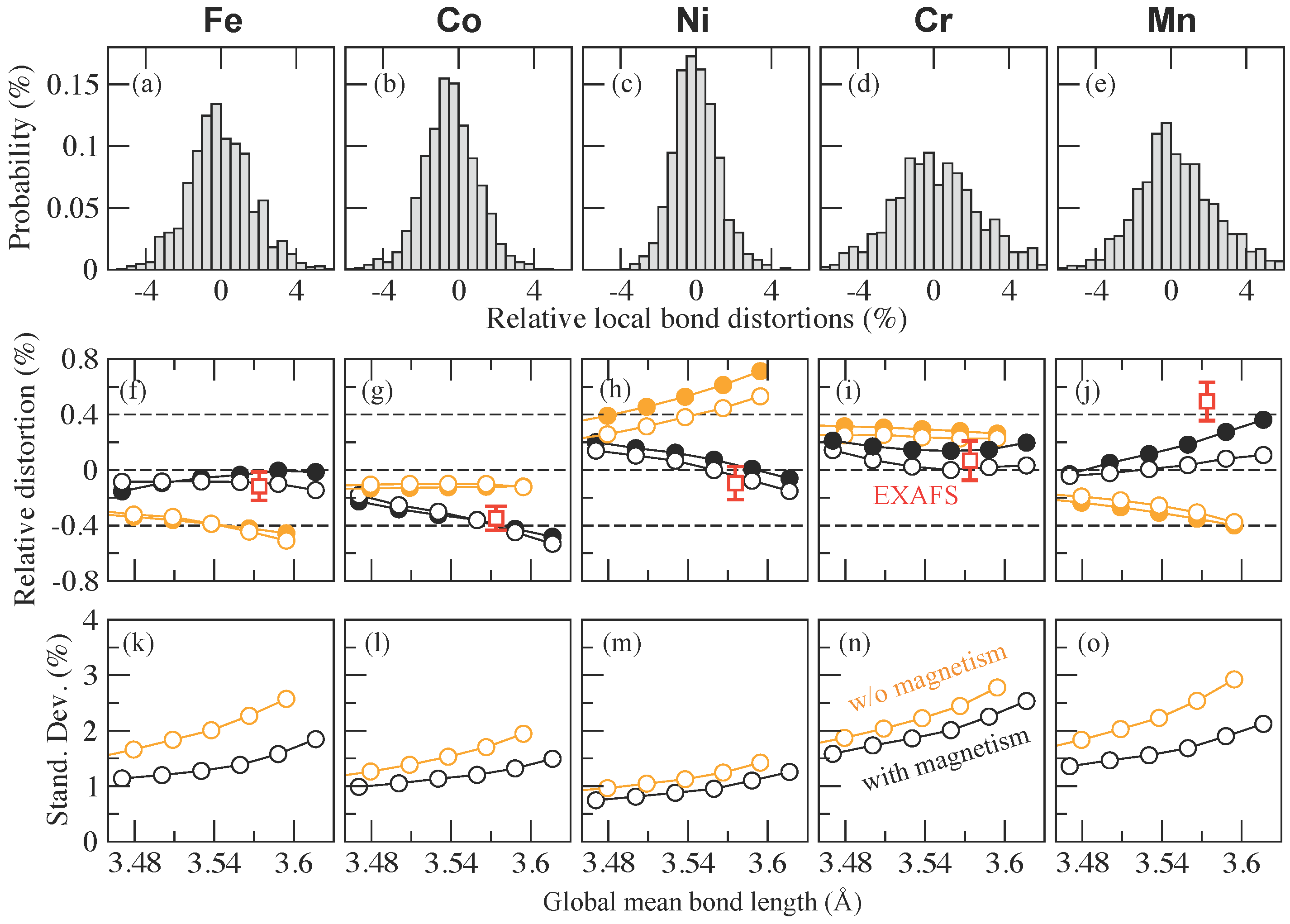{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Murty, B.S.; Yeh, W.J.; Ranganathan, S. High-Entropy Alloys; Elsevier: London, UK, 2014. [Google Scholar]
- Zhang, Y.; Zuo, T.T.; Tang, Z.; Gao, M.C.; Dahmen, K.A.; Liaw, P.K.; Lu, Z.P. Microstructures and properties of high-entropy alloys. Prog. Mater. Sci. 2014, 61, 1–93. [Google Scholar] [CrossRef]
- Gao, M.; Yeh, J.; Liaw, P.; Zhang, Y. High-Entropy Alloys: Fundamentals and Applications; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Gludovatz, B.; Hohenwarter, A.; Catoor, D.; Chang, E.H.; George, E.P.; Ritchie, R.O. A fracture-resistant high-entropy alloy for cryogenic applications. Science 2014, 345, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Ma, H.; Spolenak, R. Ultrastrong ductile and stable high-entropy alloys at small scales. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Pradeep, K.G.; Deng, Y.; Raabe, D.; Tasan, C.C. Metastable high-entropy dual-phase alloys overcome the strength–ductility trade-off. Nature 2016, 534, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Pradeep, K.; Tasan, C.; Raabe, D. A novel, single phase, non-equiatomic FeMnNiCoCr high-entropy alloy with exceptional phase stability and tensile ductility. Scr. Mater. 2014, 72, 5–8. [Google Scholar] [CrossRef]
- Deng, Y.; Tasan, C.C.; Pradeep, K.G.; Springer, H.; Kostka, A.; Raabe, D. Design of a twinning-induced plasticity high entropy alloy. Acta Mater. 2015, 94, 124–133. [Google Scholar] [CrossRef]
- Zhang, Y.; Zuo, T.; Cheng, Y.; Liaw, P.K. High-entropy alloys with high saturation magnetization, electrical resistivity, and malleability. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Sales, B.C.; Stocks, G.M.; Samolyuk, G.D.; Daene, M.; Weber, W.J.; Zhang, Y.; Bei, H. Tailoring the physical properties of Ni-based single-phase equiatomic alloys by modifying the chemical complexity. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Körmann, F.; Ma, D.; Belyea, D.D.; Lucas, M.S.; Miller, C.W.; Grabowski, B.; Sluiter, M.H. “Treasure maps” for magnetic high-entropy-alloys from theory and experiment. Appl. Phys. Lett. 2015, 107, 142404. [Google Scholar] [CrossRef]
- Huang, S.; Vida, Á.; Molnár, D.; Kádas, K.; Varga, L.K.; Holmström, E.; Vitos, L. Phase stability and magnetic behavior of FeCrCoNiGe high-entropy alloy. Appl. Phys. Lett. 2015, 107, 251906. [Google Scholar] [CrossRef]
- Ma, D.; Grabowski, B.; Körmann, F.; Neugebauer, J.; Raabe, D. Ab initio thermodynamics of the CoCrFeMnNi high entropy alloy: Importance of entropy contributions beyond the configurational one. Acta Mater. 2015, 100, 90–97. [Google Scholar] [CrossRef]
- Labusch, R. A statistical theory of solid solution hardening. Phys. Status Solidi 1970, 41, 659–669. [Google Scholar] [CrossRef]
- Labusch, R. Statistical theories of solid solution hardening (Concentration of solute atoms, interaction range between solute atoms and distortion, and interaction strength of solid solution hardening). Acta Metall. 1972, 20, 917–927. [Google Scholar] [CrossRef]
- Varvenne, C.; Luque, A.; Curtin, W.A. Theory of strengthening in fcc high entropy alloys. Acta Mater. 2016, 118, 164–176. [Google Scholar] [CrossRef]
- Fleischer, R. Solution hardening. Acta Metall. 1961, 9, 996–1000. [Google Scholar] [CrossRef]
- Wang, S. Atomic Structure Modeling of Multi-Principal-Element Alloys by the Principle of Maximum Entropy. Entropy 2013, 15, 5536–5548. [Google Scholar] [CrossRef]
- Egami, T.; Ojha, M.; Khorgolkhuu, O.; Nicholson, D.; Stocks, G. Local electronic effects and irradiation resistance in high-entropy alloys. JOM 2015, 67, 2345–2349. [Google Scholar] [CrossRef]
- Niu, C.; Zaddach, A.; Oni, A.; Sang, X.; Hurt, J., III; LeBeau, J.M.; Koch, C.C.; Irving, D.L. Spin-driven ordering of Cr in the equiatomic high entropy alloy NiFeCrCo. Appl. Phys. Lett. 2015, 106, 161906. [Google Scholar] [CrossRef]
- Tamm, A.; Aabloo, A.; Klintenberg, M.; Stocks, M.; Caro, A. Atomic-scale properties of Ni-based FCC ternary, and quaternary alloys. Acta Mater. 2015, 99, 307–312. [Google Scholar] [CrossRef]
- Troparevsky, M.C.; Morris, J.R.; Daene, M.; Wang, Y.; Lupini, A.R.; Stocks, G.M. Beyond Atomic Sizes and Hume-Rothery Rules: Understanding and Predicting High-Entropy Alloys. JOM 2015, 67, 2350–2363. [Google Scholar] [CrossRef]
- Li, X.; Tian, F.; Schönecker, S.; Zhao, J.; Vitos, L. Ab initio-predicted micro-mechanical performance of refractory high-entropy alloys. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Li, W.; Lu, S.; Tian, F.; Shen, J.; Holmström, E.; Vitos, L. Temperature dependent stacking fault energy of FeCrCoNiMn high entropy alloy. Scr. Mater. 2015, 108, 44–47. [Google Scholar] [CrossRef]
- Tian, F.; Delczeg, L.; Chen, N.; Varga, L.K.; Shen, J.; Vitos, L. Structural stability of NiCoFeCrAl x high-entropy alloy from ab initio theory. Phys. Rev. B 2013, 88, 085128. [Google Scholar] [CrossRef]
- Körmann, F.; Ruban, A.V.; Sluiter, M.H. Long-ranged interactions in bcc NbMoTaW high-entropy alloys. Mater. Res. Lett. 2016. [Google Scholar] [CrossRef]
- Yeh, J.W.; Chang, S.Y.; Hong, Y.D.; Chen, S.K.; Lin, S.J. Anomalous decrease in X-ray diffraction intensities of Cu–Ni–Al–Co–Cr–Fe–Si alloy systems with multi-principal elements. Mater. Chem. Phys. 2007, 103, 41–46. [Google Scholar] [CrossRef]
- Guo, W.; Dmowski, W.; Noh, J.Y.; Rack, P.; Liaw, P.K.; Egami, T. Local atomic structure of a high-entropy alloy: An X-ray and neutron scattering study. Metall. Mater. Trans. A 2013, 44, 1994–1997. [Google Scholar] [CrossRef]
- Otto, F.; Dlouhỳ, A.; Pradeep, K.; Kuběnová, M.; Raabe, D.; Eggeler, G.; George, E. Decomposition of the single-phase high-entropy alloy CrMnFeCoNi after prolonged anneals at intermediate temperatures. Acta Mater. 2016, 112, 40–52. [Google Scholar] [CrossRef]
- Newville, M. IFEFFIT: Interactive XAFS analysis and FEFF fitting. J. Synchrotron Radiat. 2001, 8, 322–324. [Google Scholar] [CrossRef] [PubMed]
- Newville, M. Fundamentals of XAFS. Rev. Mineral. Geochem. 2014, 78, 33–74. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Zunger, A.; Wei, S.H.; Ferreira, L.; Bernard, J.E. Special quasirandom structures. Phys. Rev. Lett. 1990, 65, 353. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.C.; Niu, C.; Jiang, C.; Irving, D.L. Applications of Special Quasi-random Structures to High-Entropy Alloys. In High-Entropy Alloys: Fundamentals and Applications; Springer: Cham, Switzerland, 2016; pp. 333–368. [Google Scholar]
- Körmann, F.; Grabowski, B.; Dutta, B.; Hickel, T.; Mauger, L.; Fultz, B.; Neugebauer, J. Temperature Dependent Magnon-Phonon Coupling in bcc Fe from Theory and Experiment. Phys. Rev. Lett. 2014, 113, 165503. [Google Scholar] [CrossRef] [PubMed]
- Körmann, F.; Hickel, T.; Neugebauer, J. Influence of magnetic excitations on the phase stability of metals and steels. Curr. Opin. Solid. St. Mater. 2016, 20, 77–84. [Google Scholar] [CrossRef]
- Körmann, F.; Ma, P.W.; Dudarev, S.L.; Neugebauer, J. Impact of magnetic fluctuations on lattice excitations in fcc nickel. J. Phys. Condens. Matter 2016, 28, 076002. [Google Scholar] [CrossRef] [PubMed]
- Alling, B.; Körmann, F.; Grabowski, B.; Glensk, A.; Abrikosov, I.; Neugebauer, J. Strong impact of lattice vibrations on electronic and magnetic properties of paramagnetic Fe revealed by disordered local moments molecular dynamics. Phys. Rev. B 2016, 93, 224411. [Google Scholar] [CrossRef]
- Uesugi, T.; Higashi, K. First-principles studies on lattice constants and local lattice distortions in solid solution aluminum alloys. Comput. Mater. Sci. 2013, 67, 1–10. [Google Scholar] [CrossRef]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, H.S.; Ma, D.; Leyson, G.P.; Grabowski, B.; Park, E.S.; Körmann, F.; Raabe, D. Lattice Distortions in the FeCoNiCrMn High Entropy Alloy Studied by Theory and Experiment. Entropy 2016, 18, 321. https://doi.org/10.3390/e18090321
Oh HS, Ma D, Leyson GP, Grabowski B, Park ES, Körmann F, Raabe D. Lattice Distortions in the FeCoNiCrMn High Entropy Alloy Studied by Theory and Experiment. Entropy. 2016; 18(9):321. https://doi.org/10.3390/e18090321
Chicago/Turabian StyleOh, Hyun Seok, Duancheng Ma, Gerard Paul Leyson, Blazej Grabowski, Eun Soo Park, Fritz Körmann, and Dierk Raabe. 2016. "Lattice Distortions in the FeCoNiCrMn High Entropy Alloy Studied by Theory and Experiment" Entropy 18, no. 9: 321. https://doi.org/10.3390/e18090321
APA StyleOh, H. S., Ma, D., Leyson, G. P., Grabowski, B., Park, E. S., Körmann, F., & Raabe, D. (2016). Lattice Distortions in the FeCoNiCrMn High Entropy Alloy Studied by Theory and Experiment. Entropy, 18(9), 321. https://doi.org/10.3390/e18090321