Recent Synthetic Approaches Toward Non-anomeric Spiroketals in Natural Products


Abstract
:
Introduction

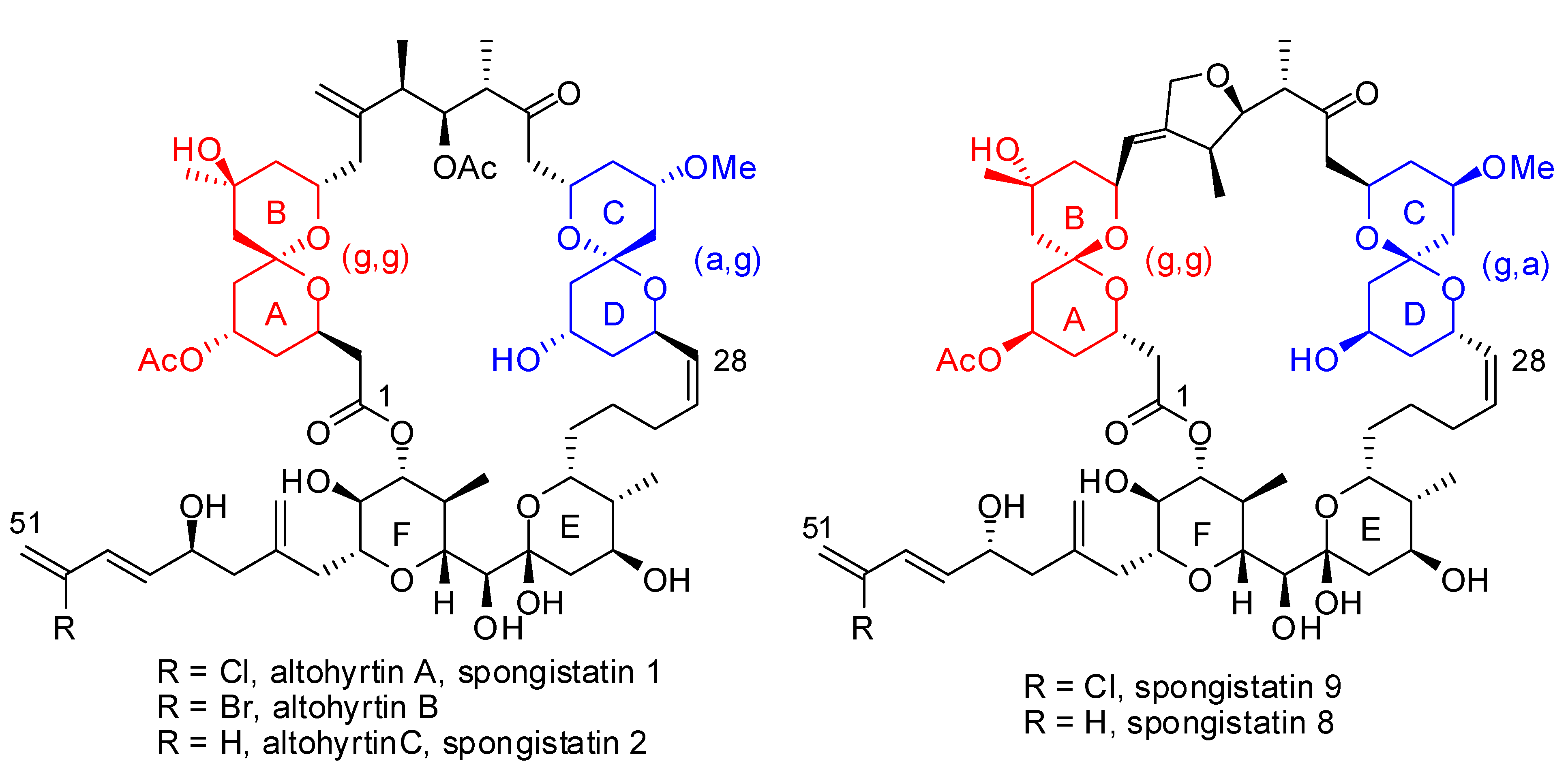
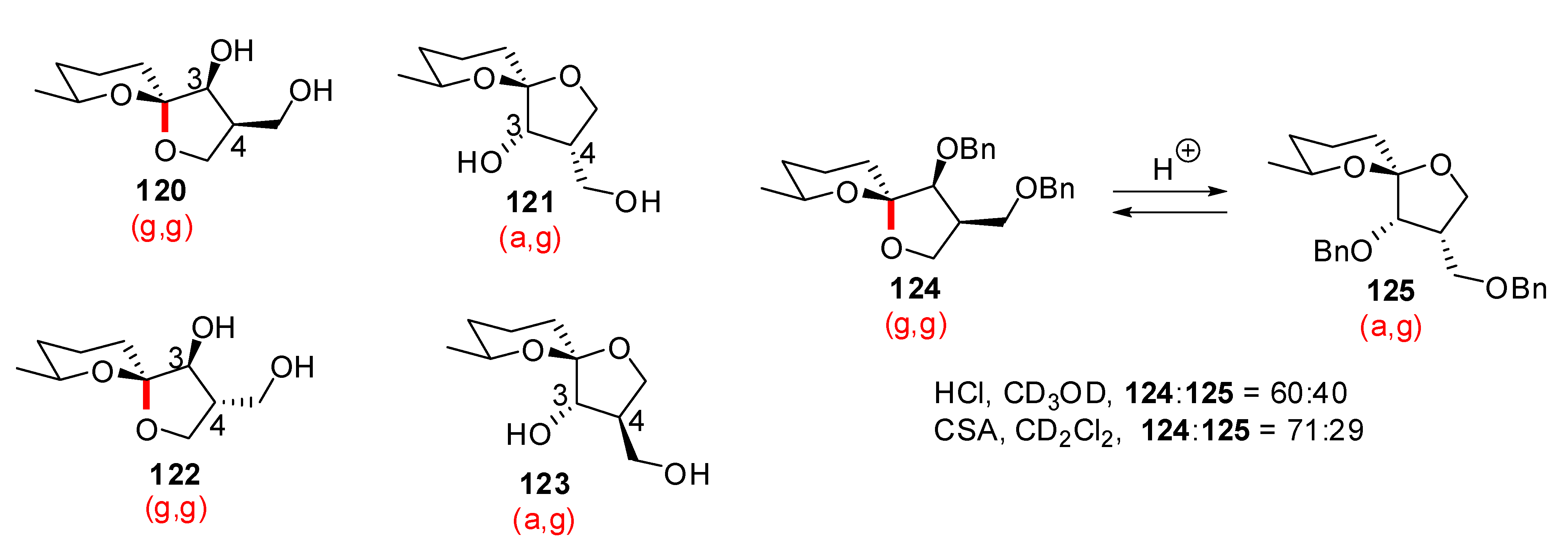
2. Relative stability of spiroketal stereoisomers and conformers


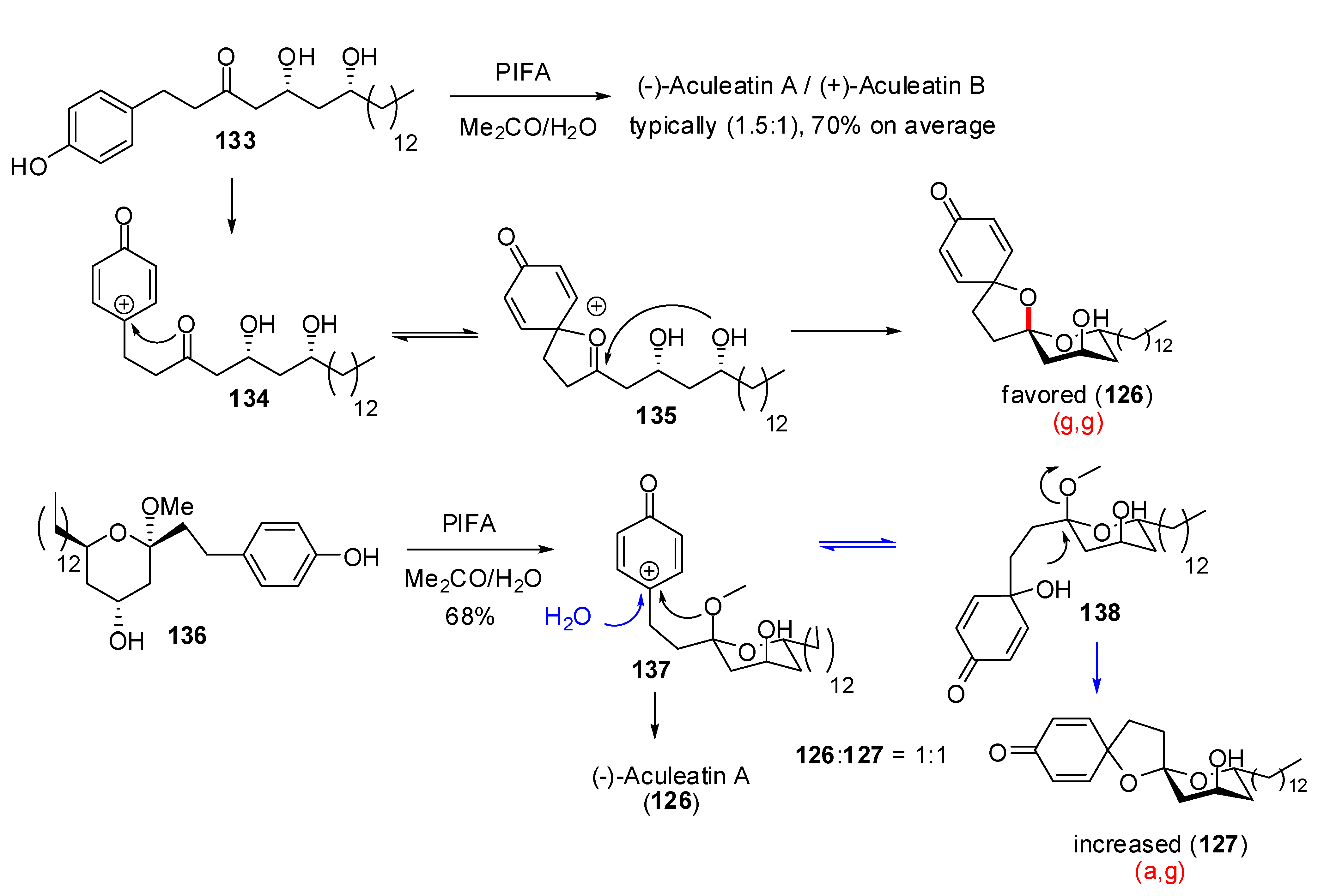
 ), (g,
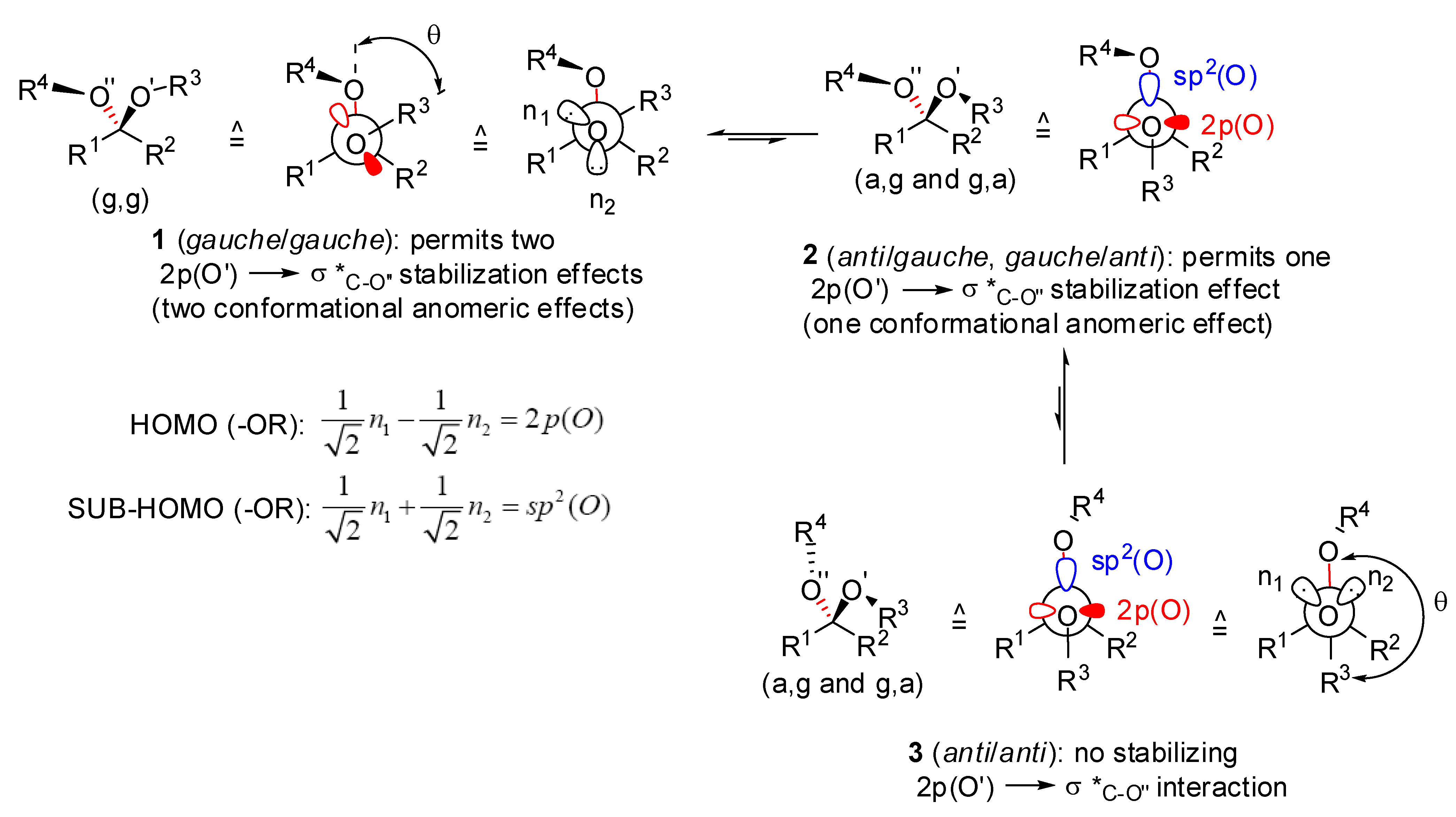
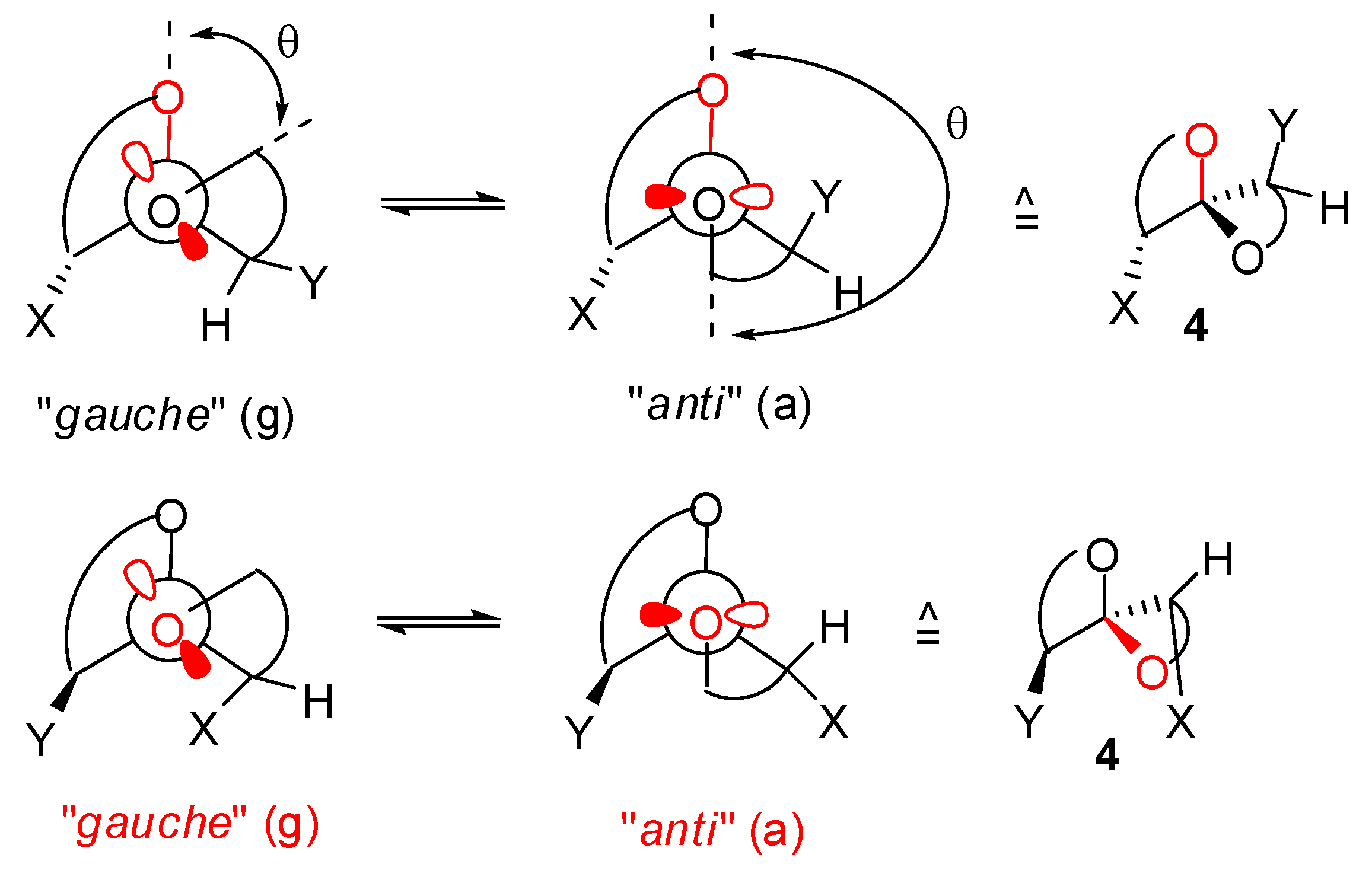
), (g,  ), (a, ) and (a, ) shown for 4 might have different relative stabilities. Ignoring differential solvation effects, steric factors, gauche effects, electrostatic effects and intramolecular hydrogen bridging due to substituents X and Y, one expects that (g, )-conformers are more stable than both (g, )- and (a, )-conformers, themselves expected to be more stable than the corresponding (a, )-conformers for stereoelectronic reasons (see above). In the cases of [5,6]- and [6,6]-spiroketals, the tetrahydropyran ring usually prefers chair conformations. Their interconversions are not degenerate upon substitution as equatorial substituents in one chair occupy axial positions (destabilizing gauche interactions) in the other. This might affect stability difference between (g,g), (g,a), (a,g) and (a,a)-conformers and favor one of the two possible diastereoisomeric spiroketals (4 vs 5), should they equilibrate by acetal epimerization (Figure 3). If substituents X and Y are part of a bridge in a tricyclic structure, any of the (g,g), (g,a), (a,a) or (a,a) conformations might be obtained by the adequate choice of the nature of the bridge and relative configuration of the substituted centers. Such bridging might also favor other conformations than chairs for their tetrahydropyran moieties.
), (a, ) and (a, ) shown for 4 might have different relative stabilities. Ignoring differential solvation effects, steric factors, gauche effects, electrostatic effects and intramolecular hydrogen bridging due to substituents X and Y, one expects that (g, )-conformers are more stable than both (g, )- and (a, )-conformers, themselves expected to be more stable than the corresponding (a, )-conformers for stereoelectronic reasons (see above). In the cases of [5,6]- and [6,6]-spiroketals, the tetrahydropyran ring usually prefers chair conformations. Their interconversions are not degenerate upon substitution as equatorial substituents in one chair occupy axial positions (destabilizing gauche interactions) in the other. This might affect stability difference between (g,g), (g,a), (a,g) and (a,a)-conformers and favor one of the two possible diastereoisomeric spiroketals (4 vs 5), should they equilibrate by acetal epimerization (Figure 3). If substituents X and Y are part of a bridge in a tricyclic structure, any of the (g,g), (g,a), (a,a) or (a,a) conformations might be obtained by the adequate choice of the nature of the bridge and relative configuration of the substituted centers. Such bridging might also favor other conformations than chairs for their tetrahydropyran moieties.
3. Synthesis of naturally occurring [6,6]-spiroketals
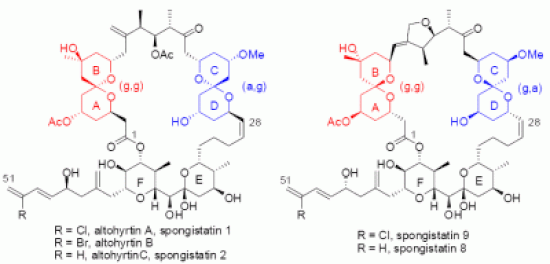
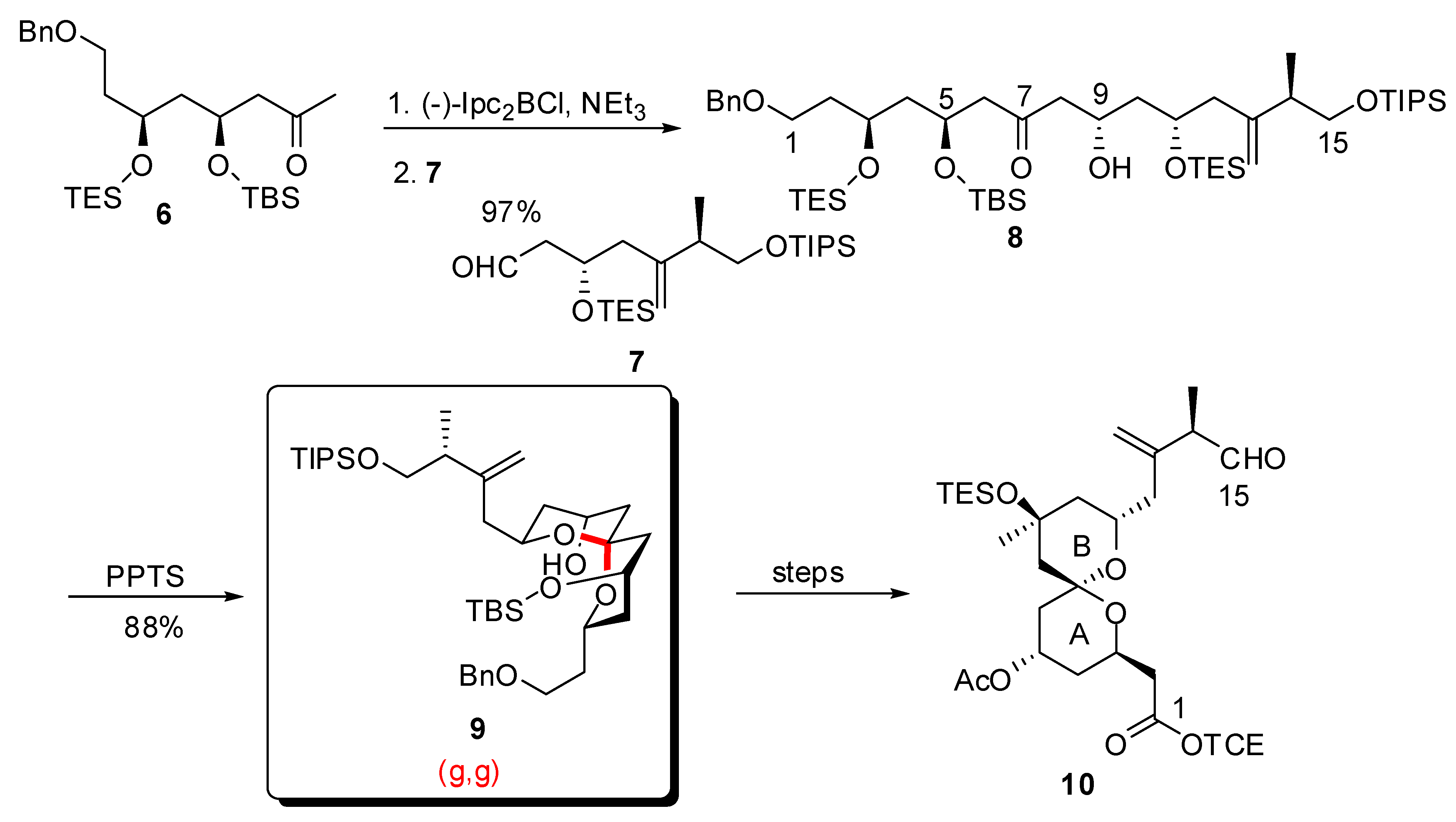
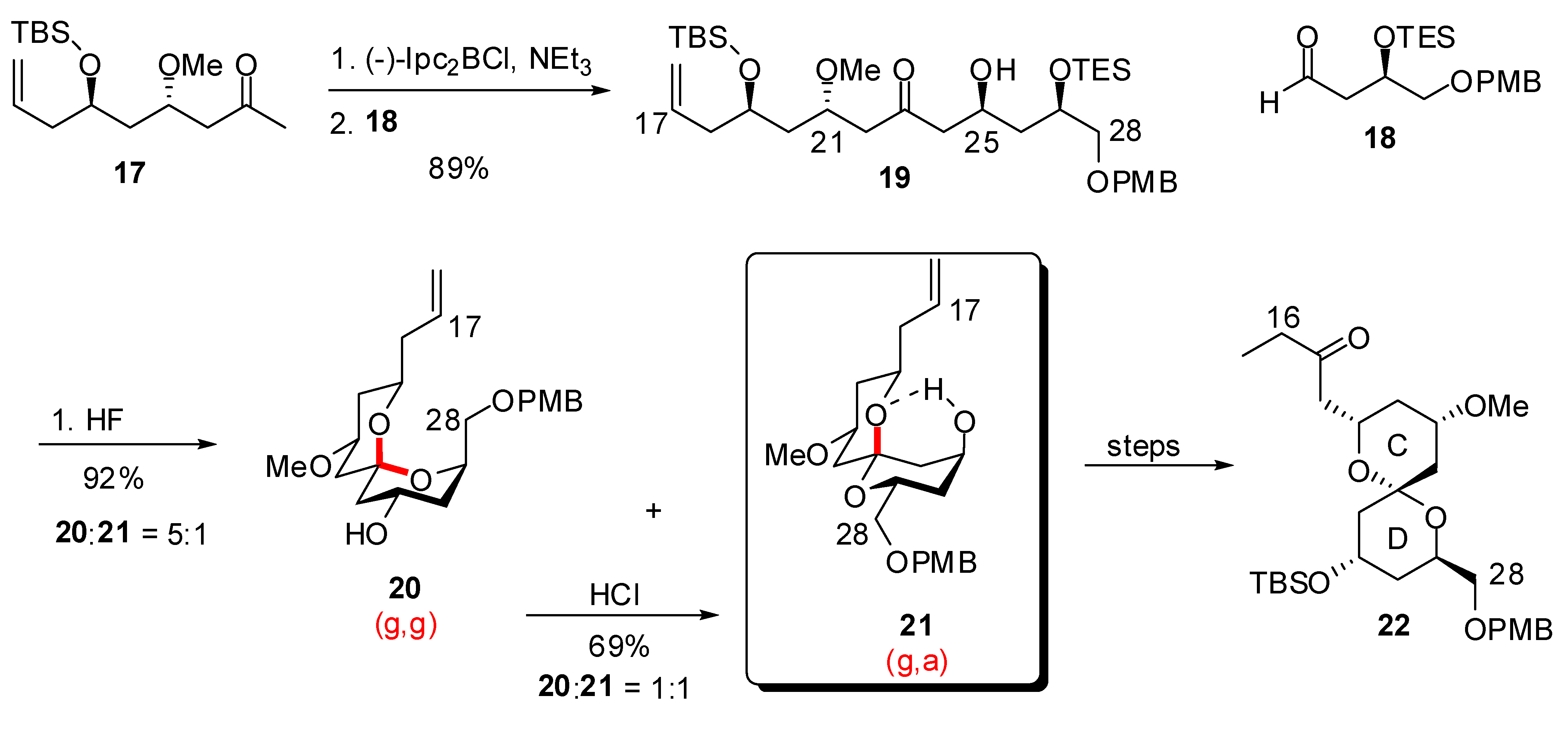
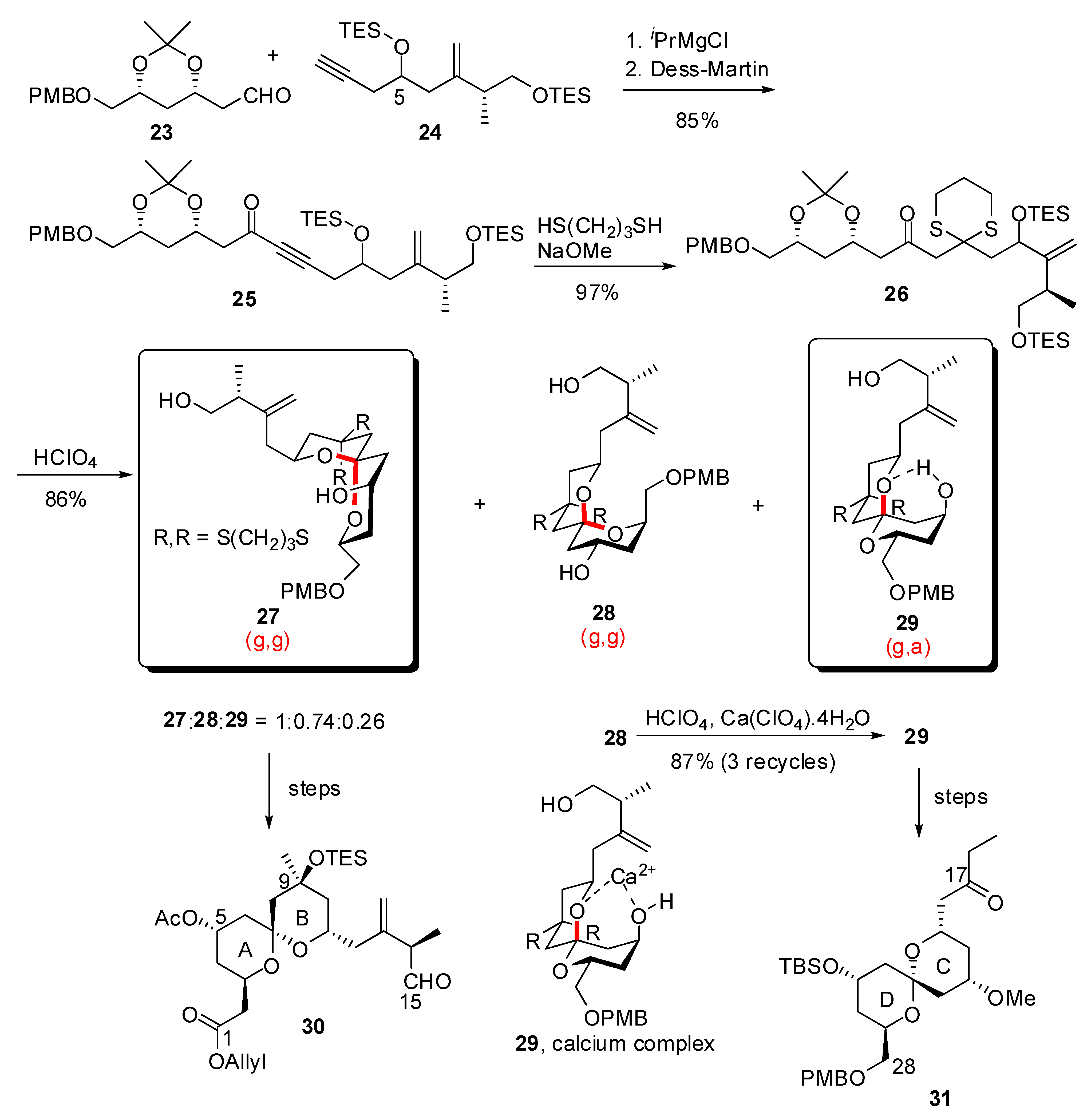
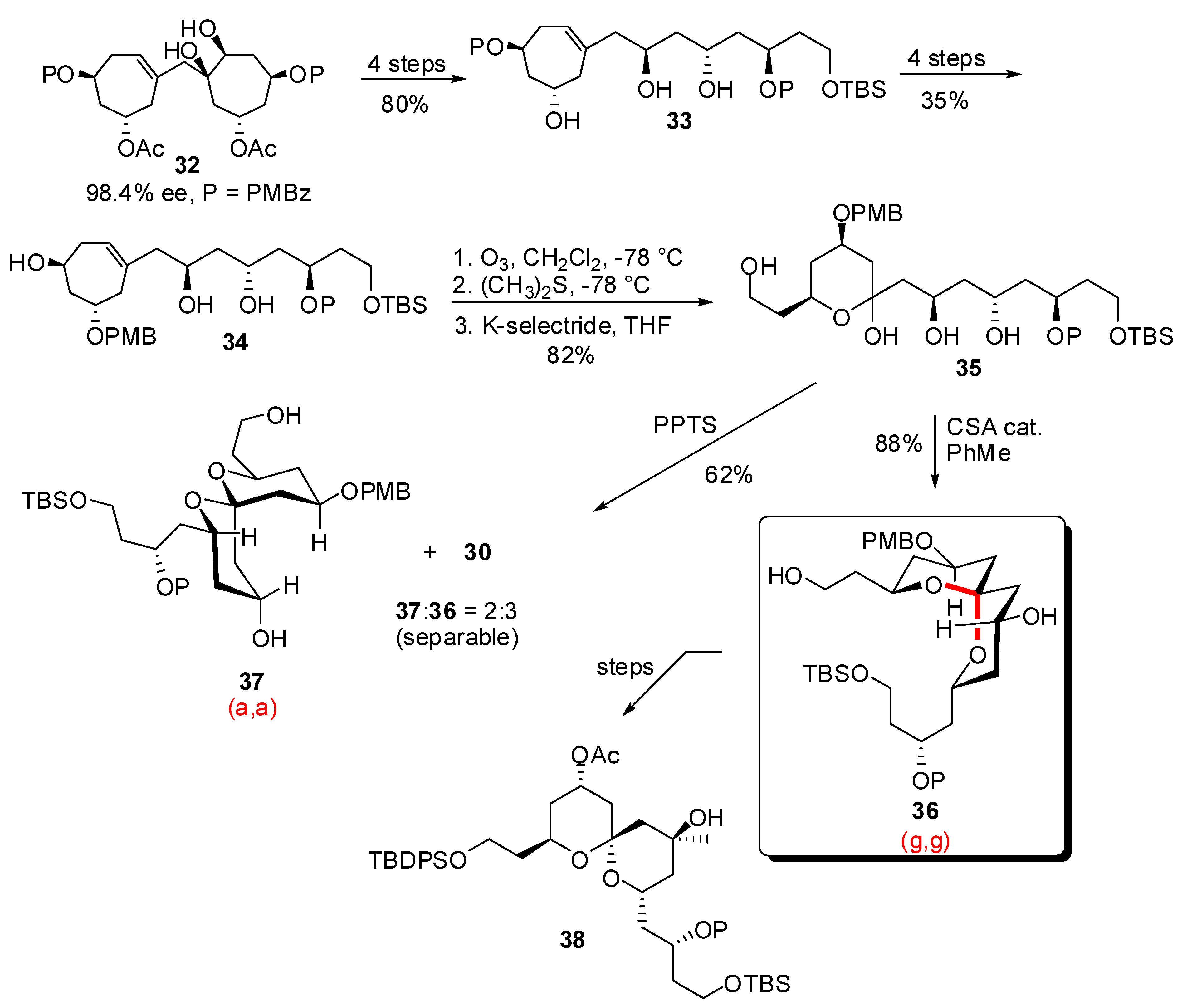
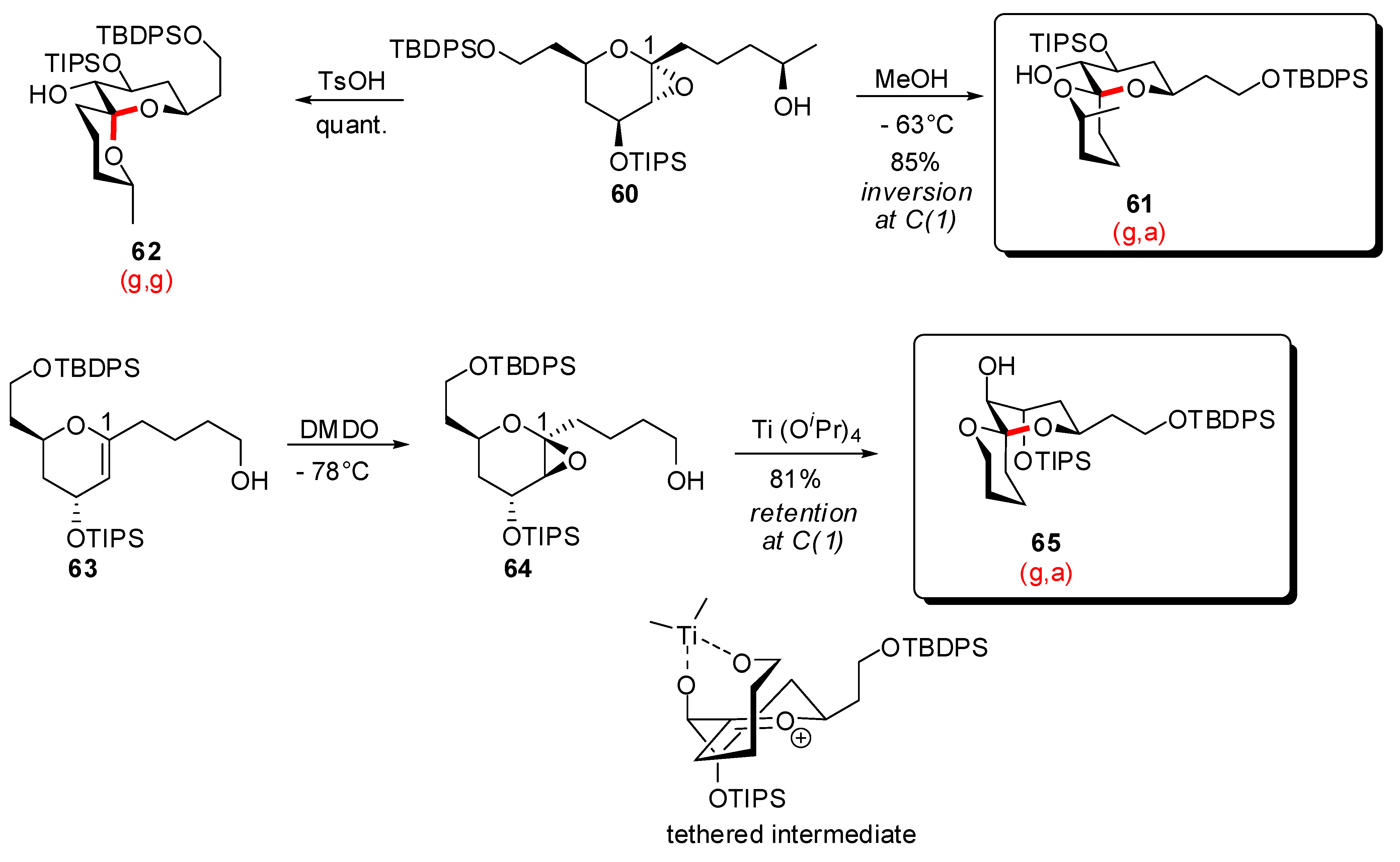
3.1. AB and CD spiroketals of spongistatins/altohyrtins






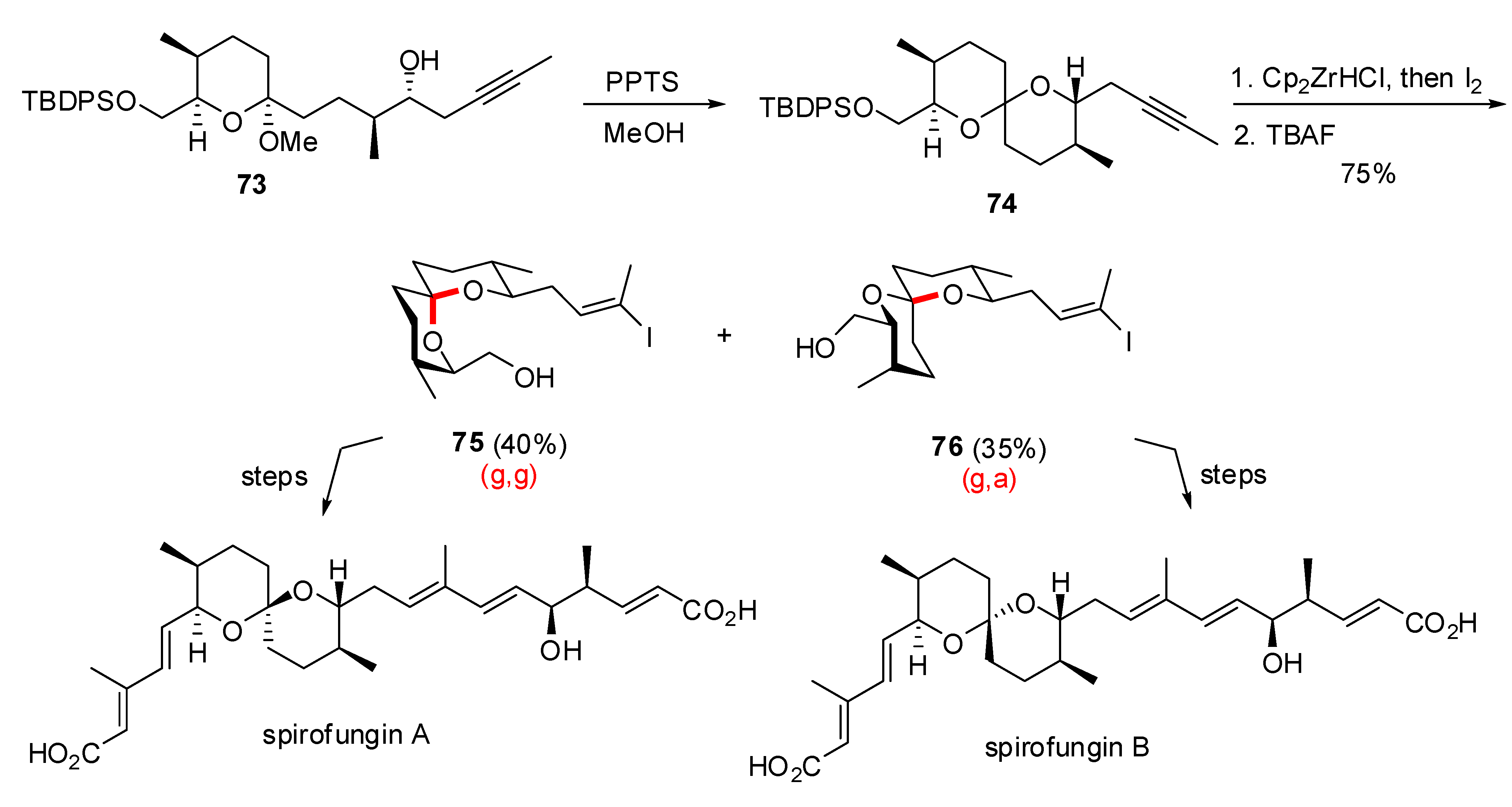
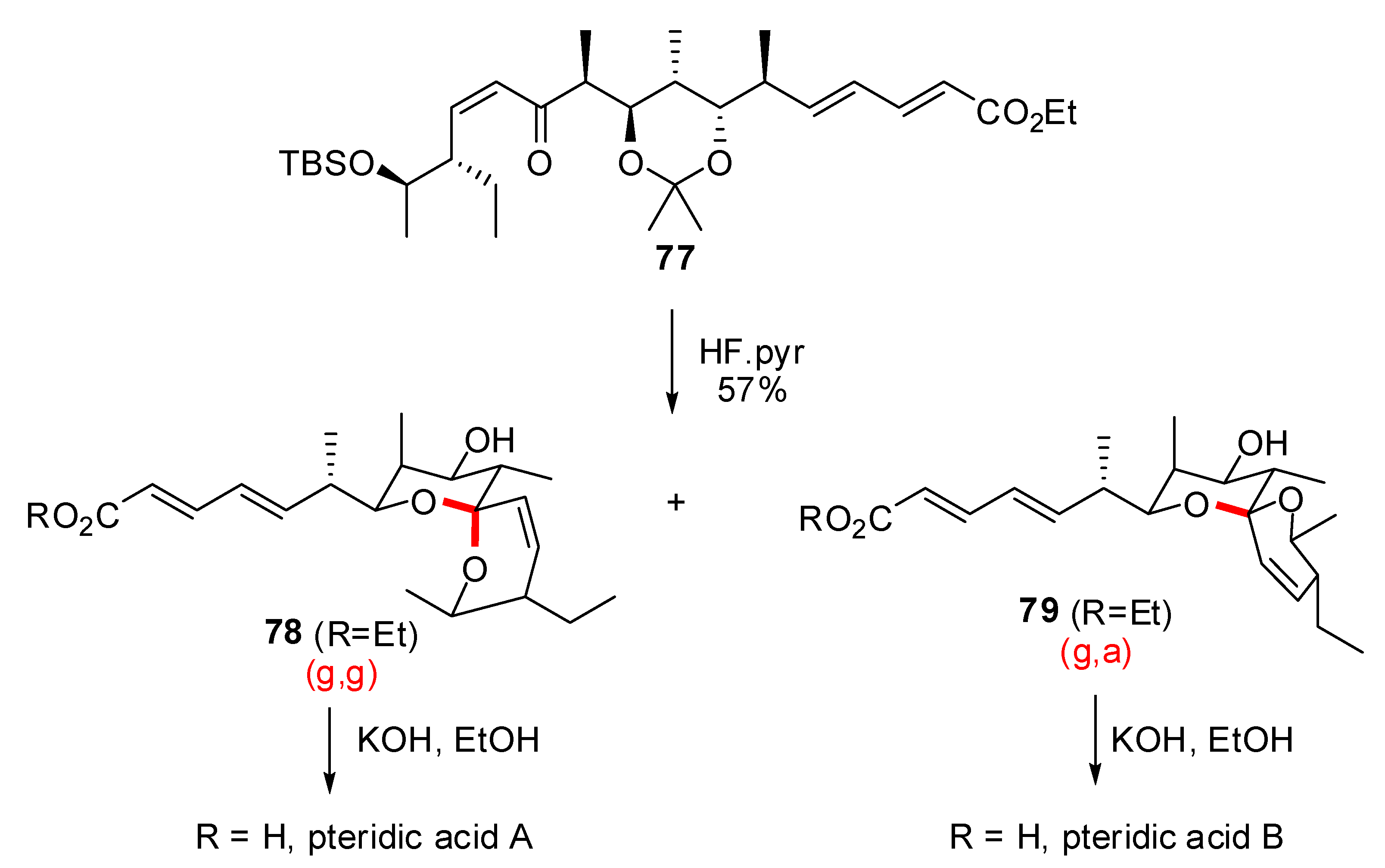
3.2. Synthesis of other nonanomeric [6,6]-spiroketals related to natural products








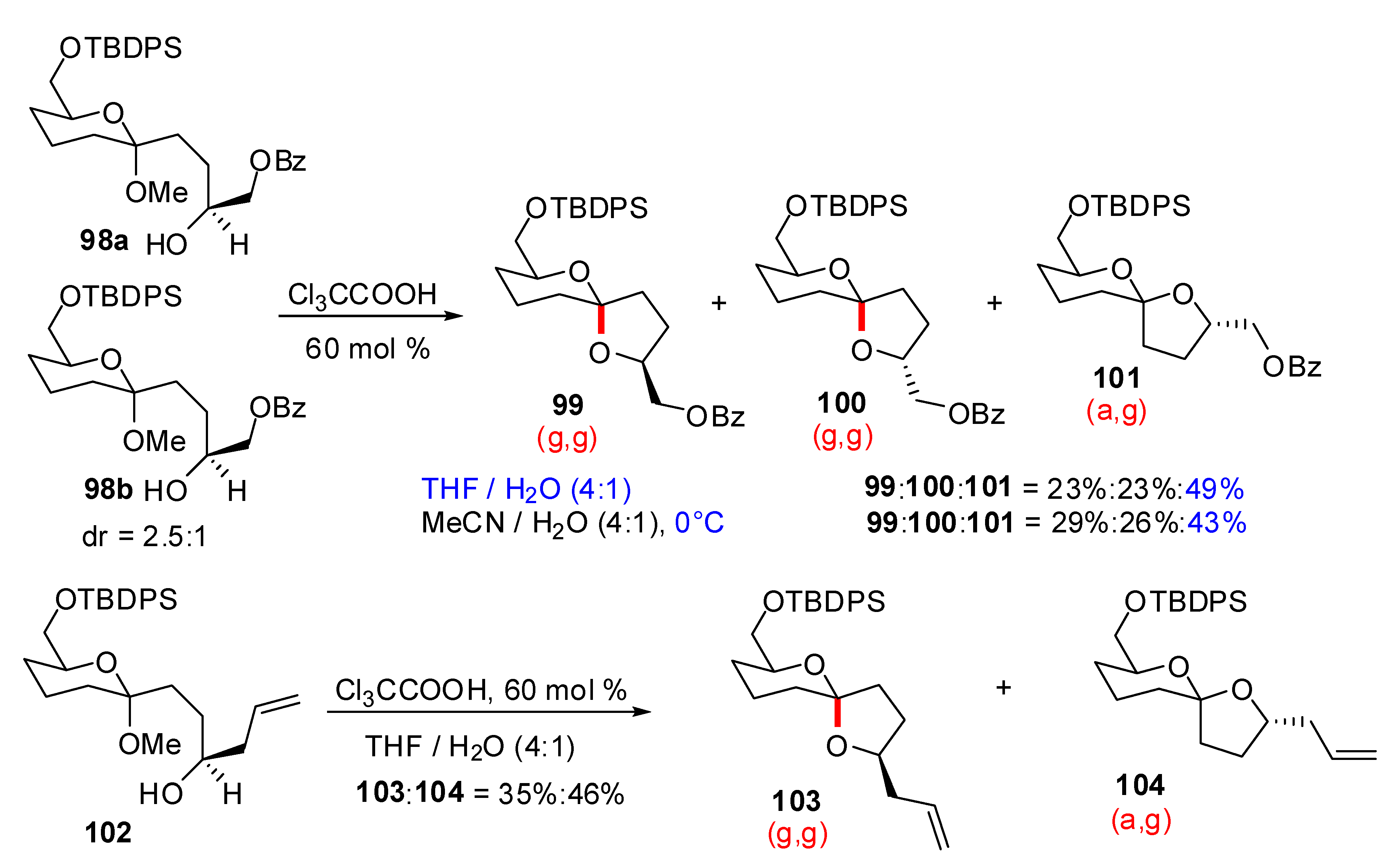
4. Synthesis of naturally occurring nonanomeric [6,5]-spiroketals













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
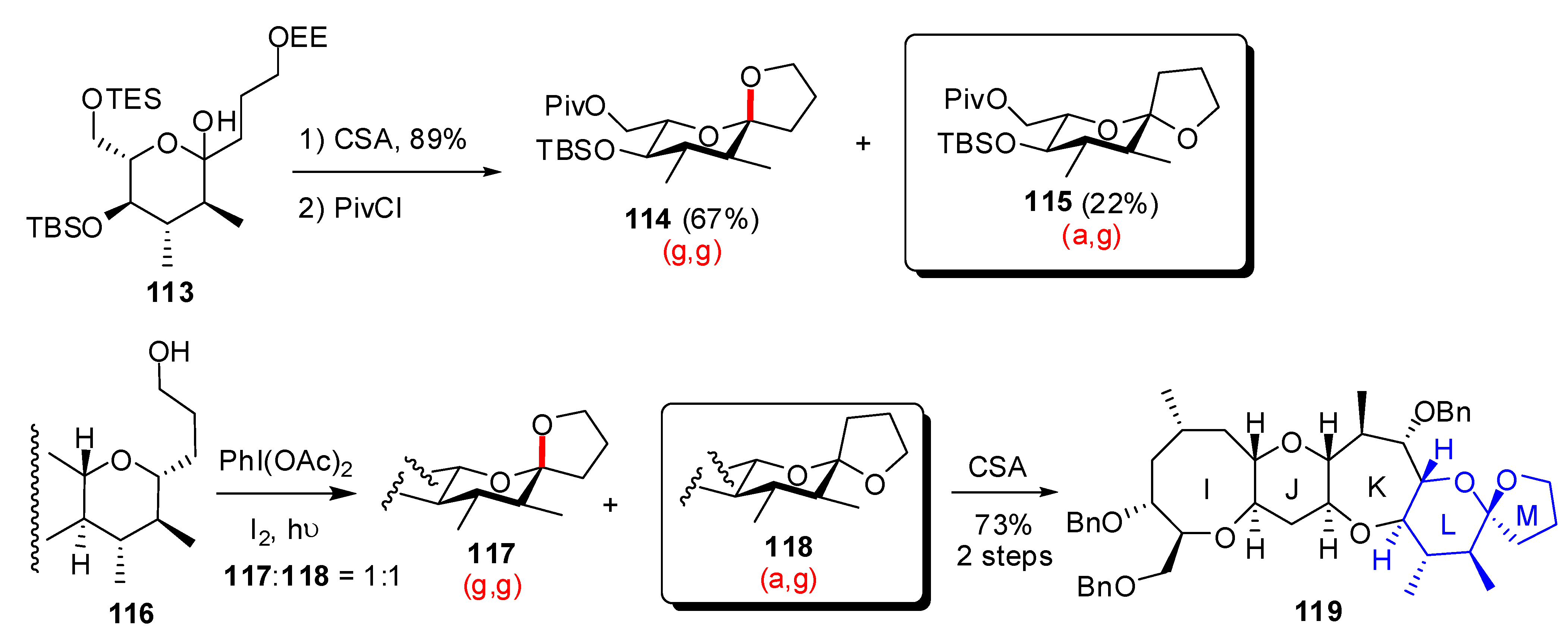
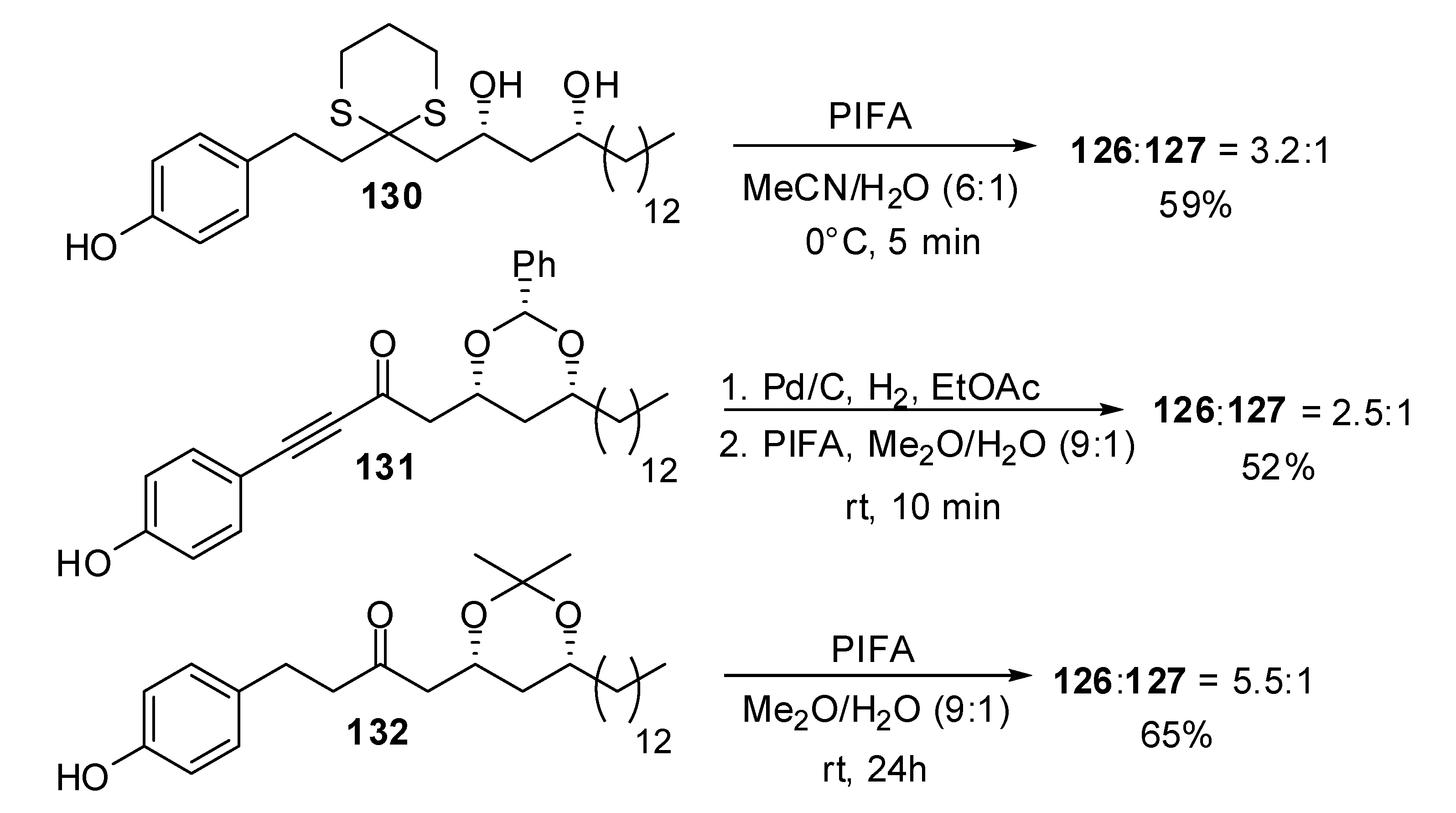
| Conditions | 114 (%) | 113 (%) | 127 (%) |
|---|---|---|---|
| MeCN, 0°C, 1h | 0 | 33 | 15 |
| MeCN/H2O (6:1), 0°C, 1h | 0 | 29 | 44 |
| Acetone, rt, 20 min | 11 | 40 | 20 |
| Acetone/H2O (9:1), rt, 20 min | 19 | 43 | 27 |

Conclusions
References and notes
- For reviews, see e.g.: Boivin, T.L.B. Synthetic routes to tetrahydrofuran, tetrahydropyran, and spiroketal units of polyether antibiotics and a survey of spiroketals of other natural products. Tetrahedron 1987, 43, 3309–3362. [Google Scholar] Perron, F.; Albizati, K.F. Chemistry of spiroketals. Chem. Rev. 1989, 89, 1617–1661. [Google Scholar] Vaillancourt, V.; Pratt, N.E.; Perron, F; Albizati, K.F. The total synthesis of spiroketal-containing natural products. Total Synth. Nat. Prod. 1992, 533–691. [Google Scholar] Haddad, N.; Abramovich, Z.; Ruhman, I. Novel stereoselective synthesis of spiroethers and spiroketals. Recent Res. Dev. Org. Chem. 1997, 1, 35–42. [Google Scholar]
- Pettit, G.R.; Cichacz, Z.A.; Gao, F.; Herald, C.L.; Boyd, M.R.; Schmidt, J.M.; Hooper, J.N.A. Isolation and structure of spongistatin 1. J. Org. Chem. 1993, 58, 1302–1304. [Google Scholar] Pettit, G.R.; Cichacz, Z.A.; Gao, F.; Herald, C.L.; Boyd, M.R. Isolation and structure of the remarkable human cancer cell growth inhibitors spongistatins 2 and 3 from and eastern Indian ocean Spongia sp. J. Chem. Soc. Chem. Commun. 1993, 1166–1168. [Google Scholar] Kobayashi, M.; Aoki, S.; Sakai, H.; Kawazoe, K.; Kihara, N.; Sasaki, T.; Kitagawa, I. Altohyrtin A, a potent anti-tumor macrolide from the okinawan marine sponge Hyrtios altum. Tetrahedron Lett. 1993, 34, 2795–2798. [Google Scholar] Kobayashi, M.; Aoki, S.; Sakai, H.; Kihara, N.; Sasaki, T.; Kitagawa, I. Altohyrtins B and C and 5-desacetylaltohyrtin A, potent cytotoxic macrolide congeners of altohyrtin A, from the Okinawan marine sponge sponge Hyrtios altum. Chem. Pharm. Bull. 1993, 41, 989–991. [Google Scholar] Kobayashi, M.; Aoki, S.; Kitagawa, I. Absolute stereostructures of altohyrtin A and its congeners, potent cytotoxic macrolides from the Okinawan marine sponge Hyrtios altum. Tetrahedron Lett. 1994, 35, 1243–1246. [Google Scholar] Kobayashi, M.; Aoki, S.; Gato, K.; Kitagawa, I. Marine natural products. XXXVIII. Absolute stereostructures of altohyrtins A, B, and C and 5-desacetylaltohyrtin A, potent cytotoxic macrolides, from the Okinawan marine sponge Hyrtios altum. Chem. Pharm. Bull. 1996, 44, 2142–2149. [Google Scholar] Fusetani, N.; Shinoda, K.; Matsunaga, S. Bioactive marine metabolites. 48. Cinachyrolide A: a potent cytotoxic macrolide possessing two spiro ketals from marine sponge Cinachyra sp. J. Am. Chem. Soc. 1993, 115, 3977–3981. [Google Scholar]
- For a review of methodologies developed up to 2004, see: Aho, J.E.; Pihko, P.M.; Rissa, T.K. Nonanomeric spiroketals in natural products: structures, sources, and synthetic strategies. Chem. Rev. 2005, 105, 4406–4440. [Google Scholar] [CrossRef]
- Schleyer, P.V.R.; Jemmis, J.E.; Spitznagel, G.W. Do anomeric effects involving the second-row substituents, chlorine, mercapto, and phosphino exist? Stabilization energies and structural preferences. J. Am. Chem. Soc. 1985, 107, 6393–6394. [Google Scholar] Apeloig, Y.; Schleyer, P.V.R.; Pople, J.A. Molecular orbital theory of the electronic structure of molecules. 35. β -Substituent effects on the stabilities of ethyl and vinyl cations. Comparison with isoelectronic methyl boranes. The relative importance of hyperconjugative and inductive effects. J. Am. Chem. Soc. 1997, 99, 5901–5909. [Google Scholar] Beckhaus, H.-D.; Dogan, B.; Verevkin, S.; Hädrich, J.; Rückardt, C. Dependence of anomeric stabilization on structure in acetals. Angew. Chem. Int. Ed. Engl. 1990, 29, 320–321. [Google Scholar] Hati, S.; Datta, D. Anomeric effect and hardness. J. Org. Chem. 1992, 57, 6056–6057. [Google Scholar] Available online: http://webbook.nist.gov.
- Jungius, C.L. Rearrangement between some isomers glucosederivatives and the muta-rotation of the kinds of sugar [machine translation]. Z. Phys. Chem. 1905, 52, 97–108. [Google Scholar]
- Edward, J.T. Stability of glycosides to acid hydrolysis. Chem. Ind. (London). 1955, 1102–1104. [Google Scholar]
- Lemieux, R.U.; Chü, N.J. Abstr. Papers Am. Chem. Soc. Meeting 1958. 31 N. Lemieux, R.U. Effects of unshared pairs of electrons and their salvation on conformational equilibria. Pure Appl. Chem. 1971, 25, 527–548. [Google Scholar] Jeffrey, G.A.; Pople, J.A.; Radom, L. Application of ab initio molecular orbital theory to the anomeric effect. Comparison of theoretical predictions and experimental data on conformations and bond lengths in some pyranoses and methyl pyranosides. Carbohydr. Res. 1972, 25, 117–131. [Google Scholar]
- Wolfe, S.; Whangbo, M.-H.; Mitchell, D.J. Molecular orbitals from group orbitals. Part VII. On the magnitudes and origins of the “anomeric effects”, “reverse anomeric effects”, and C-X and C-Y bond lengths in X-CH2-YH. Carbohydr. Res. 1979, 69, 1–26. [Google Scholar] Fuchs, B.; Elleincweig, A. Structure and conformation of heterocycles. Generalized anomeric effects in 1,4-dioxanes bearing polar substituents. Nouv. J. Chim. 1979, 3, 145–147. [Google Scholar]
- Dewar, M.J.S. Chemical implication of σ conjugation. J. Am. Chem. Soc. 1984, 106, 669–682. [Google Scholar] Cramer, C.J. Hyperconjugation as it affects conformational analysis. THEOCHEM 1996, 370, 135–146. [Google Scholar]
- Salzner, U.; Schleyer, P.V.R. Generalized anomeric effects and hyperconjugation in CH2(OH)2, CH2(SH)2, CH2(SeH)2 and CH2(TeH)2. J. Am. Chem. Soc. 1993, 115, 10231–10236. [Google Scholar] [CrossRef]
- Deslongchamps, P.; Rowan, D.D.; Pothier, N.; Sauve, T.; Saunders, J.K. 1,7-dioxaspiro-[5.5]undecanes. An excellent system for the study of stereoelectronic effects (anomeric and exo-anomeric effects) in acetals. Can. J. Chem. 1981, 59, 1105–1121. [Google Scholar] [CrossRef]
- Dubois, J.E.; Cossé-Barbi, A.; Watson, D.G. When local crowding reinforces an anomeric effect. Tetrahedron Lett. 1989, 30, 167–170. [Google Scholar] Salzner, U.; Schleyer, P.V.R. Ab initio examination of anomeric effects in tetrahydropyrans, 1,3-dioxanes and glucose. J. Org. Chem. 1994, 59, 2138–2155. [Google Scholar]
- Bailey, W.F.; Eliel, E.L. Conformational analysis. XXIX. 2-Substituted and 2,2-disubstituted 1,3-dioxanes. Generalized and reverse anomeric effects. J. Am. Chem. Soc. 1974, 96, 1798–1806. [Google Scholar] Praly, J.P.; Lemieux, R.U. Influence of solvent on the magnitude of the anomeric effect. Can. J. Chem. 1987, 65, 213–223. [Google Scholar] Wiberg, K.B.; Marquez, M. The energy components of the anomeric effect for 2-methoxytetrahydropyran. An experimental comparison of the gas phase and solutions. J. Am. Chem. Soc. 1994, 116, 2197–2198. [Google Scholar] Molteni, C.; Parrinello, M. Glucose in aqueous solution by first principles molecular dynamics. J. Am. Chem. Soc. 1998, 120, 2168–2171. [Google Scholar] Woodcock, H.L.; Moran, D.; Pastor, R.W.; Mackerell, A.D., Jr.; Brooks, B.R. Ab initio modeling of glycosyl torsions and anomeric effects in a model carbohydrate: 2-ethoxy tetrahydropyran. Biophys. J. 2007, 93, 1–10. [Google Scholar]
- Booth, H.; Khedhair, K.A. Endo-anomeric and exo-anomeric effects in 2-substituted tetrahydropyrans. J. Chem. Soc., Chem. Comm. 1985, 467–468.
- Tvaroska, I.; Carver, J.P. The anomeric and exo-anomeric effects of the hydroxyl group and the stereochemistry of the hemiacetal linkage. Carbohydr. Res. 1998, 309, 1–9. [Google Scholar] [CrossRef]
- Benedict, W.S.; Gailar, N.; Plyer, E.K. Rotation-vibration spectra of deuteriated water vapour. J. Chem. Phys. 1956, 24, 1139–1165. [Google Scholar] Clough, S.A.; Beers, Y.; Klein, G.P.; Rothman, L.S. Dipole moment of water from Stark measurements of water, monodeuterated water and deuterated water. J. Chem. Phys. 1973, 59, 2254–2259. [Google Scholar] Bicerano, J.; Marynick, D.S.; Lipscomb, W.N. Basis set and electron correlation effects on the total electron density in water, hydrogen sulphide and borane. J. Chem. Phys. 1978, 100, 732–739. [Google Scholar] Wiberg, K.B.; Rablen, P.R. Comparison of atomic charges derived via different procedures. J. Comput. Chem. 1993, 14, 1504–1518. [Google Scholar] Martin, F.; Zipse, H. Charge distribution in the water molecule – a comparison of methods. J. Comput. Chem. 2004, 26, 97–105. [Google Scholar]
- Bock, H.; Mollère, P.; Becker, G.; Fritz, G. Photoelectron spectra and molecular properties. XX. Dimethyl ether, methoxysilane, and disiloxane. J. Organomet. Chem. 1973, 61, 113–125. [Google Scholar] Tsuboyama, A.; Takeshita, K.; Konada, S.; Kimura, M. Ab initio gradient calculation of the molecular structures of dimethyl ether and dimethyl sulphide. Bull. Chem. Soc. Jpn. 1984, 57, 3589–3590. [Google Scholar] Clark, S.A.C.; Bawagan, A.O.; Brian, C.E. The valence orbital momentum distribution binding enegy spectra of dimethyl ether by electron momentum spectroscopy: an investigation of the methyl inductive effect. Chem. Physics 1989, 137, 407–426. [Google Scholar] Tsuzuki, S.; Uchimaru, T.; Tanabe, K.; Yliniemela, A. Comparison of atomic charge distributions obtained from different procedures: basis set and electron correlation effects. THEOCHEM 1996, 365, 81–88. [Google Scholar]
- Vila, A.; Mosquera, R.A. Atoms in molecules interpretation of the anomeric effect in the O-C-O unit. J. Comput. Chem. 2007, 28, 1516–1530. [Google Scholar] Trapp, M.L.; Watts, J.K.; Weinberg, N; Pinto, B.M. Component analysis of the X-C-Y anomeric effect (X = O, S; Y = F, OMe, NHMe) by DFT molecular orbital calculations and natural bond orbital analysis. Can. J. Chem. 2006, 84, 692–701. [Google Scholar]
- Zefirov, N.S.; Samoshin, V.V.; Subbotin, O.A.; Baranenkov, V.I.; Wolfe, S. The gauche effect on the nature of the interactions between electronegative substituents in trans-1,2-disubstituted cyclohexanes. Tetrahedron 1978, 34, 2953–2959. [Google Scholar] Juaristi, E. The attractive and repulsive gauche effect. J. Chem. Educ. 1979, 56, 438–441. [Google Scholar] Wiberg, K.B.; Murcko, M.A.; Laidig, K.E.; MacDougall, P.J. Origin of the gauche effect in substituted ethanes and ethenes. J. Phys. Chem. 1990, 94, 6956–6959. [Google Scholar] Juaristi, E.; Antunez, S. Conformational analysis of 5-substituted 1,3-dioxanes. 6. Study of the attractive gauche effect in O-C-C-O. Tetrahedron 1992, 48, 5941–5950. [Google Scholar] Svenson, S.; Schaefer, A.; Iuhrhop, J.H. Conformational effects of 1,3-syn-diaxial repulsion and 1,2-gauche attraction between hydroxyl groups in nanomolecular N-octyl-d-hexonamide solutions. A 13-C and 1-H NMR spectroscopy study. J. Chem. Soc., Perkin Trans. 2 1994, 1023–1028. [Google Scholar] Gil, F.P.S.C.; Da Costa, A.M.A.; Teixeira-Dias, J.J.C. Conformational analysis of CmH2m-OCH2CH2OH (m = 1-4): the role of the CH…O intermolecular interactions. J. Phys. Chem. 1995, 99, 8066–8070. [Google Scholar] Sasanuma, Y.; Sugita, K. The attractive gauche effect in ethylene oxides. Polym. J. (Toxyo, Japan). 2006, 38, 983–988. [Google Scholar]
- Cramer, C.J.; Truhlar, D.G.; French, A.D. Exo-anomeric effects on energies and geometries of different conformations of glucose and related systems in the gas phase and aqueous solution. Carbohydr. Res. 1997, 298, 1–14. [Google Scholar] Plavec, J.; Thibaudeau, C.; Chattopadhyaya, J. How do the energetics of the stereoelectronic gauche and anomeric effects modulate the conformation of nucleosides and nucleotides? Pure Appl. Chem. 1996, 68, 2137–2144. [Google Scholar]
- Sellers, H.; Saebø, S.; Pulay, P. The ring puckering potential of oxetane: local correlation results. Chem. Phys. Lett. 1986, 132, 29–31. [Google Scholar] Sellers, H.; Almlöf, J.; Saebø, S.; Pulay, P. Ring puckering potential of oxetane: TZ + nP/MP4 (SDQ) results. J. Phys. Chem. 1987, 91, 4216–4218. [Google Scholar] Mastryukov, V.S.; Boggs, J.E. Structure and conformation of some saturated four membered rings, CH2CH2CH2X. THEOCHEM 1995, 338, 235–248. [Google Scholar]
- Engerholm, G.G.; Luntz, A.C.; Gwinn, W.D.; Harris, D.O. Ring puckering in five-membered rings. II. Microwave spectrum, dipole moment and barrier to pseudorotation in tetrahydrofuran. J. Chem. Phys. 1969, 50, 2446–2457. [Google Scholar] Infarnet, Y.; Dunlan, J.C.; Delmau, J.; Huet, J. Conformation of methyltetrahydrofurans. Compt. Red. Acad. Sci. Ser. C 1969, 269, 1415–1418. [Google Scholar] Luger, P.; Buschmann, J. Twist conformation of tetrahydrofuran in the crystal form. Angew. Chem. 1983, 95, 423–424. [Google Scholar] Rayón, V.M.; Sordo, J.A. Pseudorotation motion in tetrahydrofuran: An ab initio study. J. Chem. Phys. 2005, 122, 204303-1-8. [Google Scholar]
- Lambert, J.B.; Keske, R.G.; Weary, D.K. Conformational characterization of simple group VI heterocycles. J. Am. Chem. Soc. 1967, 89, 5921–5924. [Google Scholar] Lambert, J.B.; Mixan, C.E.; Johnson, D.H. Conformational analysis of selenanes and telluranes. J. Am. Chem. Soc. 1973, 95, 4634–4639. [Google Scholar] Freeman, F.; Kasner, M.L.; Hehre, W.J. An ab initio theory and density functional theory (DFT) study of conformers of tetrahydro-2H-pyran. J. Phys. Chem. A. 2001, 105, 10123–10132. [Google Scholar] Ionescu, A.R.; Bérces, A.; Zgierski, M.Z.; Whitfield, D.M.; Nakada, T. Conformational pathways of saturated six-membered rings. A static and dynamical density functional study. J. Phys. Chem. A. 2005, 109, 8096–8105. [Google Scholar]
- For reviews, see e.g.: Yeung, K.-S.; Paterson, I. Advances in the total synthesis of biologically important marine macrolides. Chem. Rev. 2005, 105, 4237–4313. [Google Scholar] Pietruska, J. Spongistatins, cynachyrolides, or altohyrtins? Marine macrolides in cancer therapy. Angew. Chem. Int. Ed. 1998, 37, 2629–2636. [Google Scholar] Morris, J.C.; Nicholas, G.M.; Phillips, A.J. Marine natural products: synthetic aspects. Nat. Prod. Rep. 2007, 24, 87–108. [Google Scholar] Gerber-Lemaire, S.; Vogel, P. Spongistatins : biological activity and synthetic studies. Compt. Rend. Acad. Sci. 2008, in press. [Google Scholar]
- Paterson, I.; Chen, D.Y.-K.; Coster, M.J.; Aceña, J.L.; Bach, J.; Gibson, K.R.; Keown, L.E.; Oballa, R.M.; Trieselmann, T.; Wallace, D.J.; Hodgson, A.P.; Norcross, R.D. Stereocontrolled total synthesis of (+)-altohyrtin A/spongistatin 1. Angew. Chem., Int. Ed. 2001, 40, 4055–4060. [Google Scholar] [CrossRef]
- Paterson, I.; Coster, M.J.; Chen, D.Y.-K.; Oballa, R.M.; Wallace, D.J.; Norcross, R.D. The stereocontrolled total synthesis of altohyrtin A/spongistatin 1: the AB-spiroacetal segment. Org. Biomol. Chem. 2005, 3, 2399–2409. [Google Scholar] Paterson, I.; Coster, M.; Chen, D. Y.-K.; Gibson, K.R.; Wallace, D. The stereocontrolled total synthesis of altohyrtin A/spongistatin 1: the CD-spiroacetal segment. Org. Biomol. Chem. 2005, 3, 2410–2419. [Google Scholar] Paterson, I.; Coster, M.J.; Chen, D.Y.-K.; Acena, J.L.; Bach, J.; Keown, L.; Trieselmann, T. The stereocontrolled total synthesis of altohyrtin A/spongistatin 1: the southern hemisphere EF segment. Org. Biomol. Chem. 2005, 3, 2420–2430. [Google Scholar] Paterson, I.; Chen, D.Y.-K.; Coster, M.J.; Acena, J.L.; Bach, J.; Wallace, D. The stereocontrolled total synthesis of altohyrtin A/spongistatin 1: fragment couplings, completion of the synthesis, analogue generation and biological evaluation. Org. Biomol. Chem. 2005, 3, 2431–2440. [Google Scholar]
- Ball, M.; Gaunt, M.J.; Hook, D.F.; Jessiman, A.S.; Kawahara, S.; Orsini, P.; Scolaro, A.; Talbot, A.C.; Tanner, H.R.; Yamanoi, S.; Ley, S.V. Total synthesis of spongistatin 1: a synthetic strategy exploiting its latent pseudo-symmetry. Angew. Chem., Int. Ed. Engl. 2005, 44, 5433–5438. [Google Scholar] [CrossRef]
- O’Brien, M.; Diéguez-Vásquez, A.; Hsu, D.S.; Kraus, H.; Sumino, Y.; Ley, S.V. Azeotropic reflux chromatography: an efficient solution to difficult separation in the scale-up synthesis of spongistatin 1. Org. Biomol. Chem. 2008, 6, 1159–1164. [Google Scholar]
- Favre, S.; Gerber-Lemaire, S.; Vogel, P. New efficient route to an advanced precursor of the AB spiroketal of spongistatins. Org. Lett. 2007, 9, 5107–5110. [Google Scholar] [CrossRef]
- Gerber-Lemaire, S.; Vogel, P. An expeditive asymmetric synthesis of polyfunctional 1,7-dioxaspiro[5.5]undecanes. Eur. J. Org. Chem. 2004, 5040–5046. [Google Scholar] [CrossRef]
- Doubský, J.; Saman, D.; Zedník, J.; Vasíckova, S.; Koutek, B. A convenient access to thermodynamically nonstabilised spiroketal isomers: the first synthesis of (Z)-7-methyl-1,6-dioxaspiro[4,5]decane. Tetrahedron Lett. 2005, 46, 7923–7926. [Google Scholar] [CrossRef]
- Corbet, M.; Bourdon, B.; Gueyrard, D.; Goekjian, P.G. A Julia olefination approach to the synthesis of functionalized enol ethers and their transformation into carbohydrate-derived spiroketals. Tetrahedron Lett. 2008, 49, 750–754. [Google Scholar] [CrossRef]
- Conway, J.C.; Urch, C.J.; Quayle, P.; Xu, J. Spiroketalization reactions on a carbohydrate template. Synlett 2006, 5, 776–780. [Google Scholar]
- Potuzak, J.S.; Moilanen, S.B.; Tan, D.S. Stereocontrolled synthesis of spiroketals via a remarkable methanol-induced kinetic spirocyclization reaction. J. Am. Chem. Soc. 2005, 127, 13796–13797. [Google Scholar] [CrossRef]
- Moilanen, S.B.; Potuzak, J.S.; Tan, D.S. Stereocontrolled synthesis of spiroketals via Ti(Oi-Pr)4-mediated kinetic spirocyclization of glycal epoxides with retention of configuration. J. Am. Chem. Soc. 2006, 128, 1792–1793. [Google Scholar] [CrossRef]
- Fuwa, H.; Sasaki, M. An efficient strategy for the synthesis of endocyclic enol ethers and its application to the synthesis of spiroacetals. Org. Lett. 2008, 10, 2549–2552. [Google Scholar] [CrossRef]
- Shimizu, T.; Satoh, T.; Murakoshi, K.; Sodeoka, M. Asymmetric total synthesis of (-)-spirofungin A and (+)-spirofungin B. Org. Lett. 2005, 7, 5573–5576. [Google Scholar] [CrossRef]
- Paterson, I.; Anderson, E.A.; Findlay, A.D.; Knappy, C.S. Total synthesis of pteridic acids A and B. Tetrahedron 2008, 64, 4768–4777. [Google Scholar] [CrossRef]
- Nakahata, T.; Fujimura, S.; Kuwahara, S. Total synthesis of pteridic acids A and B. Chem. Eur. J. 2006, 12, 4584–4593. [Google Scholar] [CrossRef]
- Takaoda, L.R.; Buckmelter, A.J.; La Cruz, T.E.; Rychnovsky, S.D. Rational Synthesis of Contra-Thermodynamic Spiroacetals by Reductive Cyclizations. J. Am. Chem. Soc. 2005, 127, 528–529. [Google Scholar] [CrossRef]
- For a recent review on synthetic approaches to Pectenotoxins see: Halim, R.; Brimble, M.A. Synthetic studies towards the pectenotoxins: a review. Org. Biomol. Chem. 2006, 4, 4048–4058. [Google Scholar] For recents synthetic efforts toward the spiroketal of Pectenotoxins see: O’Connor, P.D.; Knight, C.K.; Friedrich, D.; Peng, X.; Paquette, L.A. Pectenotoxin-2 Synthetic Studies. 3. Assessment of the Capacity for Stereocontrolled Cyclization To Form the Entire C1-C26 Subunit Based upon the Double Bond Geometry Across C15-C16. J. Org. Chem. 2007, 72, 1747–1754. [Google Scholar] Halim, R.; Brimble, M.A.; Merten, J. Synthesis of the ABC tricyclic fragment of the pectenotoxins via stereocontrolled cyclization of a c-hydroxyepoxide appended to the AB spiroacetal unit. Org. Biomol. Chem. 2006, 4, 1387–1399. [Google Scholar] Halim, R.; Brimble, M.A.; Merten, J. Synthesis of the ABC Fragment of the Pectenotoxins. Org. Lett. 2005, 7, 2659–2662. [Google Scholar] Bondar, D.; Liu, J.; Müller, T.; Paquette, L.A. Pectenotoxin-2 Synthetic Studies. 2. Construction and Conjoining of ABC and DE Eastern Hemisphere Subtargets. Org. Lett. 2005, 7, 1813–1816. [Google Scholar]
- For a recent review on Attenol see: Kiyota, H. Synthesis of Marine Natural Products with Bicyclic and/or Spirocyclic Acetals. Top. Heterocycl. Chem. 2006, 5, 65–95. [Google Scholar]
- Rychnovsky, S.D.; Jay, P.; Bowers, J.P.; LePage, T.J. Conformation and reactivity of anomeric radicals. J. Am. Chem. Soc. 1992, 114, 8375–8384. [Google Scholar] [CrossRef]
- Still, W.C.; Sreekumar, C. Alpha.-alkoxyorganolithium reagents. A new class of configurationally stable carbanions for organic synthesis. J. Am. Chem. Soc. 1980, 102, 1201–1202. [Google Scholar] [CrossRef]
- La Cruz, T.E.; Rychnovsky, S.D. A Reductive Cyclization Approach to Attenol A. J. Org. Chem. 2007, 72, 2602–2611. [Google Scholar] [CrossRef]
- Vellucci, D.; Rychnovsky, S.D. Diastereoselective Synthesis of the Pectenotoxin 2 Non-Anomeric AB Spiroacetal. Org. Lett. 2007, 9, 711–714. [Google Scholar] [CrossRef]
- Pihko, P.M.; Aho, J.E. Access to Both Anomers of Pectenotoxin Spiroketals by Kinetic Spiroketalization. Org. Lett. 2004, 6, 3849–3852. [Google Scholar] [CrossRef]
- Castagnolo, D.; Breuer, I.; Pihko, P.M. Direct Kinetic Formation of Nonanomeric [6.5]-Spiroketals in Aqueous Media. J. Org. Chem. 2007, 72, 10081–10087. [Google Scholar] [CrossRef]
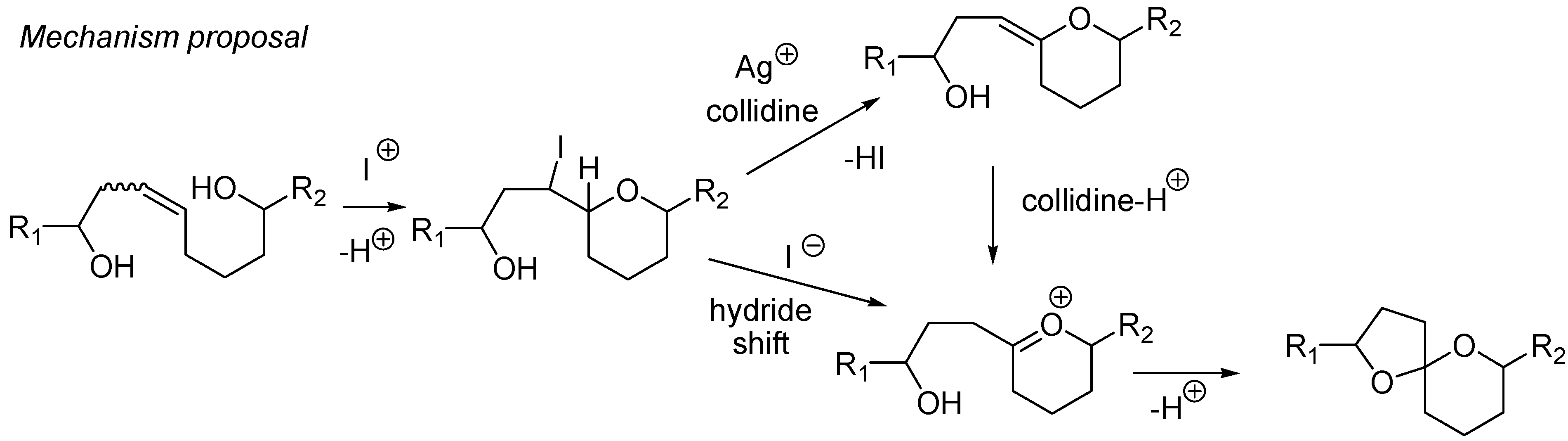
- Tony, K.A.; Li, X.; Dabideen, D.; Li, J.; Mootoo, D.R. An iodoetherification–dehydroiodination strategy for the synthesis of complex spiroketals from dihydroxyalkene precursors. Org. Biomol. Chem. 2008, 6, 1165–1169. [Google Scholar]
- Bedford, S.B.; Bell, E.; Bennet, F.; Haynes, C.J.; Knight, D.W.; Shaw, D.E. Model studies of the overall 5-endo-trig iodocyclization of homoallylic alcohols. J. Chem. Soc., Perkin Trans. 1 1999, 2143–2153. [Google Scholar]
- For reviews on the synthesis of marine polycyclic ethers, including ciguatoxins, see: Nakata, T. Total Synthesis of Marine Polycyclic Ethers. Chem. Rev. 2005, 105, 4314–4347. [Google Scholar] Sasaki, M. Recent Advances in Total Synthesis of Marine Polycyclic Ethers. Top. Heterocycl. Chem. 2006, 5, 149–178. [Google Scholar]
- Domon, D.; Fujiwara, K.; Ohtaniuchi, Y.; Takezawa, A.; Takeda, S; Kawasaki, H.; Murai, A.; Kawai, H.; Suzuki, T. Synthesis of the C42–C52 part of ciguatoxin CTX3C. Tetrahedron Lett. 2005, 46, 8279–8283. [Google Scholar] Domon, D.; Fujiwara, K.; Murai, A.; Kawai, H.; Suzuki, T. Convergent synthesis of the IJKLM-ring part of ciguatoxin CTX3C. Tetrahedron Lett. 2005, 46, 8285–8288. [Google Scholar]
- Beder, T.; Schuhmann, T.; Magull, J.; Grond, S.; von Zezschwitz, P. Comprehensive Study of Okaspirodiol: Characterization, Total Synthesis, and Biosynthesis of a New Metabolite from Streptomyces. J. Org. Chem. 2006, 71, 7125–7132. [Google Scholar] [CrossRef]
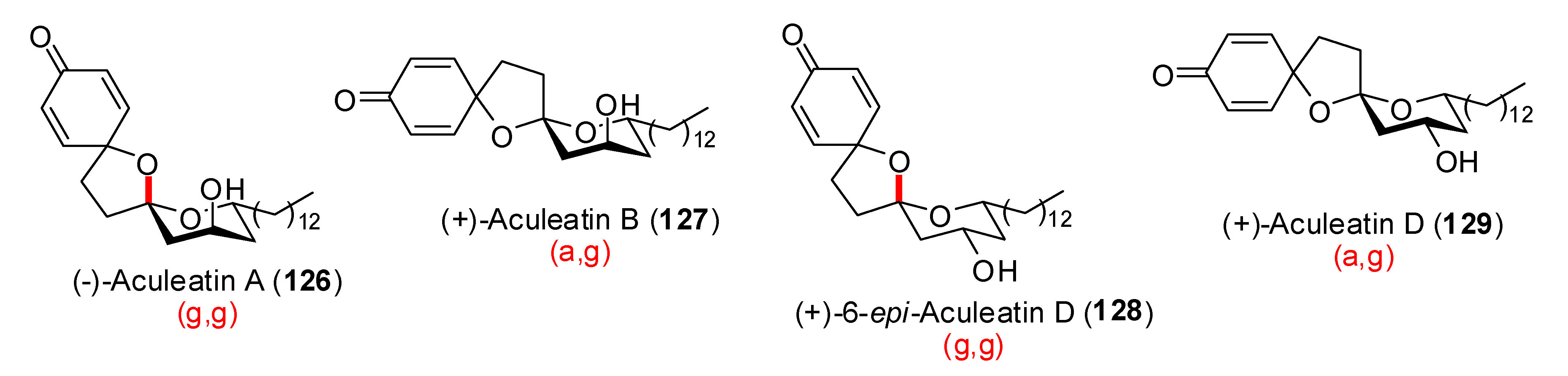
- Salim, A.A.; Su, B.-N.; Chai, H.-B.; Riswan, S.; Kardono, L.B.S.; Ruskandi, A.; Farnsworth, N.R.; Swanson, S.M.; Kinghorn, A.D. Dioxadispiroketal compounds and a potential acyclic precursor from Amomum aculeatum. Tetrahedron Lett. 2007, 48, 1849–1853. [Google Scholar] [CrossRef]
- Peuchmaur, M.; Wong, Y.-S. Studies towards the Synthesis of Aculeatin C. Synlett 2007, 2902–2906. [Google Scholar]
- Heilmann, J.; Brun, R.; Mayr, S.; Rali, T.; Sticher, O. Minor cytotoxic and antibacterial compounds from the rhizomes of Amomum aculeatum. Phytochemistry 2001, 57, 1281–1285. [Google Scholar] [CrossRef]
- Heilmann, J.; Mayr, S.; Brun, R.; Rali, T.; Sticher, O. Antiprotozoal Acivity and Cytotoxicity of Novel 1,7-Dioxadispiro[5.1.5.2]pentadeca-9,12.dien-11-one Derivatives from Amomum aculeatum. Helv. Chim. Acta 2000, 83, 2939–2945. [Google Scholar] [CrossRef]
- Wong, Y-S. Synthesis of (±)-aculeatins A and B. Chem. Commun. 2002, 686–687. [Google Scholar] [CrossRef]
- Falomir, E.; lvarez-Bercedo, P.; Carda, M.; Marco, J.A. Enantioselective synthesis and absolute configurations of aculeatins A and B. Tetrahedron Lett. 2005, 46, 8407–8410. [Google Scholar] [CrossRef]
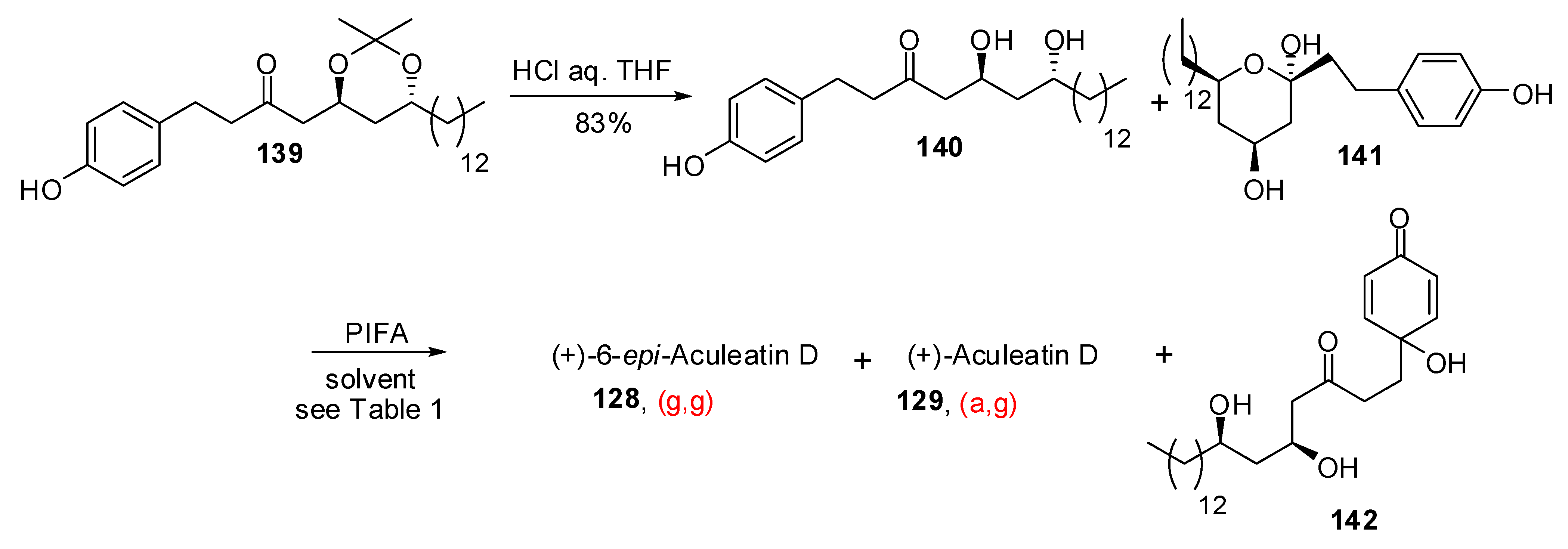
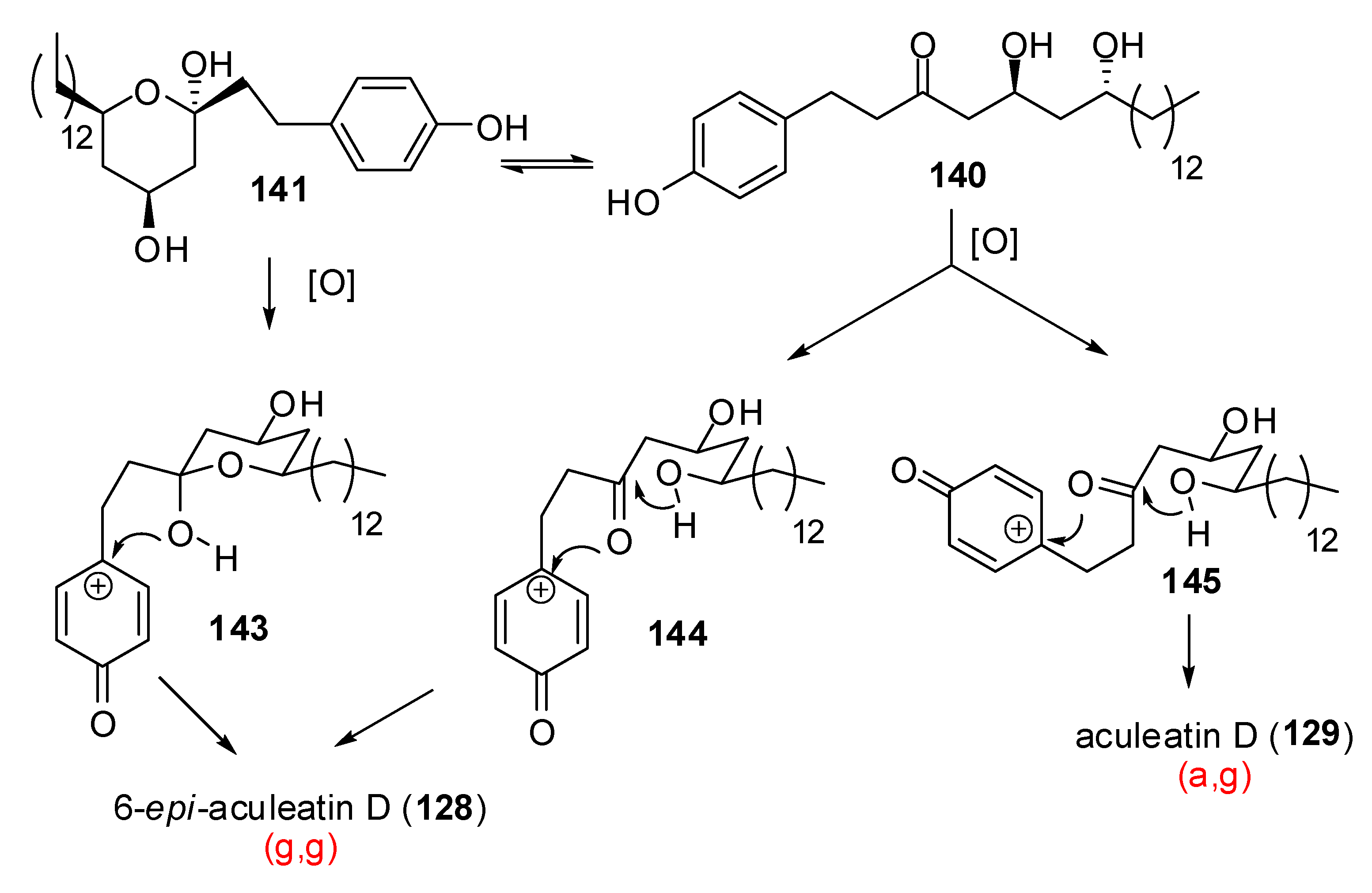
- Alvarez-Bercedo, P.; Falomir, E.; Carda, M.; Marco, J.A. Enantioselective synthesis and absolute configurations of aculeatins A, B, D, and 6-epi-aculeatin D. Tetrahedron 2006, 62, 9641–9649. [Google Scholar] [CrossRef]
- Chandrasekhar, S.; Rambabu, C.; Shyamsunder, T. Total synthesis of aculeatins A and B via a tethered oxa-Michael approach. Tetrahedron Lett. 2007, 48, 4683–4685. [Google Scholar] [CrossRef]
- Peuchmaur, M.; Wong, Y.-S. Diastereodivergent Strategies for the Synthesis of Homochiral Aculeatins. J. Org. Chem. 2007, 72, 5374–5379. [Google Scholar] [CrossRef]
- Baldwin, J.E.; Adlington, R. M.; Sham, V. W.-W.; Marquez, R.; Bulger, P.G. Biomimetic synthesis of (G)-aculeatin D. Tetrahedron 2005, 61, 2353–2363. [Google Scholar] [CrossRef]
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Favre, S.; Vogel, P.; Gerber-Lemaire, S. Recent Synthetic Approaches Toward Non-anomeric Spiroketals in Natural Products. Molecules 2008, 13, 2570-2600. https://doi.org/10.3390/molecules13102570
Favre S, Vogel P, Gerber-Lemaire S. Recent Synthetic Approaches Toward Non-anomeric Spiroketals in Natural Products. Molecules. 2008; 13(10):2570-2600. https://doi.org/10.3390/molecules13102570
Chicago/Turabian StyleFavre, Sylvain, Pierre Vogel, and Sandrine Gerber-Lemaire. 2008. "Recent Synthetic Approaches Toward Non-anomeric Spiroketals in Natural Products" Molecules 13, no. 10: 2570-2600. https://doi.org/10.3390/molecules13102570
APA StyleFavre, S., Vogel, P., & Gerber-Lemaire, S. (2008). Recent Synthetic Approaches Toward Non-anomeric Spiroketals in Natural Products. Molecules, 13(10), 2570-2600. https://doi.org/10.3390/molecules13102570