All commercially available reagents were used without further purification and solvents were dried according to standard procedures. Column chromatography was carried out on Silica Gel 60 (Merck, 0.040-0.063 mm, 230-400 mesh). Analytical thin-layer chromatography (TLC) was performed on Merck silica gel 60 F254 analytical plates; detection was carried out with either UV (254 nm), or spraying with a solution of phosphomolybdic acid, and with a basic solution of KMnO4, with subsequent heating. NMR spectra were recorded at room temperature on a Varian Mercury Plus 400 FT NMR spectrometer (1H at 400.13 MHz and 13C at 100.6 MHz), in CDCl3 as the solvent (unless otherwise noted) with tetramethylsilane as internal reference. For those fully assigned 1H- and 13C-NMR spectra standard NMR (COSY, DEPT, HSQC) experiments were conducted. Optical rotations were measured with a P3002 Krüss polarimeter in chloroform at 25 oC. All moisture-sensitive reactions were performed under a nitrogen atmosphere. Microwave experiments were conducted using a focused microwave system (CEM Discover). Reactions were performed in a glass vessel (10 mL) sealed with a septum. At the end of the reaction the vessels together with their contents were cooled rapidly using a stream of compressed air. The melting points were determined on the Kofler block and are uncorrected.
General procedure for preparation of trichloroacetimidates
To a solution of allyl alcohol in dry dichloromethane were added 1,8-diazabicyclo[5,4,0]undec-7-ene (DBU, 2 eq) and trichloroacetonitrile (2 eq) at 0 oC. The reaction mixture was stirred at 0 oC for 1 h. The insoluble material was removed by filtration and the filtrate was concentrated under reduced pressure to give a residue, which was purified by chromatography on silica gel (cyclohexane-ethyl acetate) to afford corresponding imidates 1a-1d, 6, 7, 8.
O-Allyl-2,2,2-trichloroacetimidate: Allyl alcohol (0.50 g, 8.61 mmol), DBU (2.57 mL, 17.22 mmol), trichloroacetonitrile (1.73 mL, 17.22 mmol) in CH
2Cl
2 (20 mL) afforded after flash chromatography (cyclohexane-ethyl acetate, 10:1) compound
1a (1.56 g, 89.5%) as a colorless oil;
1H-NMR: δ 4.81 (2H, m, CH
2), 5.31 (1H, dd,
J=10.5 Hz,
J=1.3 Hz, CH
2=), 5.44 (1H, ddd,
J=17.2 Hz,
J=3.1 Hz,
J=1.5 Hz, CH
2=), 6.03 (1H, ddd,
J=17.2 Hz,
J=10.5 Hz,
J=5.4 Hz, CH=), 8.32 (1H, bs, NH);
13C-NMR: δ 69.6, 109.7, 118.5, 131.4, 162.5; Anal. Calcd. for C
5H
6Cl
3NO (202.47): C 29.66, H 2.99, N 6.91; found C 29.53, H 2.87, N 6.74. The procedure and
1H-NMR spectroscopic data were previously reported [
10].
13C-NMR data have not been reported before [
10].
O-(But-3-en-2-yl)-2,2,2-trichloroacetimidate (
1b): But-3-en-2-ol (0.50 g, 6.93 mmol), DBU (1.45 mL, 9.70 mmol, 1.4 eq), trichloroacetonitrile (1.04 mL, 10.4 mmol, 1.5 eq) in CH
2Cl
2 (25 mL) afforded (1.30 g, 87%) of compound
1b after flash chromatography (cyclohexane-ethyl acetate, 5:1) as a pale yellow oil;
1H-NMR: δ 1.44 (3H, d,
J=6.5 Hz, CH
3), 5.20 (1H, d,
J=10.6 Hz, H
4), 5.36 (1H, d,
J=17.3 Hz, H
4), 5.49 (1H, m, H
3), 5.94 (1H, m, H
2), 8.29 (1H, bs, NH);
13C-NMR: δ 19.4, 75.7, 91.8, 115.9, 136.8, 161.8.; Anal. Calcd. for C
6H
8Cl
3NO (216.49): C 33.29, H 3.72, N 6.47; found C 33.10, H 3.45, N 6.28. The procedure and
1H-NMR spectroscopic data have been reported [
18].
13C-NMR data have not previously been reported [
18].
2,2,2-Trichloro-O-(hept-1-en-3-yl)acetimidate (
1c): Hept-1-en-3-ol (0.50 g, 4.38 mmol), DBU (1.31 mL, 8.76 mmol), trichloroacetonitrile (0.88 mL, 8.76 mmol) in CH
2Cl
2 (20 mL) afforded after flash chromatography (cyclohexane-ethyl acetate, 10:1) compound
1c (1.11 g, 98%) as a pale yellow oil;
1H-NMR: δ 0.90 (3H, t,
J=6.9 Hz, CH
3), 1.38 (4H, m, 2 x CH
2), 1.74 (2H, m, CH
2), 5.21 (1H, dd,
J2,1=10.6 Hz,
J1,1=0.7 Hz, H
1), 5.36 (2H, m, H
1, H
3), 5.80 (1H, m, H
2), 8.27 (1H, s, NH);
13C-NMR: δ 14.2, 22.6, 27.3, 34.0, 79.7 92.1, 116.8, 135.8, 162.2; Anal. Calcd for C
9H
14Cl
3NO
2 (258.57): C 41.76, H 5.41, N 5.41; found C 41.65, H 5.21, N 5.33. The procedure and
1H-NMR spectroscopic data were reported [
10].
13C-NMR data have not previously been reported [
10].
2,2,2-Trichloro-O-(oct-1-en-3-yl)acetimidate (1d): Oct-1-en-3-ol (0.50 g, 3.90 mmol), DBU (1.17 mL, 7.8 mmol), trichloroacetonitrile (0.78 mL, 7.80 mmol) in CH2Cl2 (20 mL) afforded after flash chromatography (cyclohexane-ethyl acetate, 10:1) compound 1d (0.90 g, 85%) as a pale yellow oil; 1H-NMR: δ 0.88 (3H, t, J=7.1 Hz, CH3), 1.29-1.47 (6H, m, 3 x CH2), 1.65-1.82 (2H, m, CH2), 5.21 (1H, m, H1), 5.30 (2H, m, H1, H3), 5.86 (1H, m, H2), 8.26 (1H, bs, NH); 13C-NMR: δ 14.2, 79.7, 22.7, 24.8, 31.7, 34.3, 92.1, 116.7, 136.0, 162.2; Anal. Calcd. for C10H16Cl3NO (272.60): C 44.06, H 5.91, N 5.14; found C 43.95, H 5.77, N 5.01.
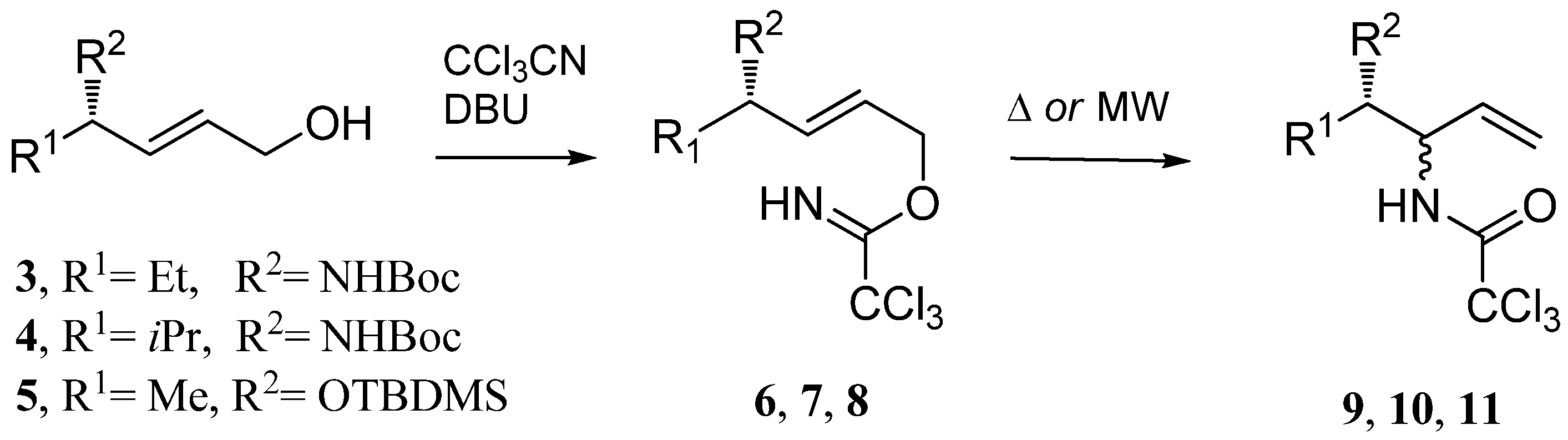
tert-Butyl N-[(3S,4E)-6-(trichloroacetimidyloxy)hex-4-en-3-yl]carbamate (
6): Compound
3 (0.30 g, 1.393 mmol), DBU (0.42 mL, 2.79 mmol), trichloroacetonitrile (0.28 mL, 2.79 mmol) in CH
2Cl
2 (15 mL) afforded after flash chromatography (cyclohexane-ethyl acetate, 3:1) compound
6 (0.40 g, 80%) as a colorless oil;
1H-NMR: δ 0.92 (3H, t,
J=7.4 Hz, CH
3), 1.44 (9H, s, 3 x CH
3), 1.53 (2H, m, CH
2), 4.08 (1H, m, H
3), 4.45 (1H, bs, NH), 4.77 (2H, m, H
6), 5.79 (2H, m, H
4, H
5), 8.29 (1H, bs, NH);
13C-NMR: δ 10.1, 28.2, 28.4 (3x), 53.1, 68.9, 79.4, 123.4, 135.8, 155.4, 162.5; Anal. Calcd. for C
13H
21Cl
3N
2O
3 (359.68): C 43.37, H 5.83, N 7.78; found C 43.01, H 5.64, N 7.64.
1H and
13C-NMR spectroscopic data have not previously been reported [
12].
tert-Butyl N-[(3S,4E)-6-(trichloroacetimidyloxy)-2-methylhex-4-en-3-yl]carbamate (
7): Compound
4 (0.10 g, 0.436 mmol), DBU (0.13 mL, 0.87 mmol), trichloroacetonitrile (0.087 mL, 0.87 mmol) in CH
2Cl
2 (10 mL) afforded after flash chromatography (cyclohexane-ethyl acetate, 3:1) compound
7 (0.12 g, 74%) as white crystals; m.p. 42 – 43
oC;
1H-NMR: δ 0.89 (6H, m, 2 x CH
3), 1.44 (9H, s, 3 x CH
3), 1.78 (1H, m, CH), 4.04 (1H, m, H
3), 4.52 (1H, m, NH), 4.80 (2H, m, H
6), 5.79 (2H, m, H
4, H
5), 8.30 (1H, bs, NH);
13C-NMR: δ 18.1, 18.7, 28.4, (3 x C), 32.4, 56.9, 68.9, 79.4, 91.4, 123.9, 134.5, 155.5, 162.4; Anal. Calcd. for C
14H
23Cl
3N
2O
3 (373.71): C 44.99, H 6.20, N 7.49; found C 44.78, H 6.05, N 7.21.
1H- and
13C-NMR spectroscopic data have not previously been reported [
12].
O-[(4S,2E)-4-(tert-Butyldimethylsilyoxy)pent-2-enyl]-2,2,2-trichloroacetimidate (
8): Compound
5 (0.35 g, 1.62 mmol), DBU (0.48 mL, 3.24 mmol), trichloroacetonitrile (0.325 mL, 3.24 mmol) in CH
2Cl
2 (18 mL) afforded compound
8 (0.50 g, 85.5%) as a colorless oil;
1H-NMR: δ 0.05 (3H, s, CH
3), 0.06 (3H, s, CH
3), 0.89 (9H, s, 3 x CH
3), 1.23 (3H, d,
J=6.7 Hz, CH
3), 4.35 (1H, m, H
4), 4.77 (2H, m, H
1), 5.86 (2H, m, H
2, H
3), 8.28 (1H, bs, NH);
13C-NMR: δ -4.8, -4.7, 18.3, 24.1, 25.9 (3 x C), 68.4, 69.1, 121.4, 139.8, 162.5; Anal. Calcd. for C
13H
24Cl
3NO
2Si (360.78): C 43.28, H 6.71, N 3.88; found C 43.17, H 6.56, N 3.59. The procedure and
1H-NMR spectroscopic data were reported before [
14].
13C-NMR data have not previously been reported [
14].
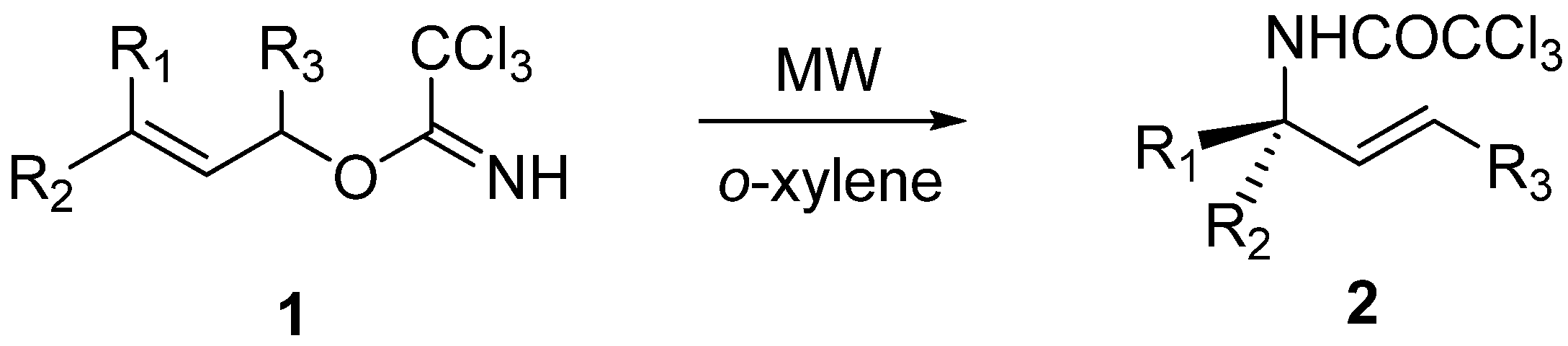
General procedure for Overman rearrangement
Conventional method (Procedure A): To a solution of imidates in dry solvent (see
Table 1 and
Table 2) was added anhydrous K
2CO
3 (1.1 eq). The reaction mixture was heated (for temperatures see
Table 1 and
Table 2). The solvent was evaporated under reduced pressure and chromatography of the residue on the silica gel (cyclohexane-ethyl acetate) afforded corresponding amides
2a-
2f,
9-11, 14 (
Table 1 and
Table 2). (
B1)
Microwave-assisted synthesis (Procedure B): To a solution of the corresponding imidate in
o-xylene in a 10 mL glass pressure microwave tube equipped with a magnetic stirrer bar was added anhydrous K
2CO
3 (1.1 eq) and the tube was closed with a silicon septum. The reaction mixture was subjected to microwave irradiation (power: 300W; for temperatures, reaction times and yields see
Table 1 and
Table 2). The solvent was removed under reduced pressure and the residue was purified by flash chromatography on silica gel (cyclohexane-ethyl acetate) to give amides
2a-
2f,
9-11, 14 (
Table 1 and
Table 2). (
B2)
N-Allyl-2,2,2-trichloroacetamide (
2a): Following general procedure A,
1a (0.30 g, 1.48 mmol), K
2CO
3 (0.23 g, 1.63 mmol) in
o-xylene (3 mL) afforded after flash chromatography (cyclohexane-ethyl acetate, 10:1) compound
2a (0.195 g, 65%).
2a: white crystals; m.p. 28 - 32
oC (Ref. [
10] m.p. 28–31
oC);
1H-NMR: δ 3.99-4.02 (2H, m, CH
2), 5.24-5.32 (2H, m, CH
2=), 5.84-5.93 (1H, m, CH=), 6.78 (1H, bs, NH);
13C-NMR: δ 43.6, 92.5, 117.8, 132.2, 161.8; Anal. Calcd. for C
5H
6Cl
3NO (202.46): C 29.66, H 2.98, N 7.90; found C 29.59, H 2.83, N 7.75. The procedure and
1H-NMR spectroscopic data were reported [
10].
13C-NMR data have not previously been reported [
10].
N-[(E)-But-2-enyl]-2,2,2-trichloroacetamide (
2b): Following general procedure A,
1b (0.10 g, 0.462 mmol), K
2CO
3 (70.2 mg, 0.508 mmol) in
o-xylene (2 mL) afforded after flash chromatography (cyclohexane-ethyl acetate, 10:1) compound
2b (0.093 g, 93%). Following general procedure B,
1b (0.30 g, 1.386 mmol), K
2CO
3 (0.21 g, 1.525 mmol) in
o-xylene (5 mL) afforded after flash chromatography (cyclohexane-ethyl acetate, 10:1) compound
2b (0.18 g, 60%).
2b: white crystals; m.p. 27 - 29
oC (Ref. [
10] m.p. 28 – 29
oC);
1H-NMR: δ 1.72 (3H, d,
J=6.5 Hz, CH
3), 3.91 (2H, m, CH
2), 5.50 (1H, m, CH=), 5.74 (1H, m, CH=), 6.68 (1H, bs, NH);
13C-NMR: δ 17.7, 43.3, 109.7, 124.8, 130.3, 161.6; Anal. Calcd. for C
6H
8Cl
3NO (216.49): C 33.26, H 3.72, N 6.47; found C 33.14, H 3.55, N 6.38. The procedure,
1H-NMR and
13C-NMR data spectroscopic data have been reported [
18].
2,2,2-Trichloro-N-[(E)-hept-2-enyl]acetamide (
2c): Following general procedure A,
1c (0.20 g, 0.773 mmol), K
2CO
3 (0.117 g, 0.85 mmol) in
o-xylene (2 mL) afforded after flash chromatography (cyclohexane-ethyl acetate, 10:1) compound
2c (0.18 g, 90%). Following general procedure B,
1c (0.40 g, 1.55 mmol), K
2CO
3 (0.236 g, 1.71 mmol) in
o-xylene (4 mL) afforded after flash chromatography (cyclohexane-ethyl acetate, 10:1) compound
2c (0.30 g, 75%).
2c: a colorless oil;
1H-NMR: δ 0.90 (3H, t,
J=7.1 Hz, CH
3), 1.28-1.39 (4H, m, 2 x CH
2), 2.02-2.07 (2H, m, CH
2), 3.92 (2H, m, CH
2); 5.44-5.51 (1H, m, CH=), 5.68-5.76 (1H, m, CH=), 6.67 (1H; bs, NH);
13C-NMR: δ 14.1, 22.4, 31.3, 32.1, 43.6, 92.8, 123.7, 135.9, 161.8; Anal. Calcd. for C
9H
14Cl
3NO
2 (258.57): C 41.77, H 5.45, N 5.41; found C 41.63, H 5.24, N 5.35. The procedure and
1H-NMR spectroscopic data were reported before [
10].
13C-NMR data have not previously been reported [
10].
(E)-2,2,2-Trichloro-N-(oct-2-enyl)acetamide (2d): Following general procedure A, 1d (0.40 g, 1.47 mmol), K2CO3 (0.224 g, 1.62 mmol) in o-xylene (4 mL) afforded after flash chromatography (cyclohexane-ethyl acetate, 10:1) compound 2d (0.37 g, 92.5%). Following general procedure B, 1d (0.40 g, 1.47 mmol), K2CO3 (0.224 g, 1.62 mmol) in o-xylene (4 mL) afforded after flash chromatography (cyclohexane-ethyl acetate, 10:1) compound 2d (0.39 g, 97.5%). 2d: a colorless oil; 1H-NMR: δ 0.87 (3H, t, J=7.0 Hz, CH3), 1.26-1.40 (6H, m, 3 x CH2), 2.04 (2H, q, J=7.0 Hz, CH2), 3.93 (2H, t, J=5.9 Hz, CH2), 5.46 (1H, m, CH=), 5.73 (1H, m, CH=), 6.69 (1H, bs, NH); 13C-NMR: δ 14.6, 22.7, 28.8, 31.6, 32.4, 43.5, 92.8, 123.6, 135.8, 161.7; Anal. Calcd. for C10H16Cl3NO (272.60): C 44.02, H 5.92, N 5.13; found C 43.90, H 5.84, N 5.04.
2,2,2-Trichloro-N-(2-methylbut-3-en-2-yl)acetamide (
2e): Following general procedure A,
1e (0.3 g, 1.30 mmol), K
2CO
3 (0.198 g, 1.43 mmol) in
o-xylene (3 mL) afforded after flash chromatography (cyclohexane-ethyl acetate, 10:1) compound
2e (0.25 g, 83%). Following general procedure B,
1e (0.5g, 2.17 mmol), K
2CO
3 (0.33 g, 2.39 mmol) in
o-xylene (5 mL) afforded after flash chromatography (cyclohexane-ethyl acetate, 10:1) compound
2e (0.40 g, 80%).
2e: white crystals; m.p. 48 – 50
oC (Ref. [
11] m.p. 49 – 50
oC);
1H-NMR: δ 1.54 (6H, s, 2 x CH
3), 5.17 (2H, m, H
4), 6.00 (1H, m, H
3), 6.59 (1H, bs, NH);
13C-NMR: δ 26.5 (2 x C), 56.1, 93.4, 113.3, 142.1, 160.4; Anal. Calcd. for C
7H
10Cl
3NO (230.52): C 36.43, H 4.32, N 6.07, found C 36.30, H 4.11, N 5.98. The
1H-NMR spectrum was previously reported [
11].
2,2,2-Trichloro-N-[(E)-pent-3-en-2-yl]acetamide (
2f): Following general procedure A,
1f (0.10 g, 0.434 mmol), K
2CO
3 (66 mg, 0.48 mmol) in
o-xylene (2 mL) afforded after flash chromatography (cyclohexane-ethyl acetate, 10:1) compound
2f (75 mg, 75%). Following general procedure B,
1f (66 mg, 0.26 mmol), K
2CO
3 (43.5 mg, 0.315 mmol) in
o-xylene (1 mL) afforded after flash chromatography (cyclohexane-ethyl acetate, 10:1) compound
2f (46 mg, 70%)
2f: white crystals; m.p. 57 – 59
oC (Ref. [
20] m.p. 60
oC);
1H-NMR (DMSO [
20]): δ 1.31 (3H, d,
J =6.8 Hz, CH
3), 1.71 (3H, m, CH
3), 4.43-4.50 (1H, m, H
2), 5.43-5.49 (1H, m, CH=), 5.65-5.75 (1H, m, CH=), 6.52 (1H, bs, NH);
13C-NMR (DMSO [
20]): δ 17.9, 20.4, 49.0, 93.0, 127.6, 130.76, 161.03; Anal. Calcd. for C
7H
10Cl
3NO (230.52): C 36.44, H 4.37, N 6.07, found C 36.35, H 4.21, N 5.97.
Methyl (Z)-2,3,4-tri-O-benzyl-6,7-dideoxy-8-(trifluoroacetimidyloxy)-α-d-gluco-oct-6-enpyranoside (13): To a suspension of NaH (0.09 g, 2.244 mmol, 60% dispersion in mineral oil, freed of oil with anhydrous THF) in dry THF (3 mL) was added allylic alcohol 12 (1.0 g, 2.04 mmol) in dry THF (5 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 10 min and then treated with gaseous trifluoroacetonitrile (15 g, 0.158 mol, prepared in situ by heating trifluoroacetamide (4.57 g, 0.040 mol) and P2O5 (11.43 g, 0.102 mol) for 2 h at 150 °C). The solid was removed by filtration and solvent evaporated under reduced pressure. The residue was purified by chromatography on silica gel (hexane-ethyl acetate, 3:1) to afford 0.95 g (79.5%) of compound 13 as a pale yellow oil; 1H-NMR: δ 3.27 (1H, dd, J4,3=9.6 Hz, J5,4=9.1 Hz, H4), 3.40 (3H, s, OCH3), 3.52 (1H, dd, J3,2=9.7 Hz, J2,1=3.6 Hz, H2), 3.99 (1H, dd, J3,2=9.7 Hz, J4,3=9.6 Hz, H3), 4.45 (1H, ddd, J5,4=9.1 Hz, J6,5=9.0 Hz, J7,5=1.0, H5), 4.57 (1H, d, J=10.8 Hz, CH2Ph), 4.57 (1H, d, J2,1=3.6 Hz, H1), 4.67 (1H, d, J=12.1 Hz, CH2Ph), 4.70 (1H, ddd, J8,8=11.9 Hz, J8,7=5.4 Hz, J8,6=1.4 Hz, H8), 4.79 (1H, d, J=10.6 Hz, CH2Ph), 4.80 (1H, d, J=12.1 Hz, CH2Ph), 4.82 (1H, J=10.6 Hz, CH2Ph), 4.96 (1H, J=10.8 Hz, CH2Ph), 5.01 (1H, ddd, J8,8=11.9 Hz, J8,7=7.4 Hz, J8,6=1.4 Hz, H8), 5.61 (1H, dddd, J7,6=11.2 Hz, J6,5=9.0 Hz, J8,6=1.4 Hz, J8,6=1.4 Hz, H6) 5.81 (1H, dddd, J7,6=11.2 Hz, J8,7=7.4 Hz, J8,7=5.4 Hz, J7,5=1.0 Hz, H7), 7.22-7.37 (15H, m, Ph), 8.20 (1H, bs, NH); 13C-NMR: δ 55.5, 62.0, 66.8, 73.4, 75.3, 75.8, 79.8, 81.6, 81.9, 98.2, 127.6, 127.7, 127.7, 2x127.8, 127.9, 2x128.0, 2x128.1, 2x128.3, 2x128.4, 2x128.5, 131.2, 138.0, 138.1, 138.6, 157.3, 157.7; Anal. Calcd. for C32H34F3NO6 (585.63): C 65.63, H 5.85, N 2.39; found C 65.59, H 5.80, N 2.31.
Methyl 2,3,4-tri-O-benzyl-6-[(trifluoroacetyl)amino]-7,8-dideoxy-d-glycero-α-d-galacto-oct-7-enpyranoside 14a,
Methyl 2,3,4-tri-O-benzyl-6-[(trifluoroacetyl)amino]-7,8-dideoxy-l-glycero-α-d-galacto-oct-7-enpyranoside (
14b): Following general procedure A,
13 (0.25 g, 0.43 mmol), K
2CO
3 (65.4 mg, 0.47 mmol) in
o-xylene (2 mL) afforded after flash chromatography (hexane-ethyl acetate, 9:1) compounds
14a and
14b (0.08 g, 32%, see Table 4). Following general procedure B,
13 (0.10g, 0.171 mmol), K
2CO
3 (26 mg, 0.188 mmol) in
o-xylene (2 mL) afforded after flash chromatography (hexane-ethyl acetate, 15:1) compounds
14a and
14b (0.07 g, 70%, see
Table 2).
14a: a colorless oil; [α]D25 = -19.6 (c 0.23); 1H-NMR: δ 3.30 (1H, dd, J5,4=10.0 Hz, J4,3=9.2 Hz, H5), 3.33 (3H, s, OCH3), 3.49 (1H, dd, J3,2=9.3 Hz, J2,1=3.6 Hz, H2), 3.76 (1H, dd, J5,4=10.0 Hz, J6,5=1.4 Hz, H5), 4.01 (1H, dd, J3,2=9.3 Hz, J4,3=9.2 Hz, H3), 4.50 (1H, d, J=10.1 Hz, CH2Ph), 4.56 (1H, d, J2,1=3.6. Hz, H1), 4.66 (1H, d, J=12.1 Hz, CH2Ph), 4.82 (1H, d, J=12.1 Hz, CH2Ph), 4.84 (1H, d, J=10.7 Hz, CH2Ph), 4.90 (1H, d, J=10.1 Hz, CH2Ph), 4.97 (1H, ddd, J6,NH=9.4 Hz, J7,6=5.4 Hz, J6,5=1.4 Hz, H6), 5.01 (1H, d, J=10.7 Hz, CH2Ph), 5.23 (1H, dd, J8cis,7=10.3 Hz, J8cis,8trans=1.6 Hz, H8cis), 5.23 (1H, dd, J8trans,7=17.1 Hz, J8trans,8cis=1.6 Hz, H8trans), 5.81 (1H, ddd, J8trans,7=17.1 Hz, J8cis,7=10.3 Hz, J7,6=5.4 Hz, H7), 6.71 (1H, d, J6,NH=9.4 Hz, NH), 7.27-7.39 (15H, m, Ph); 13C-NMR: δ 50.8, 55.4, 71.0, 73.6, 75.6, 75.9, 78.0, 80.0, 81.8, 98.1, 117.2, 127.8, 2x128.0, 4x128.1, 2x128.4, 2x128.5, 4x128.6, 133.8, 137.4, 137.9, 138.2, 156.6, 157.0; Anal. Calcd. for C32H34F3NO6 (585.63): C 65.63, H 5.85, N 2.39; found C 65.56, H 5.79, N 2.32.
14b: a colorless oil; [α]D25 = +30.5 (c 0.19); 1H-NMR: δ 3.35 (3H, s, OCH3), 3.41 (1H, dd, J5,4=10.0 Hz, J4,3=8.9 Hz, H4), 3.48 (1H, dd, J3,2=9.6 Hz, J2,1=3.5 Hz, H2), 3.81 (1H, dd, J5,4=10.0 Hz, J6,5=2.7 Hz, H5), 4.01 (1H, dd, J3,2=9.6 Hz, J4,3=8.9 Hz, H3), 4.57 (1H, J2,1=3.5 Hz, H1), 4.61 (1H, d, J=11.1 Hz, CH2Ph), 4.65 (1H, d, J=12.1 Hz, CH2Ph), 4.78 (1H, d, J=11.1 Hz, CH2Ph), 4.81 (1H, d, J=12.1 Hz, CH2Ph), 4.89 (1H, ddd, J6,NH=9.0 Hz, J7,6=8.2 Hz, J6,5=2.7 Hz, H6), 4.93 (1H, d, J=10.8 Hz, CH2Ph), 4.99 (1H, d, J=10.8 Hz, CH2Ph), 5.25 (1H, dd, J8trans,7=17.1 Hz, J8trans,8cis=1.0 Hz, H8trans), 5.29 (1H, dd, J8cis,7=10.3 Hz, J8trans,8cis=1.0 Hz, H8cis), 5.71 (1H, ddd, J8trans,7=17.1 Hz, J8cis,7=10.3 Hz, J7,6=8.2 Hz, H7), 6.70 (1H, d, J6,NH=9.0 Hz, NH), 7.27-7.39 (15H, m, Ph); 13C-NMR: δ 52.4, 55.5, 71.6, 73.8, 74.7, 76.0, 77.8, 80.1, 82.1, 98.5, 121.4, 2x127.8, 128.0, 128.1, 2x128.2, 3x128.3, 2x128.7, 4x128.8, 131.1, 2x138.1, 138.6, 156.3, 156.7; Anal. Calcd. for C32H34F3NO6 (585.63): C 65.63, H 5.85, N 2.39; found C 65.53, H 5.76, N 2.45



{kind=link}
{kind=link}
{kind=link}

