Non-Linear Dynamics Analysis of Protein Sequences. Application to CYP450

Abstract
:1. Introduction
2. Experimental Dataset
2.1. Alphabetical Dictionary
| MTIKEMPQPKTFGELKNLPLLNTDKPVQALMKIADELGEIFKFEAPGRVTRYLSSQRLIKEACDES |
| RFDKNLSQALKFVRDFAGDGLATSWTHEKNWKKAHNILLPSFSQQAMKGYHAMMVDIATQLI |
| QKWSRLNPNEEIDVADDMTRLTLDTIGLCGFNYRFNSFYRDSQHPFITSMLRALKEAMNQSKRL |
| LRLWPTAPAFSLYAKEDTVLGGEYPLEKGDELMVLIPQLHRDKTIWGDDVEEFRPERFENPSAIPQ |
| HAFKPFGNGQRACIGQQFALHEATLVLGMILKYFTLIDHENYELDIKQTLTLKPGDFHISVQSRH |
| QEAIHADVQAAE |
2.2. An Application Example: Cytochrome P450
3. Methodology
3.1. Entropy and Chaos
3.2. Kolmogorov Complexity and Turing Machine
3.3. Law-Scaling and Stochastic Process
3.4. Surrogated and Shuffled Data
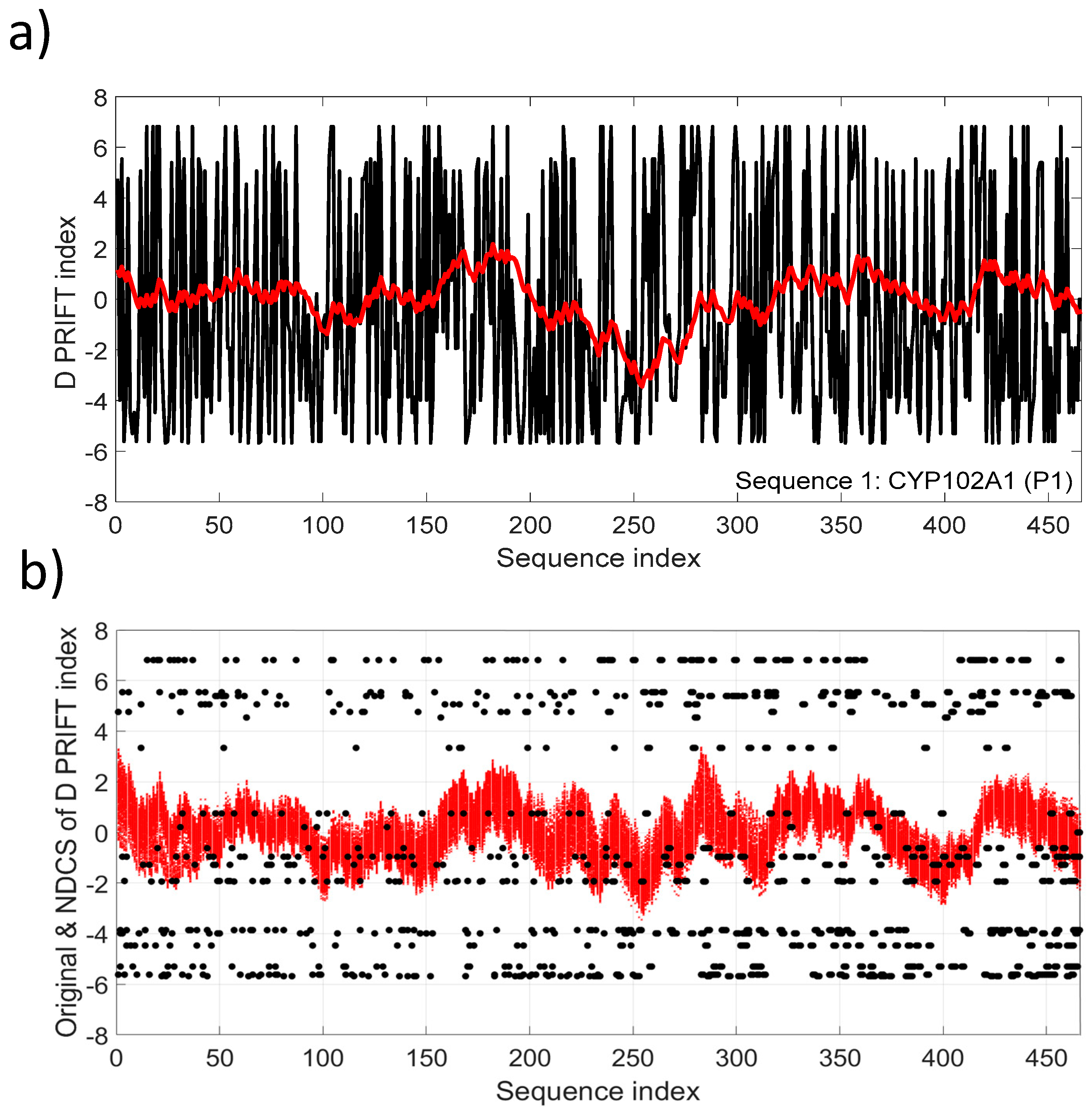
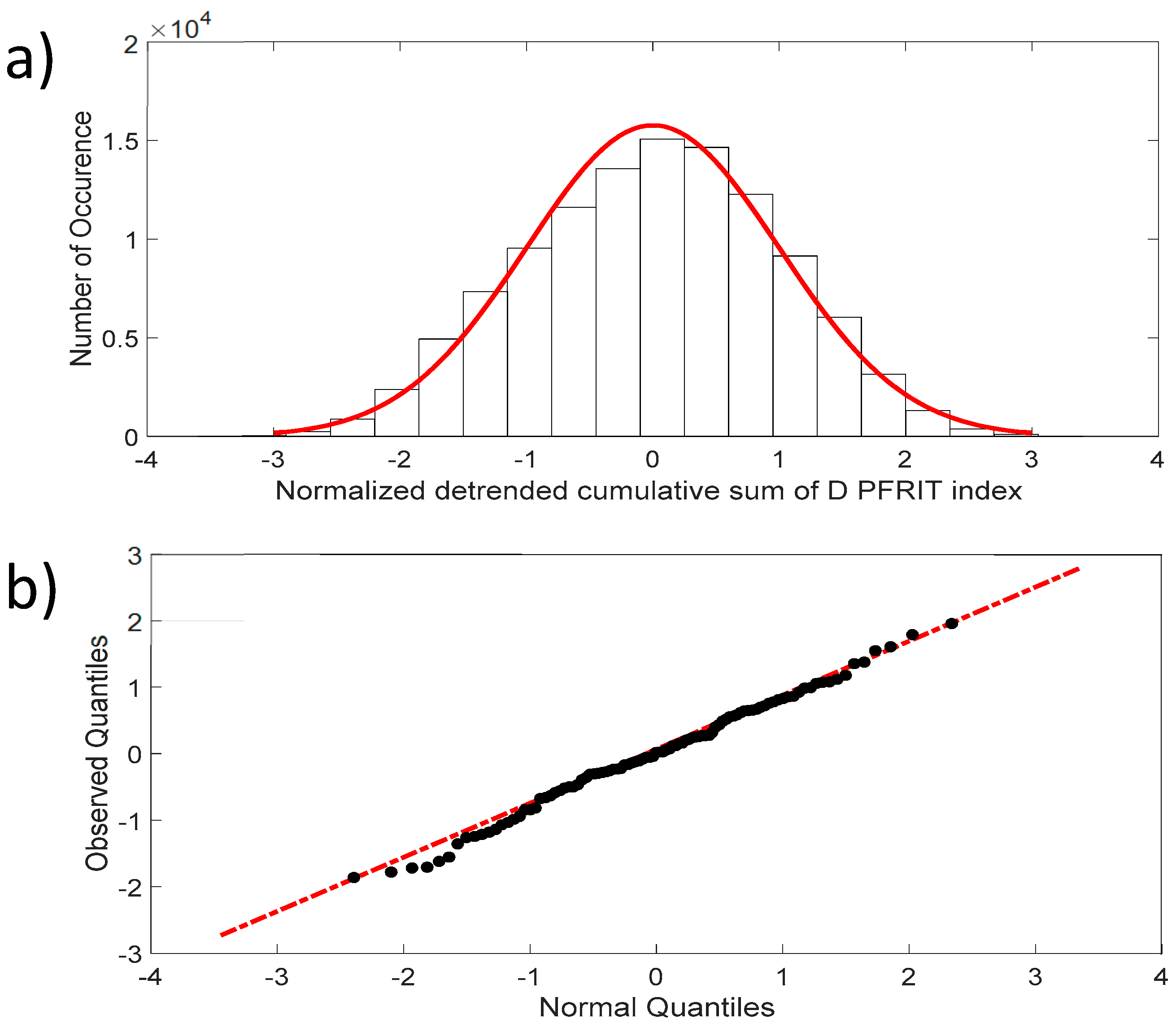
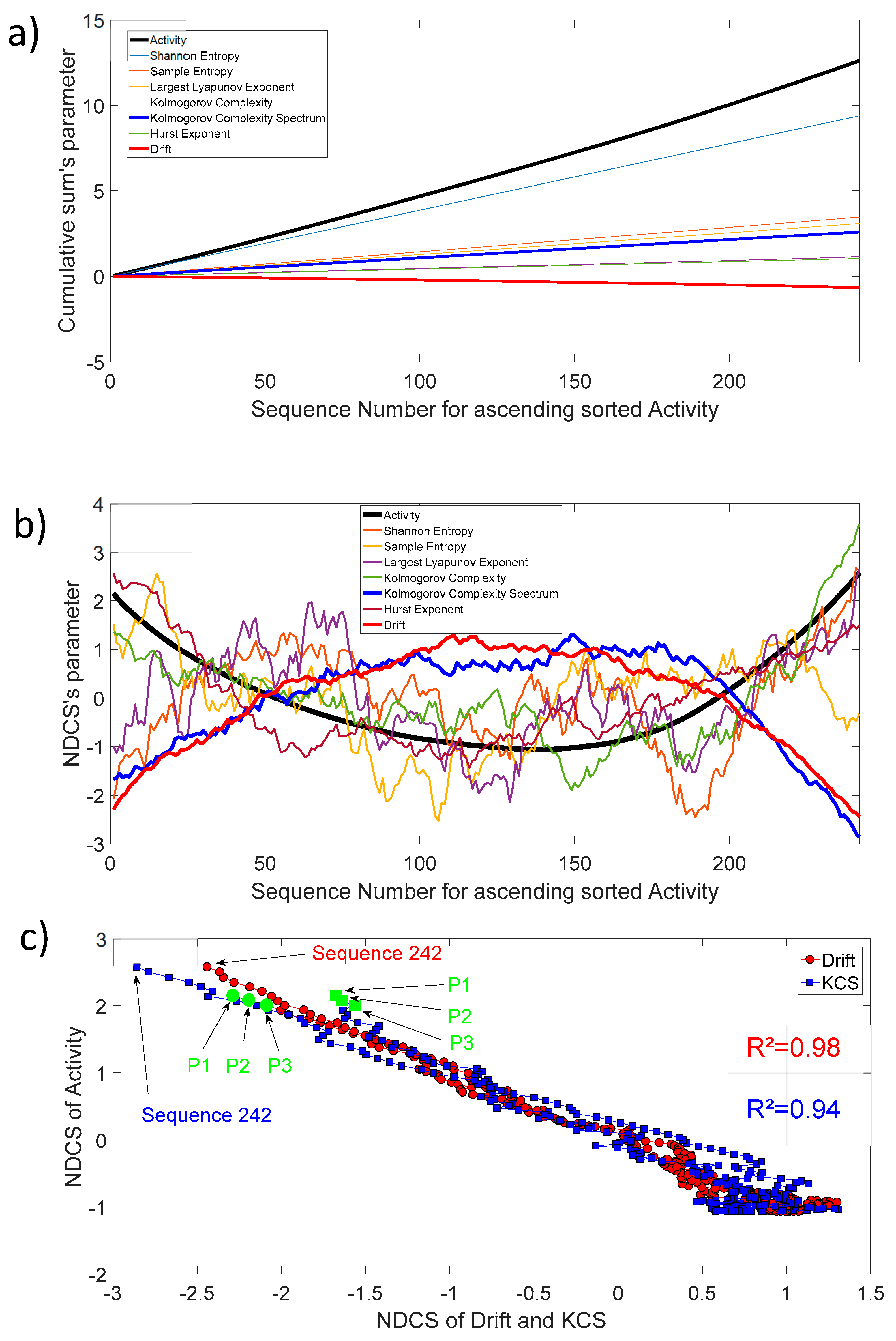
4. Normalized Detrended Cumulative Sum (NDCS) Method
5. Results and Discussion
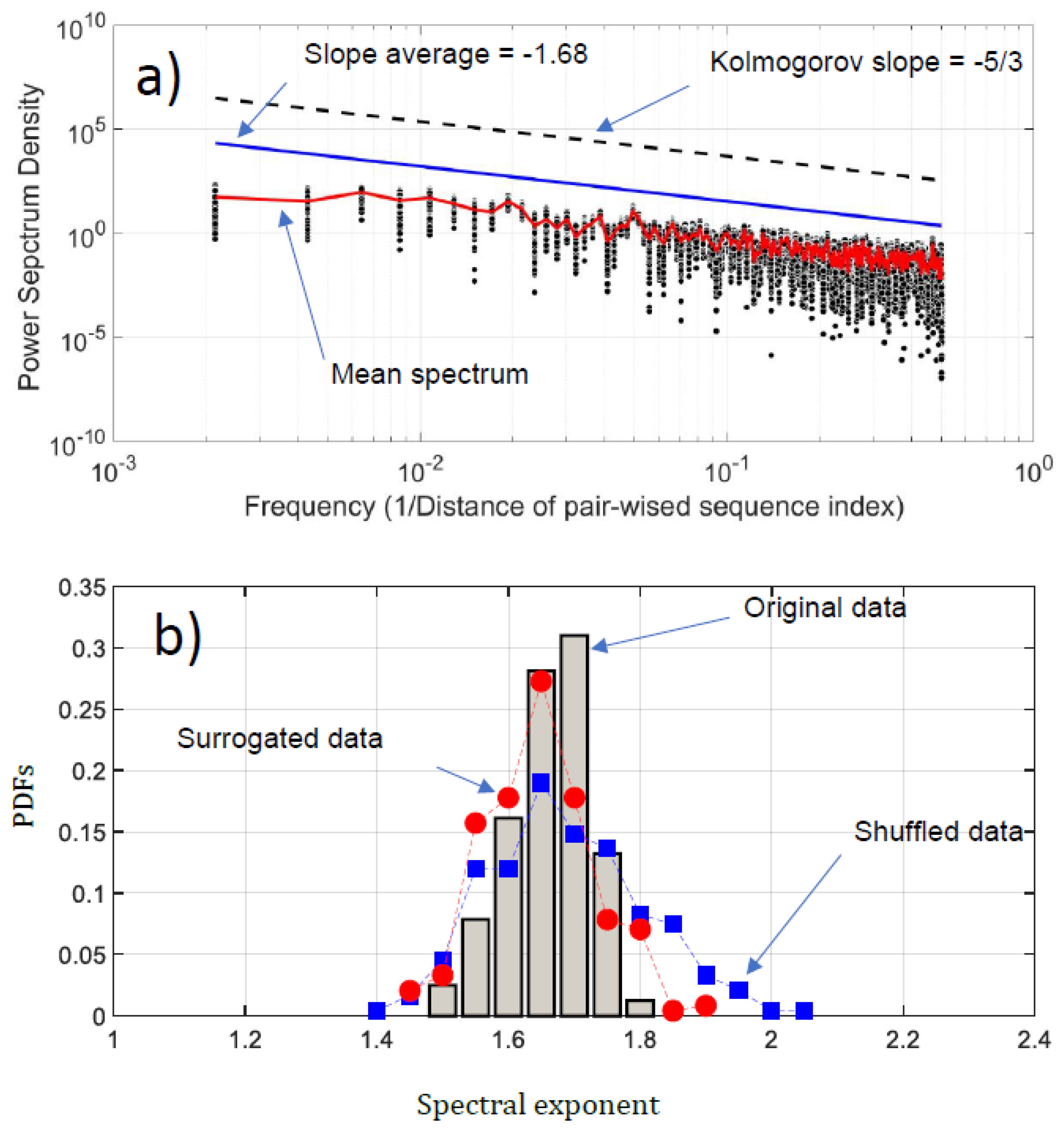
5.1. Normality and Intermittency
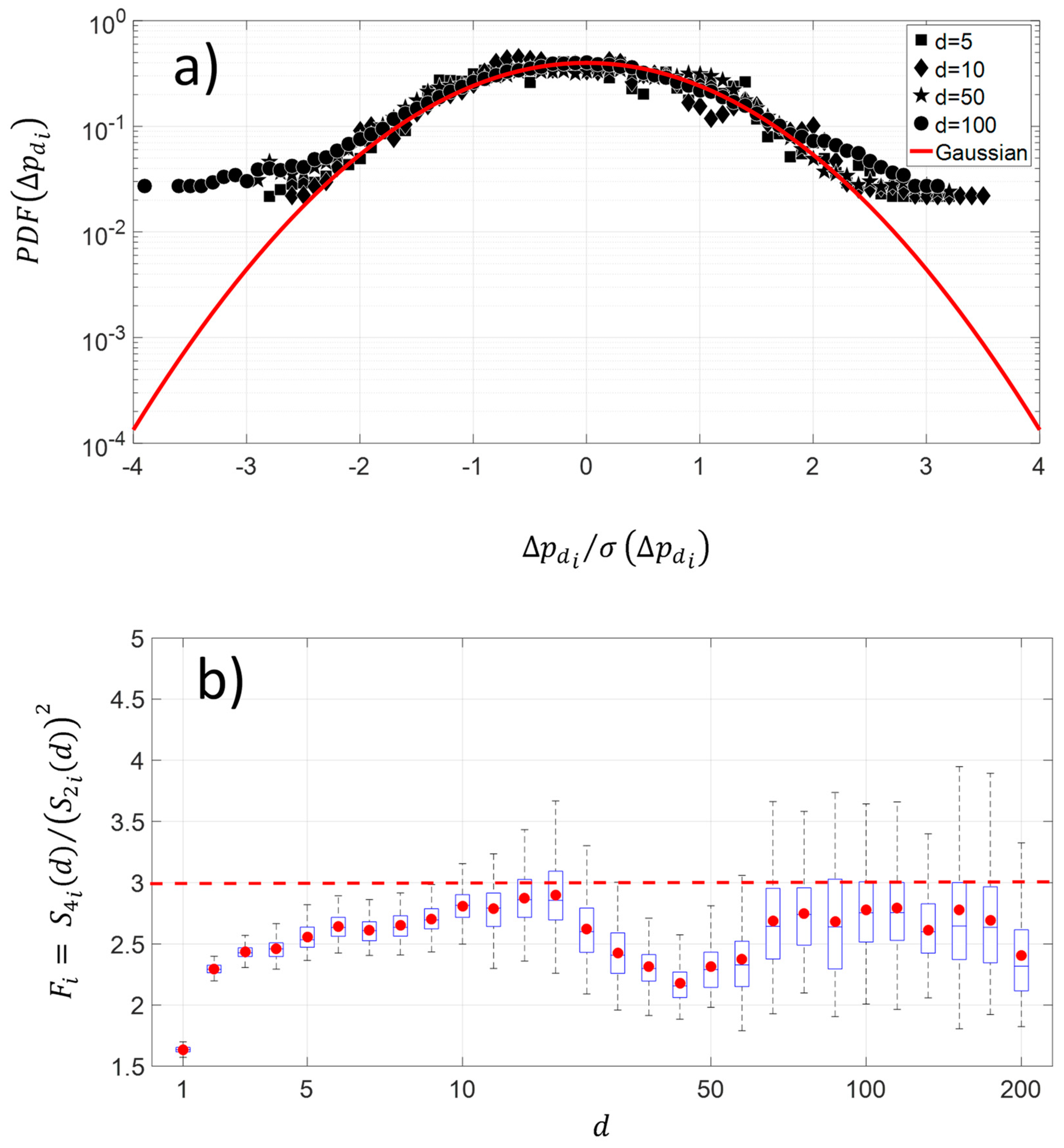
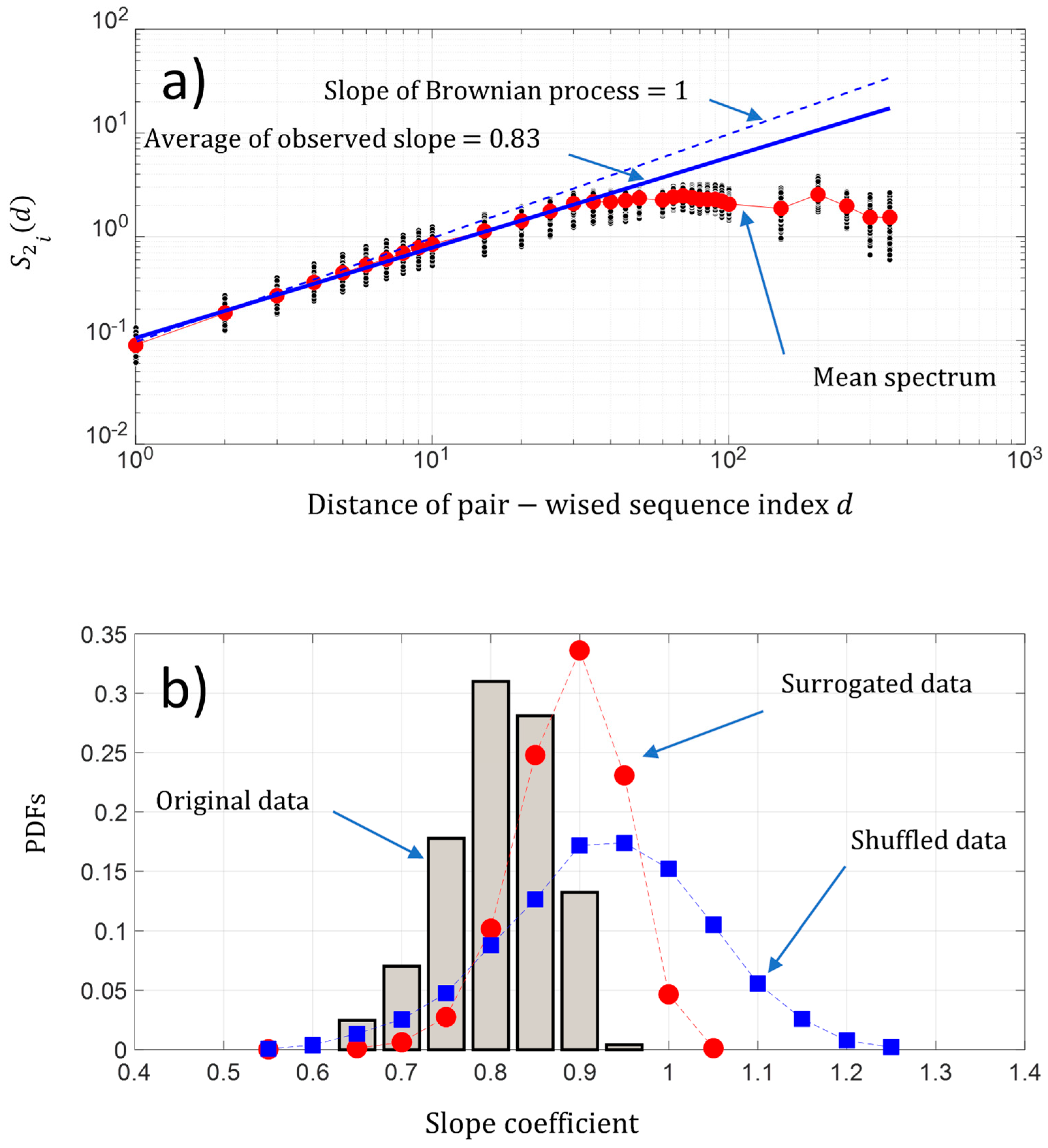
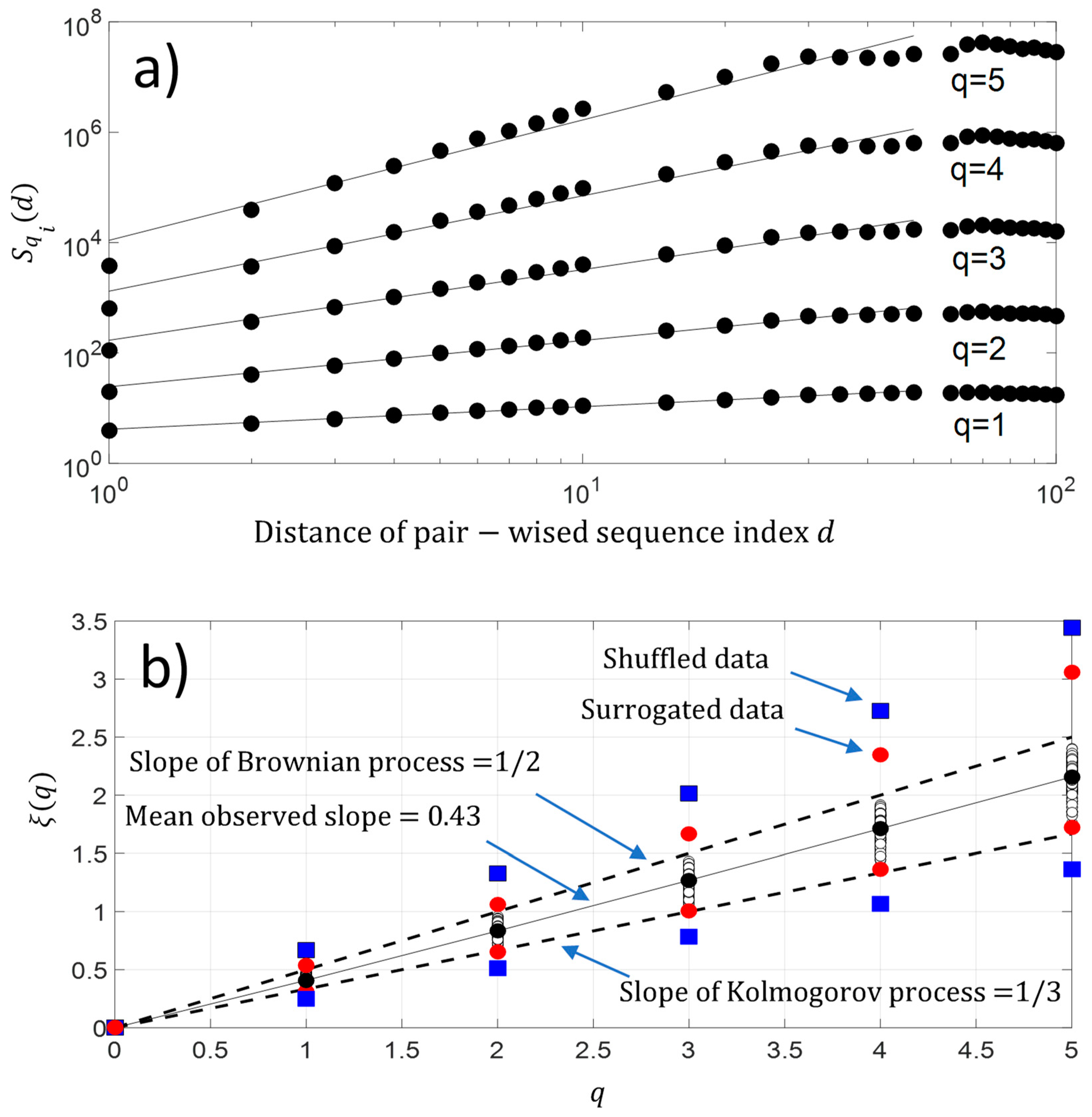
5.2. Kolmogorov’s Law and Brownian Process
- , the changes are stationary,
- , the changes are non-stationary,
- , the changes are non-stationary with stationary increments.
5.3. Entropy, Chaos, and Complexity
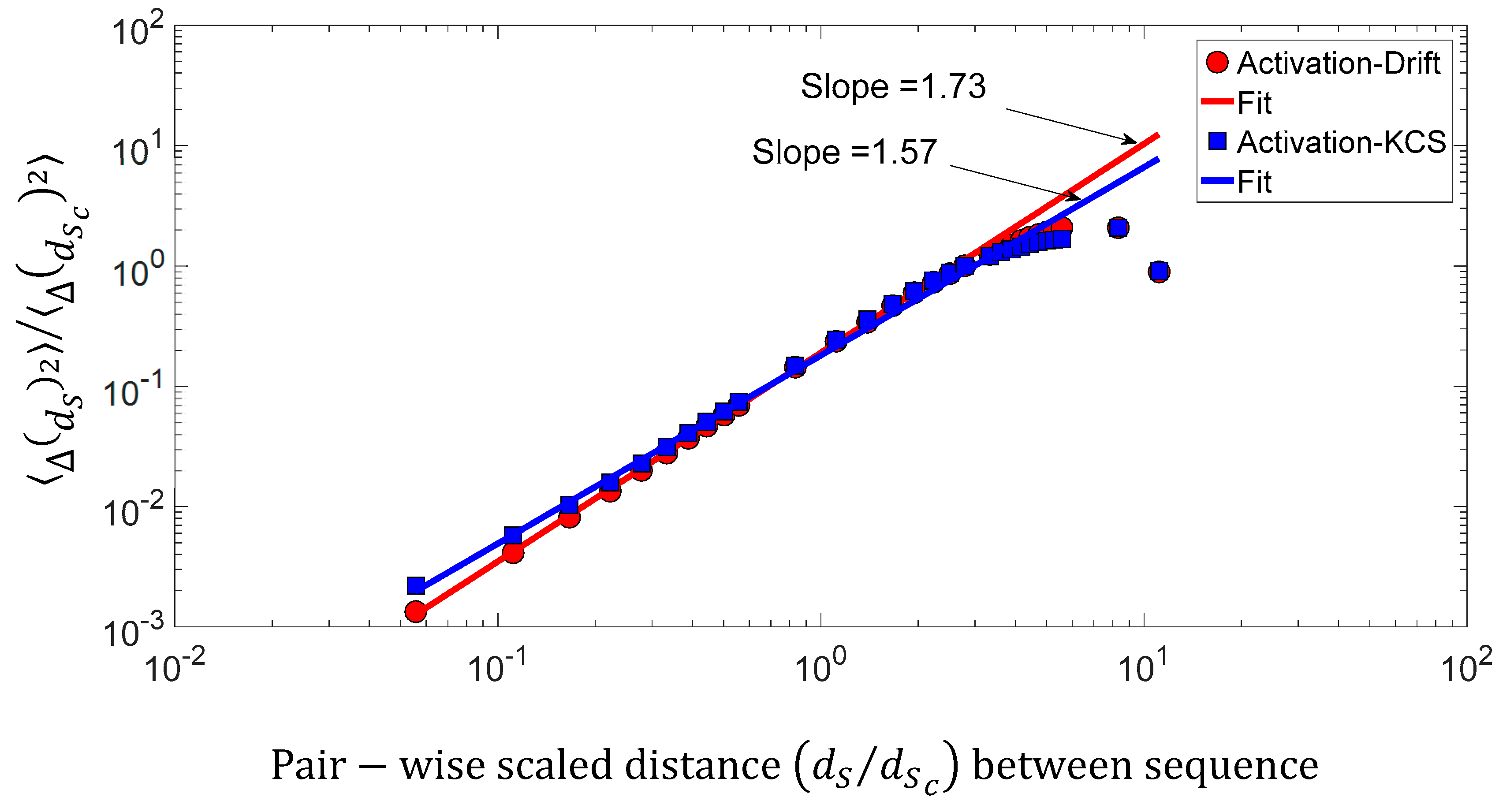
5.4. Drift (), Kolmogorov Complexity Spectrum (), and Activity (): Linear Correlation and Superdiffusive Process between Sequences
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanson, J.; Yang, Y.; Paliwal, K.; Zhou, Y. Improving protein disorder prediction by deep bidirectional long short-term memory recurrent neural networks. Bioinformatics 2016. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, A.; Telegdy, G. Modulation of active avoidance behavior of rats by ICV administration of CGRP antiserum. Peptides 1994, 15, 893–895. [Google Scholar] [CrossRef]
- Niessen, K.A.; Xu, M.; George, D.K.; Chen, M.C.; Ferré-D’Amaré, A.R.; Snell, E.H.; Cody, V.; Pace, J.; Schmidt, M.; Markelz, A.G. Protein and RNA dynamical fingerprinting. Nat. Commun. 2019, 10, 1026. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.-H.; Jin, M.-Z.; Li, S.-L.; Feng, J. A protein mapping method based on physicochemical properties and dimension reduction. Comput. Biol. Med. 2015, 57, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gök, M.; Koçal, O.H.; Genç, S. Prediction of Disordered Regions in Proteins Using Physicochemical Properties of Amino Acids. Int. J. Pept. Res. Ther. 2016, 22, 31–36. [Google Scholar] [CrossRef]
- Wang, Y.; You, Z.H.; Yang, S.; Li, X.; Jiang, T.H.; Xi, Z.X. A High Efficient Biological Language Model for Predicting Protein–Protein Interactions. Cells 2019, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Plötz, T.; Fink, G.A. Pattern recognition methods for advanced stochastic protein sequence analysis using HMMs. Pattern Recognit. 2006, 39, 2267–2280. [Google Scholar] [CrossRef]
- Chattopadhyay, A.K.; Nasiev, D.; Flower, D.R. A statistical physics perspective on alignment-independent protein sequence comparison. Bioinformatics 2015, 31, 2469–2474. [Google Scholar] [CrossRef] [PubMed]
- Vinga, S. Information theory applications for biological sequence analysis. Brief. Bioinform. 2014, 15, 376–389. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, J.; Hua, W.; Ouyang, P. Algorithm, applications and evaluation for protein comparison by Ramanujan Fourier transform. Mol. Cell. Probes 2015, 29, 396–407. [Google Scholar] [CrossRef]
- Czerniecka, A.; Bielińska-Wąż, D.; Wąż, P.; Clark, T. 20D-dynamic representation of protein sequences. Genomics 2016, 107, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-J.; Feng, C.-Q.; Lai, H.-Y.; Chen, W.; Hao, L. Predicting protein structural classes for low-similarity sequences by evaluating different features. Knowl. Based Syst. 2019, 163, 787–793. [Google Scholar] [CrossRef]
- Yang, L.; Wei, P.; Zhong, C.; Meng, Z.; Wang, P.; Tang, Y.Y. A Fractal Dimension and Empirical Mode Decomposition-Based Method for Protein Sequence Analysis. Int. J. Pattern Recognit. Artif. Intell. 2019. [Google Scholar] [CrossRef]
- Yu, J.F.; Cao, Z.; Yang, Y.; Wang, C.L.; Su, Z.D.; Zhao, Y.W.; Wang, J.H.; Zhou, Y. Natural protein sequences are more intrinsically disordered than random sequences. Cell. Mol. Life Sci. 2016, 73, 2949–2957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, C.; Liu, F.; Tan, H.; Song, D.; Shu, W.; Li, W.; Zhou, Y.; Bo, X.; Xie, Z. Deep Learning and Its Applications in Biomedicine. Genom. Proteom. Bioinform. 2018, 16, 17–32. [Google Scholar] [CrossRef]
- Li, Y.; Drummond, D.A.; Sawayama, A.M.; Snow, C.D.; Bloom, J.D.; Arnold, F.H. A diverse family of thermostable cytochrome P450s created by recombination of stabilizing fragments. Nat. Biotechnol. 2007, 25, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Ogata, H.; Kanehisa, M. Aaindex: Amino Acid Index Database. Nucleic Acids Res. 1999, 27, 368–369. [Google Scholar] [CrossRef]
- Kawashima, S.; Pokarowski, P.; Pokarowska, M.; Kolinski, A.; Katayama, T.; Kanehisa, M. Aaindex: Amino acid index database, progress report 2008. Nucleic Acids Res. 2008, 36, D202–D205. [Google Scholar] [CrossRef]
- Shannon, C.E. A Mathematical theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Richman, J.S.; Moorman, J.R. Physiological time-series analysis using approximate entropy and sample entropy. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H2039–H2049. [Google Scholar] [CrossRef] [Green Version]
- Wolf, A.; Swift, J.B.; Swinney, H.L.; Vastano, J.A. Determining Lyapunov exponents from a time series. Phys. Nonlinear Phenom. 1985, 16, 285–317. [Google Scholar] [CrossRef] [Green Version]
- Kolmogorov, A.N. The local structure of turbulence in incompressible fluid for very large Reynolds numbers. Dokl. Akad. Nauk. SSSR 1941, 30, 299–303. [Google Scholar] [CrossRef]
- Chaitin, G.J. On the Length of Programs for Computing Finite Binary Sequences: Statistical considerations. J. ACM 1969, 16, 145–159. [Google Scholar] [CrossRef]
- Lempel, A.; Ziv, J. On the Complexity of Finite Sequences. IEEE Trans. Inf. Theory 1976, 22, 75–81. [Google Scholar] [CrossRef]
- Mihailović, D.T.; Mimić, G.; Nikolić-Djorić, E.; Arsenić, I. Novel measures based on the Kolmogorov complexity for use in complex system behavior studies and time series analysis. Open Phys. 2015, 13, 1–14. [Google Scholar] [CrossRef]
- Ziv, J.; Lempel, A. Compression of individual sequences via variable-rate coding. IEEE Trans. Inf. Theory 1978, 24, 530–536. [Google Scholar] [CrossRef]
- Monin, A.S.; Yaglom, A.M. Statistical Fluid Mechanics: Mechanics of Turbulence; MIT Press: Cambridge, MA, USA, 1987; Volume 1, p. 784. [Google Scholar]
- Schreiber, T.; Schmitz, A. Surrogate time series. Phys. Nonlinear Phenom. 2000, 142, 346–382. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.; Buldyrev, S.V.; Havlin, S.; Simons, M.; Stanley, H.E.; Goldberger, A.L. Mosaic organization of DNA nucleotides. Phys. Rev. E 1994, 49, 1685–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornette, J.L.; Cease, K.B.; Margalit, H.; Spouge, J.L.; Berzofsky, J.A.; DeLisi, C. Hydrophobicity scales and computational techniques for detecting amphipathic structures in proteins. J. Mol. Biol. 1987, 195, 659–685. [Google Scholar] [CrossRef]
- Regier, P.; Briceño, H.; Boyer, J.N. Analyzing and comparing complex environmental time series using a cumulative sums approach. MethodsX 2019, 6, 779–787. [Google Scholar] [CrossRef]
- Marshak, A.; Davis, A.; Cahalan, R.; Wiscombe, W. Bounded cascade models as nonstationary multifractals. Phys. Rev. E 1994, 49, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Richardson, L.F. Atmospheric Diffusion Shown on a Distance-Neighbour Graph. Proc. R. Soc. Math. Phys. Eng. Sci. 1926, 110, 709–737. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
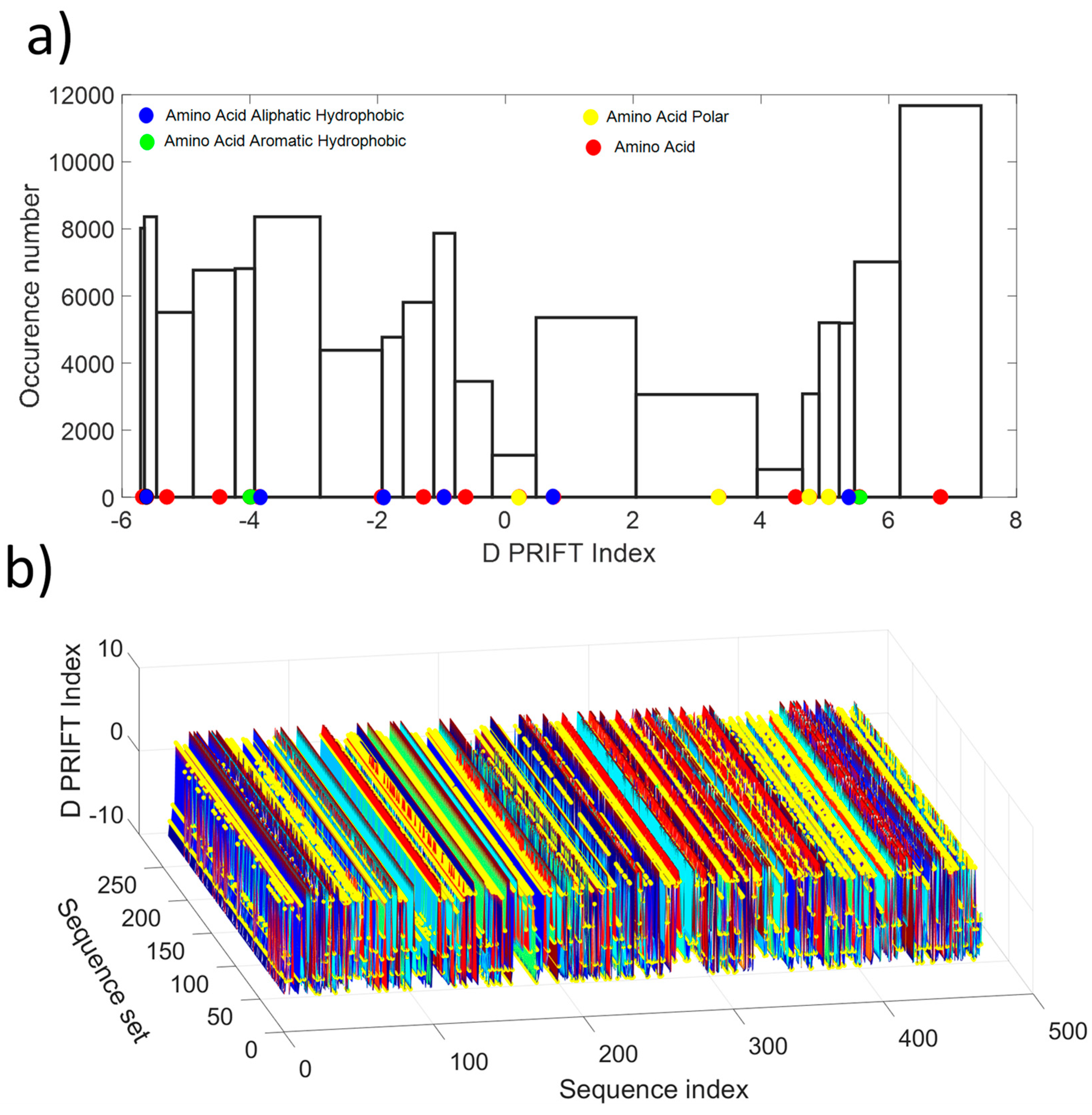
| AA Index 532 D PRIFT Index (Cornette et al. 1987) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Letter | A | C | D | E | F | G | H | I | K | L | M | N | P | Q | R | S | T | V | W | Y |
| Value Index | −5.68 | −5.62 | −5.30 | −4.47 | −3.99 | −3.86 | −1.94 | −1.92 | −1.28 | 0.96 | 0.62 | 0.21 | 0.75 | 3.34 | 4.54 | 4.76 | 5.06 | 5.39 | 5.54 | 6.81 |
| D PRIFT Index | Entropy | Chaos | Complexity | Fractal | |||
|---|---|---|---|---|---|---|---|
| Information | Regularity | ||||||
| NDCS Data | Shannon Entropy | Sample Entropy | Largest Lyapunov Exponent | Kolmogorov Complexity | Kolmogorov Complexity Spectrum | Hurst Exponent | |
| Minimum | Original | 3.671 | 1.251 | 0.930 | 0.247 | 1.008 | 0.347 |
| Surrogate | 3.514 | 1.051 | 0.730 | 0.152 | 1.046 | 0.332 | |
| Shuffled | 3.498 | 0.600 | 0.332 | 0.095 | 1.046 | 0.273 | |
| Mean | Original | 3.880 | 1.433 | 1.277 | 0.475 | 1.071 | 0.432 |
| Surrogate | 3.875 | 1.289 | 1.070 | 0.399 | 1.105 | 0.481 | |
| Shuffled | 3.911 | 1.147 | 0.911 | 0.328 | 1.103 | 0.498 | |
| Median | Original | 3.888 | 1.436 | 1.286 | 0.475 | 1.065 | 0.436 |
| Surrogate | 3.895 | 1.296 | 1.072 | 0.399 | 1.103 | 0.482 | |
| Shuffled | 3.933 | 1.154 | 0.906 | 0.323 | 1.103 | 0.498 | |
| Maximum | Original | 4.066 | 1.618 | 1.601 | 0.647 | 1.141 | 0.481 |
| Surrogate | 4.131 | 1.547 | 1.501 | 0.646 | 1.179 | 0.615 | |
| Shuffled | 4.188 | 1.604 | 1.469 | 0.627 | 1.160 | 0.690 | |
| Standard deviation | Original | 0.084 | 0.063 | 0.117 | 0.084 | 0.031 | 0.027 |
| Surrogate | 0.117 | 0.094 | 0.143 | 0.081 | 0.023 | 0.033 | |
| Shuffled | 0.130 | 0.188 | 0.220 | 0.109 | 0.022 | 0.058 | |
| 1st quartile | Original | 3.833 | 1.389 | 1.207 | 0.418 | 1.046 | 0.420 |
| Surrogate | 3.805 | 1.226 | 0.969 | 0.342 | 1.084 | 0.459 | |
| Shuffled | 3.842 | 1.017 | 0.750 | 0.228 | 1.084 | 0.459 | |
| 3rd quartile | Original | 3.940 | 1.470 | 1.351 | 0.533 | 1.103 | 0.450 |
| Surrogate | 3.963 | 1.355 | 1.160 | 0.456 | 1.122 | 0.503 | |
| Shuffled | 4.005 | 1.297 | 1.045 | 0.399 | 1.122 | 0.538 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cadet, X.F.; Dehak, R.; Chin, S.P.; Bessafi, M. Non-Linear Dynamics Analysis of Protein Sequences. Application to CYP450. Entropy 2019, 21, 852. https://doi.org/10.3390/e21090852
Cadet XF, Dehak R, Chin SP, Bessafi M. Non-Linear Dynamics Analysis of Protein Sequences. Application to CYP450. Entropy. 2019; 21(9):852. https://doi.org/10.3390/e21090852
Chicago/Turabian StyleCadet, Xavier F., Reda Dehak, Sang Peter Chin, and Miloud Bessafi. 2019. "Non-Linear Dynamics Analysis of Protein Sequences. Application to CYP450" Entropy 21, no. 9: 852. https://doi.org/10.3390/e21090852
APA StyleCadet, X. F., Dehak, R., Chin, S. P., & Bessafi, M. (2019). Non-Linear Dynamics Analysis of Protein Sequences. Application to CYP450. Entropy, 21(9), 852. https://doi.org/10.3390/e21090852