General
NMR spectra were recorded on Bruker Avance DPX 300 or DPX 400 instruments. Chemical shifts are reported in ppm using TMS as the internal standard in CDCl
3 or relative to 2.50 ppm for
1H and 39.99 ppm for
13C in DMSO-
d6 or 3.31 ppm for
1H and 49.15 ppm for
13C in CD
3OD. Structural assignments were based on
1H,
13C, DEPT135 and 2D spetra, COSY, HSQC, HMBC, NOESY. EI-Mass and ESI spectra were recorded on a Finnigan MAT 95XL spectrometer. IR spectra were obtained on a Thermo Nicolet FT-IR Nexus spectrometer using a Smart Endurance reflection cell. Silica gel Kieselgel 60G (Merck) was used for Flash Chromatography. The solvents were purified by standard methods. The preparations of compounds
1 were described elsewhere [
9,
10,
11,
12].
(2S,3S)-3-(allyloxy)butane-1,2,4-triol, (2)
This product was obtained by reduction of 1 with either LiAlH4 or NaBH4. LiAlH4 reduction: To a suspension of LiAlH4 (3.72 g, 95 %, 93 mmol) in dry diethyl ether (50 mL) was drop wise added a solution of 1 (4.36 g, 20 mmol) in 4 mL of diethyl ether at 0-5 °C. The reaction mixture was refluxed for 18 hours and then cooled in an ice bath. Then 5 mL of water was added and the mixture stirred for 20 minutes, followed by addition of a 15 % NaOH solution (12 mL) and then 10 mL of water. The resulting mixture was stirred and the granular salt formed, was separated by filtration, washed with hot THF (200 mL), and the filtrate concentrated under reduced pressure. The residue was purified by flash chromatography (CHCl3/CH3OH, 9:1 mixture) to give 0.83 g, 26 % of the pure product 2. NaBH4 reduction. Sodium borohydride (3.45 g, 93 mmol) in ethanol (50mL) was stirred for half an hour and then dropwise added a solution of 1 (4.35 g, 20 mmol) in ethanol (15 mL). The resulting solution was refluxed gently for 5 hours. The solution was cooled in an ice bath and added 10 mL of acetic acid. The mixture was stirred for 20 minutes and filtered. The solid was washed with 2x50 ml ethanol. The combined organic phase was concentrated under reduced pressure. The crude product was purified by flash chromatography using a 19:1 mixture of CH2Cl2 / MeOH as the eluent yielding 2.88 g, 88 % of the pure product, which exhibited the following spectroscopic properties: 1H-NMR (CDCl3, 400 MHz): δ = 3.45 (q, 1H, CH-OAllyl), 3.66-3.74 (m, 3H, 1H from CH2-CHOH, 2H from CH2-CH-OAll), 3.80 (dd, 1H from CH2-CHOH), 3.86 (q, 1H, CHOH), 4.03-4.20 (m, 2H, OCH2-CH=CH2), 4.32 (s, broad, 3H, OH), 5.18-5.38 (m, 2H, CH2=CH), 5.86-5.96 (m, 1H, CH=CH2) ppm.13C-NMR (CDCl3, 100MHz): δ = 60.6, 63.3, 71.6, 71.8, 79.1, 117.8, 134.5 ppm. MS (EI) m/z: 145 (M+-OH), 131 (M+-CH2OH), 101 (OH-CH2=O+CH2-CH=CH2), 61 (HOCH2CH=O+H). IR (neat): 3365, 2881, 1736, 1448 cm-1.
(S)-2-(allyloxy)-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)ethanol (3)
A solution of 2 (6.20 g, 38 mmol), 2,2-dimethoxylpropane (4.00 g, 38.5 mmol) and p-TsOH (223 mg, 1.2 mmol) in 100 mL acetone was stirred overnight at room temperature. The solvent was then removed and the residue was purified by flash chromatography using a 3:2 mixture of Et2O / n-hexane as the eluent to provide product 3 as acolorless oil (5.11 g, 85 %). Unreacted starting material 2 (1.05 g crude product) was recovered by continued elusion with a 19:1 mixture of CH2Cl2 / MeOH. Product 3 exhibited the following spectroscopic properties: 1H-NMR (CDCl3, 400MHz): δ = 1.37 (s, 3H, CH3), 1.44 (s, 1H, CH3), 2.48 (s, broad, 1H, OH), 3.49-3.53 (m, 1H, CH-OAll), 3.59 (dd, J= 10.8Hz, 12Hz, 1H, HOCH2-CHOAll), 3.73 (dd, J= 4.2Hz, 12Hz, 1H, HOCH2-CHOAll), 3.81 (dd, J= 7.2Hz, 8.4Hz, 1H, C-OCH2CHO-C), 4.03 (dd, J= 6.4Hz, 8.4Hz, 1H, C-OCH2CHO-C), 4.18-4.22 (m, 2H, OCH2-CH=CH2), 4.26-4.31 (m, 1H, C-OCH2CHO-C), 5.24-5.33 (m, 2H, CH2=CH), 5.88-5.95 (m, 1H, CH=CH2) ppm.13C-NMR (CDCl3, 100MHz): δ 25.3, 26.4, 61.6, 65.4, 71.8, 76.4, 79.1, 109.4, 117.4, 134.7 ppm. MS: (EI) m/z: 202(M+), 187 (M+-CH3), 171(M+-CH2OH), 101(C5H9O2+).
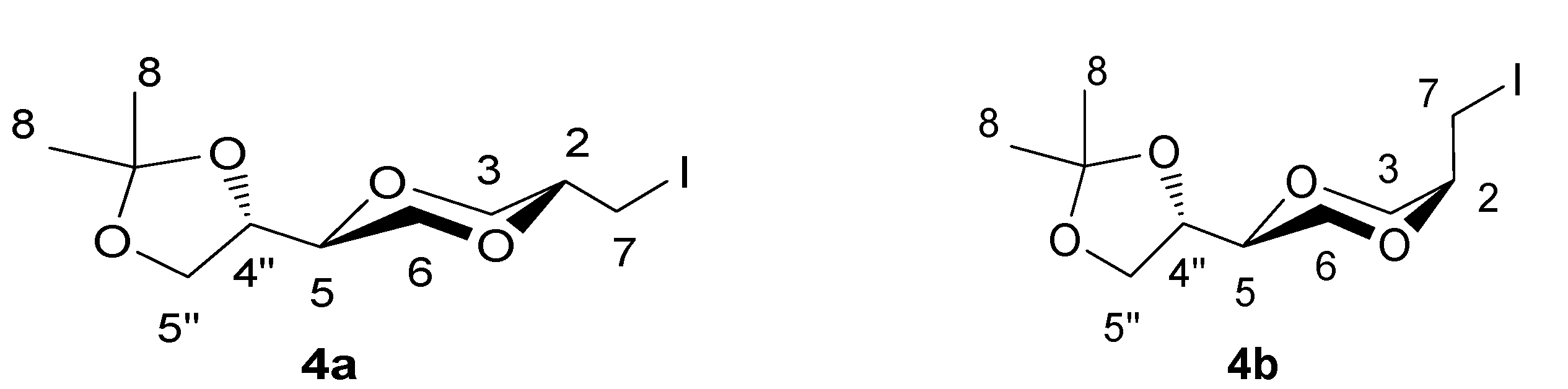
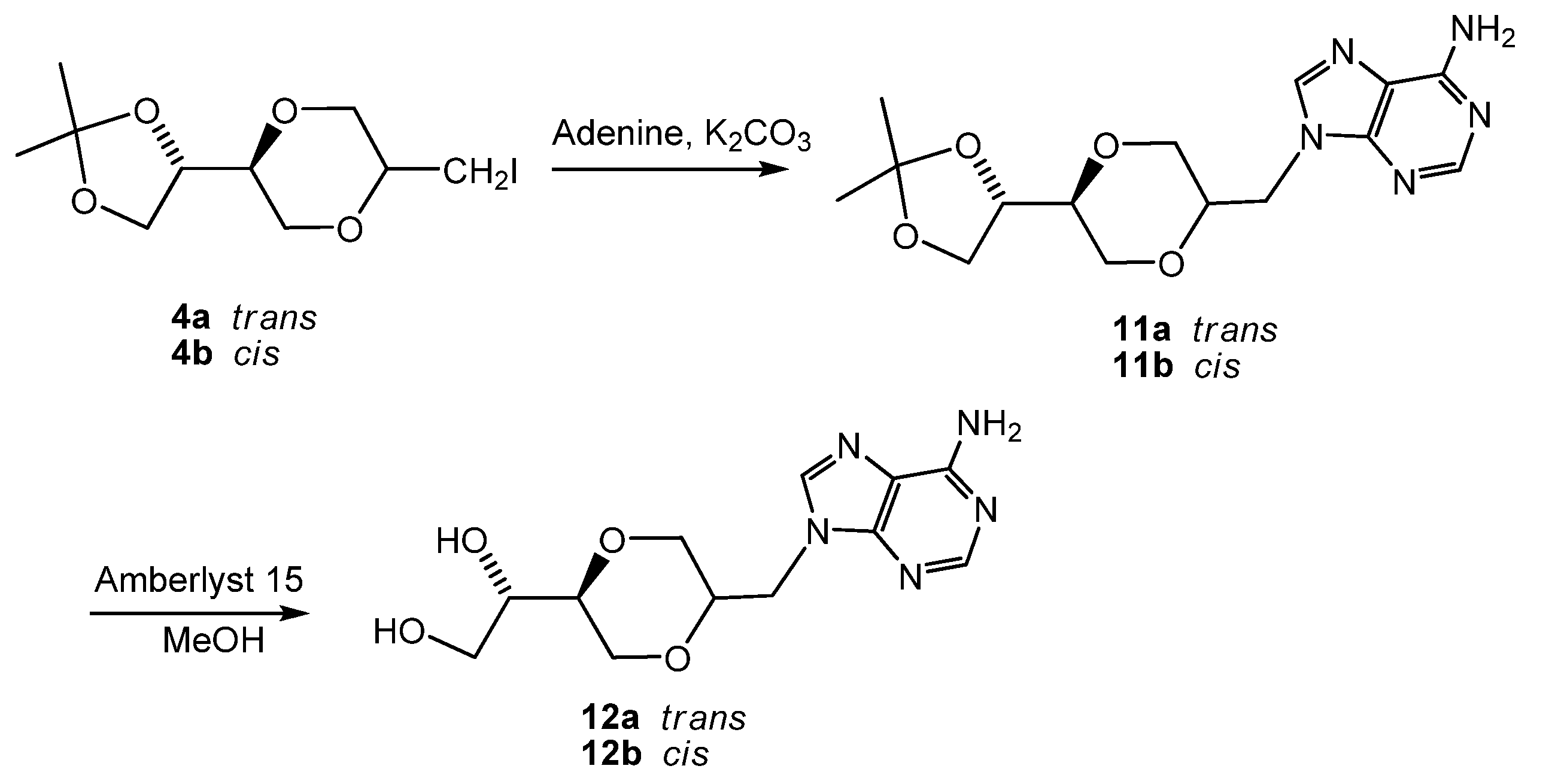
(2S,5S)-5-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]-2-iodomethyl-1,4-dioxane (4a) and (2R,5S)-5-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]-2-iodomethyl-1,4-dioxane (4b)
To a solution of 3 (3.20 g, 15.8 mmol) in dry acetonitrile (50 mL) was added NaHCO3 (4.19 g, 49.9 mmol) at -15°C. The mixture was stirred for 10 minutes and iodine (12.10 g, 47.7 mmol) was added. The reaction mixture was stirred for 68 hours with exclusion of light at -15 to -0°C. Ethyl acetate (80 mL) was added to the mixture and the solution was neutralized by saturated sodium thiosulfate solution until a colorless solution was obtained. The aqueous phase was extracted with ethyl acetate and the combined organic phase was dried over anhydrous sodium sulfate. The solution was filtered and evaporated. The residue was purified by gradient column chromatography using Et2O/n-hexane (1:4, 1:1) as eluent. The two diastereomers were separated in yields of 26.4% (4a) and 25.4% (4b). The pure compounds were white solid. Rf was 0.43 and 0.36 respectively (n-hexane/Et2O 1:1). The product 4a exhibited the following spectroscopic properties: 1H-NMR (CDCl3, 400 MHz) δ = 4.10-4.02 (m, 1H, H-4”), 4.04 (dd, 1H, J = 11.6 Hz, 2.4 Hz, H-3eq), 3.97 (dd, 1H, J= 8.0 Hz, 6.6 Hz, H-5”), 3.79 (dd, 1H, J= 8.0 Hz, 6.8 Hz, H-5”), 3.79-3.53 (m, 4H, H-2, H-5’, H-6), 3.39 (dd, 1H, J= 11.6 Hz, 10.2 Hz, H-3ax), 3.07 (d, 2H, J = 6.0 Hz, H-7), 1.42 (d, 3 H, 5JH-4” – H-8 = 0.4 Hz, H-8), 1.35 (d, 3 H, 5JH-4” – H-8 = 0.4 Hz, H-8) ppm; 13C-NMR (CDCl3, 100 MHz) δ = 109.9, 75.4, 75.0, 74.2, 70.9, 67.9, 65.3, 26.4, 25.5, 25.4 ppm; HRMS (ESI) m/z: for C10H17IO4 [M+Na]+, Calcd. 351.0069, Found 351.0063. 4b: 1H-NMR (CDCl3, 400 MHz): δ = 4.29 (dd, 1H, J = 6.8 Hz, 13.2 Hz, H-4”), 4.02 (dd, 1H, J= 8.0 Hz, 6.4 Hz, H-5”), 3.97 (dd, 1H, J = 12.0 Hz, 3.6 Hz, H-3eq), 3.84 (dd, 1H, J= 12.0 Hz, 3.0 Hz, H-3ax), 3.80-3.75 (m, 1 H, H-2), 3.75 (dd, 1H, J = 8.0 Hz, J = 6.8 Hz, H-5”), 3.67 (dd, 1H, J = 12.4 Hz, 8.0 Hz, H-6ax), 3.61-3.57 (m, 1H, H-5), 3.60 (dd, 1H, J = 12.4 Hz, 2.8 Hz, H-6eq), 3.42 (dd, 1H, J = 7.0 Hz, 12.6 Hz, H-7a), 3.40 (dd, 1H, J = 7.0 Hz, 13.0 Hz, H-7b), 1.44 (d, 3H, 5JH-4” – H-8 = 0.4 Hz, H-8), 1.38 (d, 3 H, 5JH-4” – H-8 = 0.4 Hz, H-8) ppm; 13C-NMR (CDCl3, 100MHz) δ = 109.8, 74.8, 73.4, 72.5, 66.3, 65.5, 62.3, 26.4, 25.3, 3.2 ppm; HRMS (ESI) m/z: for C10H17IO4 [M+Na]+, Calcd. 351.0069, Found 351.0079; IR (neat): 2980, 2867, 1461, 1413, 1380, 1370 cm-1.
Figure 3.
Structures 4a and 4b.
Figure 3.
Structures 4a and 4b.
(2S,5S)-5-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]-2-(uracil-1-yl-methyl)-1,4-dioxane (5a)
To a stirred suspension of uracil (0.253 g, 2.3 mmol) in dry DMF (19 mL), sodium hydride (0.065 g, 2.7 mmol) was added at room temperature. After stirring for one hour, compound 4a (0.35 g, 1.1 mmol) was added. The mixture was heated to 80°C and stirred overnight. The resulting mixture was evaporated under high vacuum. The residue was extracted with ethyl acetate. The solution was concentrated and purified by flash chromatography using ethyl acetate as the eluent. The product (201 mg) containing the inseparable byproduct 6a was obtained in 60% crude yield. The product 5a exhibited the following spectroscopic properties: 13C-NMR (CDCl3, 100 MHz): δ= 163.3, 150.9, 145.6, 109.8, 101.8, 75.2, 74.9, 73.3, 68.3, 67.4, 65.0, 48.8, 26.3, 25.2 ppm. The product contains inseparable byproduct 6a which makes the assignments of protons difficult; IR (neat) of the mixture of 5a and 6a: 3214, 3093, 2983, 2869, 1659, 1453, 1054 cm-1; HRMS (ESI) m/z: for C14H20N2O6 [M+Na]+, Calcd. 335.1219, Found 335.1222.
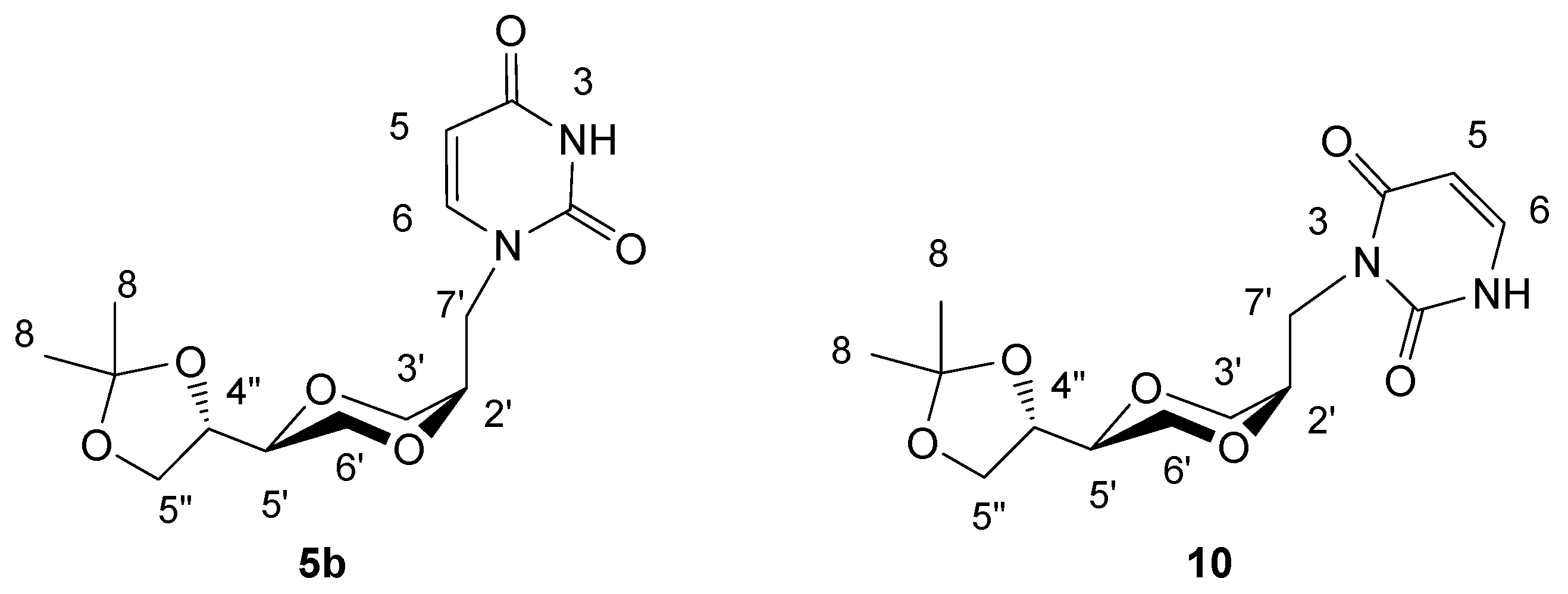
(2R,5S)-5-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]-2-(uracil-1-yl-methyl)-1,4-dioxane (5b)
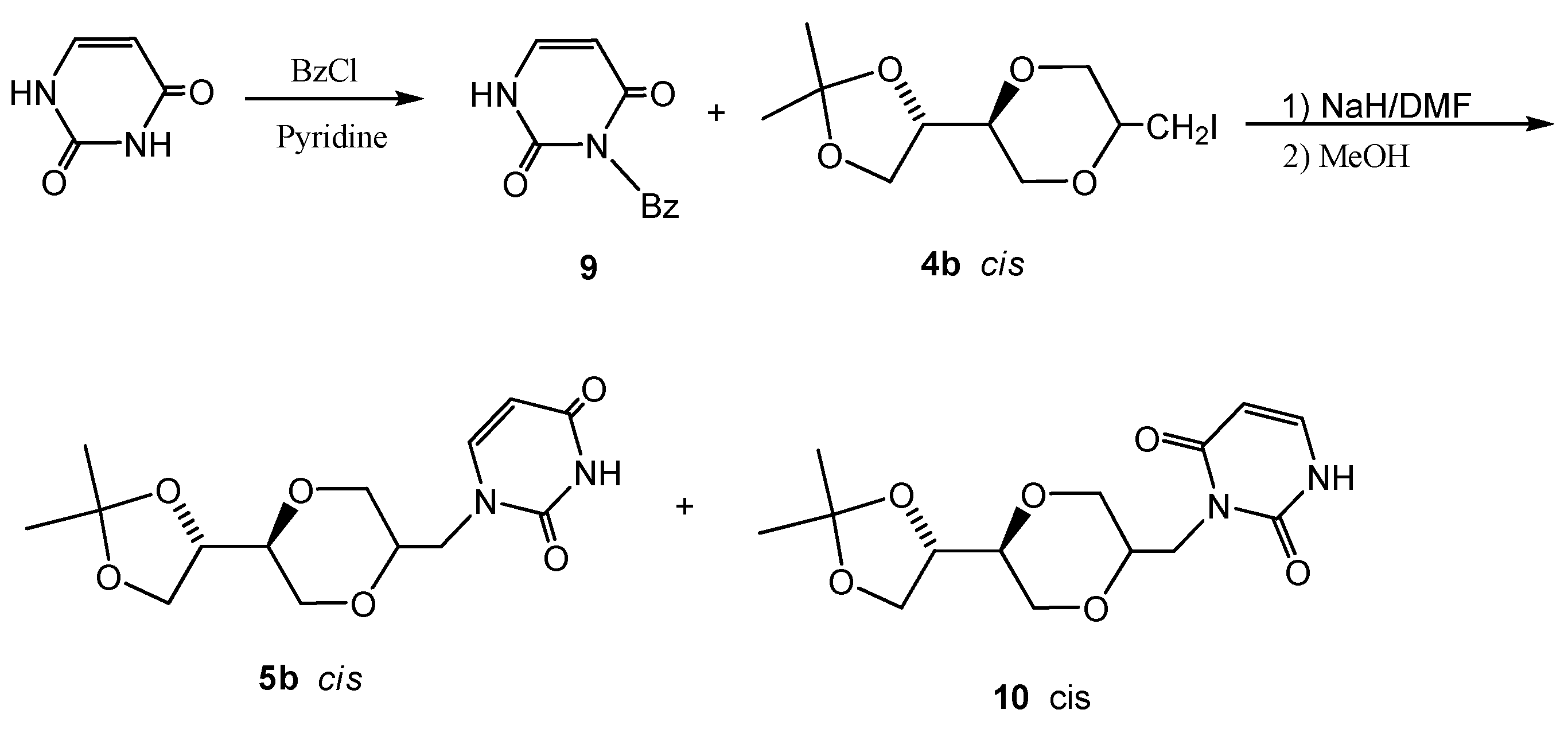
Method 1: The preparation of 5b was the same as used for the synthesis of 5a. Method 2: Sodium hydride (18 mg, 0.75 mmol) in 10 mL of dry DMF was stirred for half an hour at room temperature. N-3-benzoyluracil 9 (130 mg, 0.60 mmol) was added and stirred for an hour. To this suspension, iodide 4b (98 mg, 0.30 mmol) was added. The resulting stirred mixture was heated at 90°C overnight. The mixture was concentrated under reduced pressure to remove DMF. To the residue was added 20 mL methanol and the resulting mixture was stirred for 5 minutes. The solution was concentrated and purified by flash chromatography using CH2Cl2/CH3OH as the eluent. The obtained product (30 mg, 32 %) containing products 5b and 10 in a 5:6 ratio and was further purified by preparative TLC. The isolated product 5b exhibited the following spectroscopic properties: 1H-NMR (CDCl3, 400 MHz): δ = 1.39, 1.45 (s, 2x3H, H8), 3.60 (m, 1H, H-5’), 3.67-3.74 (m, 2H, H-6’), 3.74-3.80 (m, 2H, H-5” and H-3’), 3.82-3.86 (m, 1H, H-3’), 3.91-3.96 (m, 3H, H-7’ and H-2’), 4.05 (dd, 1H, J= 8.2 Hz, 6.6 Hz, H-5”), 4.32-4.37 (m, 1H, H-4”), 5.70 (d, 1H, J= 7.8 Hz, H-5), 7.20 (d, 1H, J= 7.8 Hz, H-6), 8.73 (brs, 1H, NH) ppm; 13C-NMR (CDCl3, 100 MHz): δ= 163.3, 150.8, 145.1, 109.9, 102.1, 74.4, 73.8, 71.0, 65.7, 64.9, 63.3, 47.6, 26.5, 25.4 ppm; IR (neat): 3097, 2985, 2874, 1682, 1652, 1455, 1124, 1060; HRMS (ESI) m/z: for C14H20N2O6 [M+Na]+, Calcd. 335.1219, Found 335.1222. The product 10 exhibited the following spectroscopic properties: 1H-NMR (CDCl3, 400 MHz): δ = 1.38, 1.45 (2×3H, CH3), 3.41 (dd, 1H, J = 4 Hz, 12 Hz), 3.62-3.67 (m, 1H), 3.70-3.75 (m, 2H), 3.87 (d, 2H), 3.97-4.08 (m, 3H), 4.23-4.28 (m, 1H), 4.81 (dd, J= 10 Hz, 14 Hz, 1H), 5.77 (dd, J= 7.6 Hz, 1.6 Hz, 1H, NCH=CH), 7.16 (dd, J= 7.6 Hz, 5.6 Hz, 1H, NCH=CH), 8.66 (d, 1H) ppm; 13C-NMR (CDCl3, 100MHz): δ= 25.3, 26.4, 39.8, 61.7, 65.4, 66.9, 69.1, 74.8, 76.0, 102.2, 109.6, 137.8, 151.8, 163.0 ppm; MS (m/z): (M+Na)+, 335.16.
Figure 5.
Structures 5b and 10.
Figure 5.
Structures 5b and 10.
(2S,5S)-5-[(1S)-1, 2-dihydroxyethyl]-2-(uracil-1-yl-methyl)-1,4-dioxane (7a)
The mixture of 5a and 6a (60mg, 5/6 ratio) was dissolved in methanol (10 mL), Amberlyst 15 (50 mg) was added and the mixture refluxed. The reaction was monitored by TLC until no more 7a was observed. The solution was filtered and the solvent was evaporated. The obtained product was further purified by flash chromatography to give pure 5a (12 mg, 34 %) and 8a (25 mg, 38 %) using CH2Cl2/MeOH (13:1) as eluent. The product 7a exhibited the following spectroscopic properties: 1H-NMR (CD3OD, 400 MHz): δ = 3.32-3.38 (m, 1H, H-3’), 3.47-3.55 (m, 2H, H-1” and H-2”), 3.55-3.67 (m, 4H, H-5’, H-2”, H-6’ and H-7’), 3.74-3.80 (m, 1H, H-2’), 3.82 (dd, 1H, J= 1.2 Hz, 10.4 Hz, H-6’), 3.88 (dd, 1H, J= 2.8 Hz, 8.4 Hz, H-3’), 3.91 (dd, 1H, J= 3 Hz, 11 Hz, H-7’), 5.61 (d, 1H, J= 8 Hz, H-5), 7.52 (d, 1H, J= 8 Hz, H-6) ppm; 13C-NMR (CD3OD, 100MHz): δ = 50.0, 63.7, 69.3, 69.7, 72.6, 74.4, 76.6, 101.8, 148.5, 153.0, 166.9 ppm; IR (neat): 3396, 2871, 1651, 1455, 1101, 1043 cm-1; HRMS (ESI) m/z: for C11H16N2O6 [M+Na]+, Calcd. 295.0906, Found 295.0911.
The product 8a exhibited the following spectroscopic properties: 1H-NMR (CD3OD, 400 MHz): δ = 3.34-3.43 (m, 2H), 3.46-3.54 (m, 4H), 3.57-3.74 (m, 8H), 3.75-3.86 (m, 6H), 3.87-3.90 (m, 1H), 3.92-3.96 (m, 1H), 4.05-4.10 (m, 1H), 5.70 (d, 1H, J= 7.8 Hz, NCH=CH), 7.52 (d, 1H, J= 7.8 Hz, NCH=CH) ppm; 13C-NMR (CD3OD, 100 MHz): δ = 42.8, 51.1, 63.7, 63.8, 69.3, 69.4, 69.7, 72.57, 72.61, 74.0, 74.4, 76.56, 76.57, 101.2, 146.8, 153.3, 165.6 ppm; MS (m/z): HRMS (ESI) m/z: for C18H28N2O10 [M+Na]+, Calcd. 455.1641, Found 455.1645.
(2R,5S)-5-[(1S)-1,2-dihydroxyethyl]-2-(-uracil-1-yl-methyl)-1,4-dioxane (7b)
The method for preparation of 7b was as same as applied for the synthesis of 7a. The product 7b exhibited the following spectroscopic properties: 1H-NMR (CD3OD, 400 MHz): δ= 3.53-3.66 (m, 3H, H-2” and H-6’), 3.60-3.70 (m, 2H, H-1” and H-5’), 3.78-3.80 (m, 2H, H-3’), 3.80-3.92 (2H, H-2’ and H-7’), 4.00 (dd, 1H, J= 11.6 Hz, 8 Hz, H-2” or H-6’), 4.23-4.31 (m, 1H, H-7’), 5.65 (d, J= 8 Hz, H-5), 7.58 (d, J= 8 Hz, H-8) ppm; 13C-NMR (CD3OD, 100 MHz): δ= 166.8, 153.0, 147.9, 102.2, 76.4, 71.8, 71.5, 66.9, 63.9, 63.8, 48.2 ppm; IR (neat): 3352, 3056, 2931, 2875, 1667, 1456, 1129, 1101 cm-1; HRMS (ESI) m/z: for C11H16N2O6 [M+Na]+, Calcd. 295.0906, Found 295.0915.
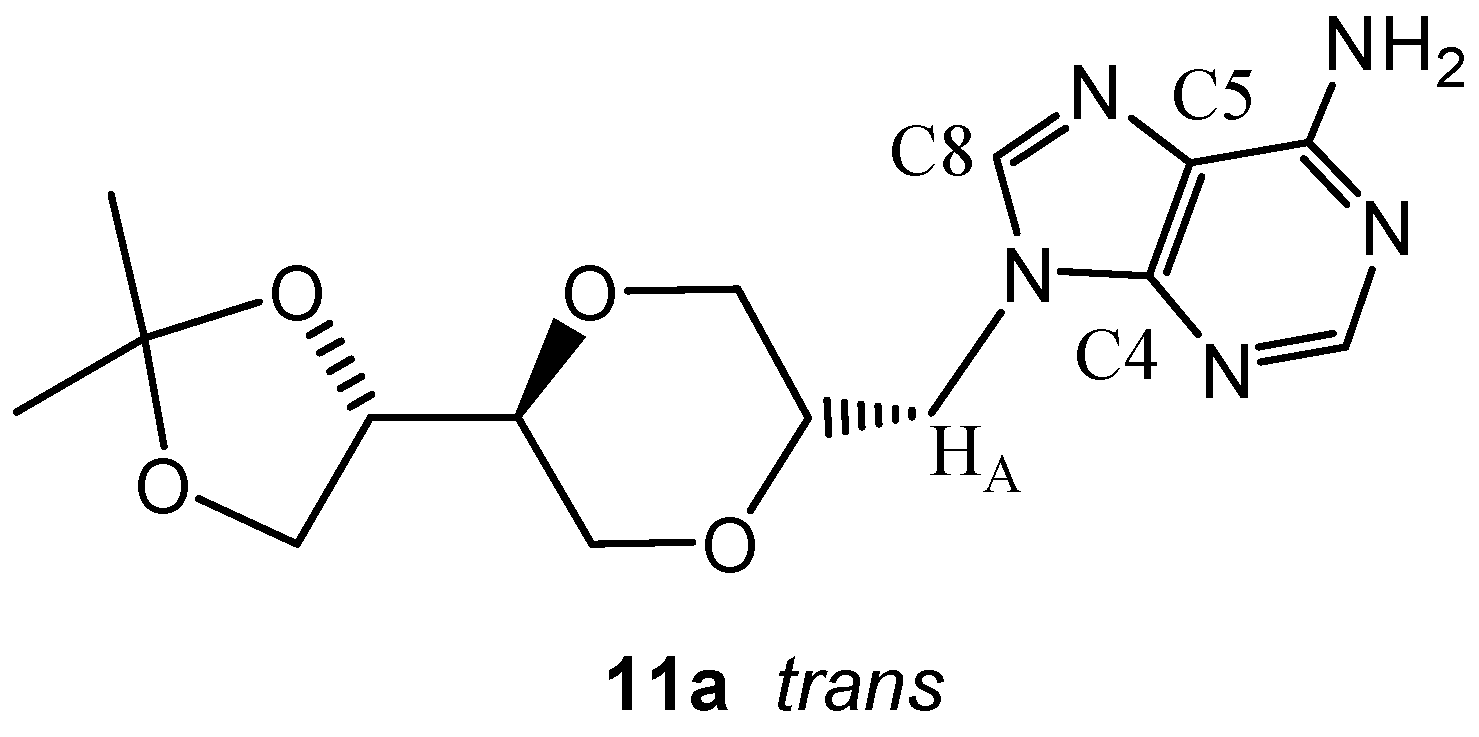
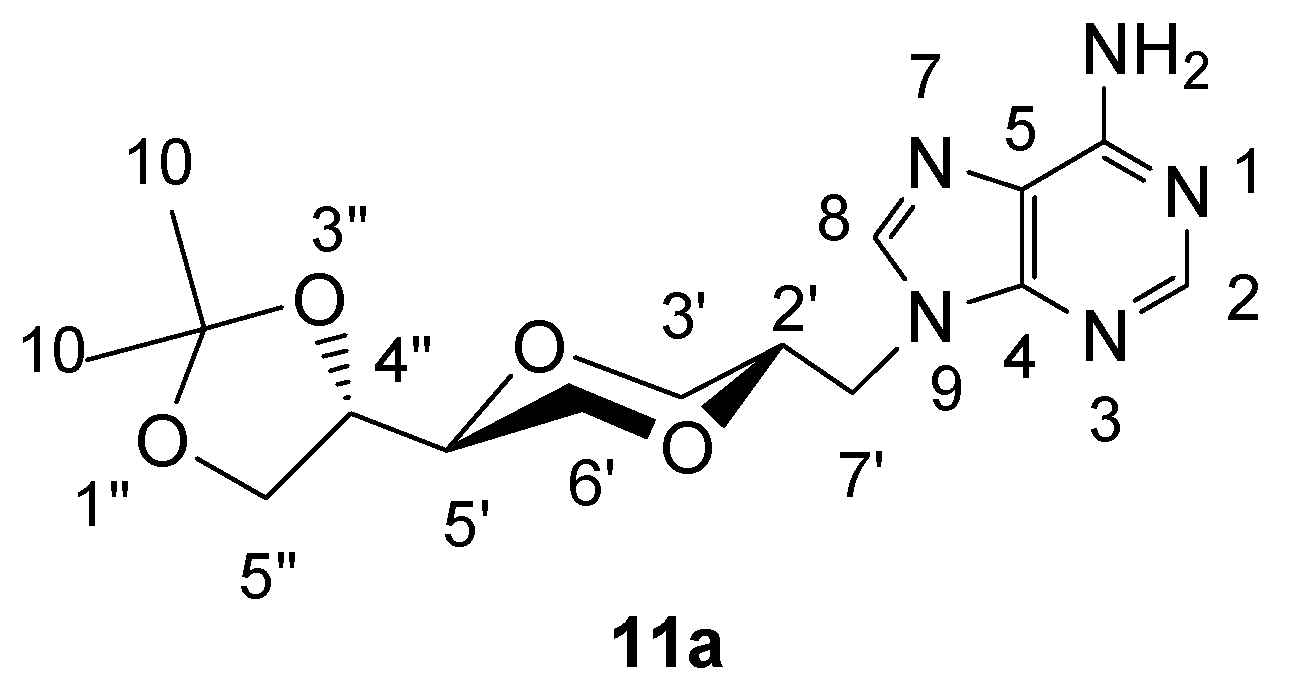
(2S,5S)-5-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]-2-(adenin-9-yl-methyl)-1,4-dioxane (11a)
A mixture of adenine (0.279 g, 2 mmol) and anhydrous potassium carbonate (0.300 g, 2.2 mmol) in dry DMF (10 mL) was heated at 120°C. After two hours, compound 4a (0.304 g, 0.9 mmol) was added to the solution. The mixture was stirred overnight at 120°C. The mixture was concentrated under reduced pressure and the residue purified by flash chromatography using a mixture of dichloromethane and methanol (14:1) as the eluent. The product (130 mg) was obtained in 42 % yield. The product 11a exhibited the following spectroscopic properties: 1H-NMR (CDCl3, 400 MHz): δ= 8.35 (s, 1H, H-2), 7.88 (s, 1H, H-8), 5.75 (s, 2H, NH2), 4.29 (dd, 1H, J= 14.6 Hz, 3.4 Hz, H-7’), 4.12 (dd, 1H, J= 14.6 Hz, 6.8 Hz, H-7’), 4.01- 4.07 (m, 1H, H-4”), 3.97 (dd, J= 11.4 Hz, 2.6 Hz, H-3’eq), 3.87-3.95 (m, 2H, H-2’, H-5”), 3.75-3.82 (m, 2H, H-5”, H-6’), 3.46-3.57 (m, 2H, H-5’, H-6’), 3.32 (dd, J= 11.4 Hz, 10.6 Hz, H-3’ax), 1.39 (s, 1H, H-10), 1.33 (s, 1H, H-10) ppm. 13C-NMR (CDCl3, 100 MHz): δ= 155.6, 153.2, 150.4, 141.6, 141.1, 119.4, 109.7, 75.1, 74.9, 73.2, 68.5, 67.5, 65.0, 44.2, 26.2, 25.3 ppm; IR (neat): 3322, 3161, 2983, 2938, 2864, 1673, 1606, 1064, 1048 cm-1; HRMS (ESI) m/z: for C15H22N5O4 M+, Calcd. 336.1671, Found 336.1676.
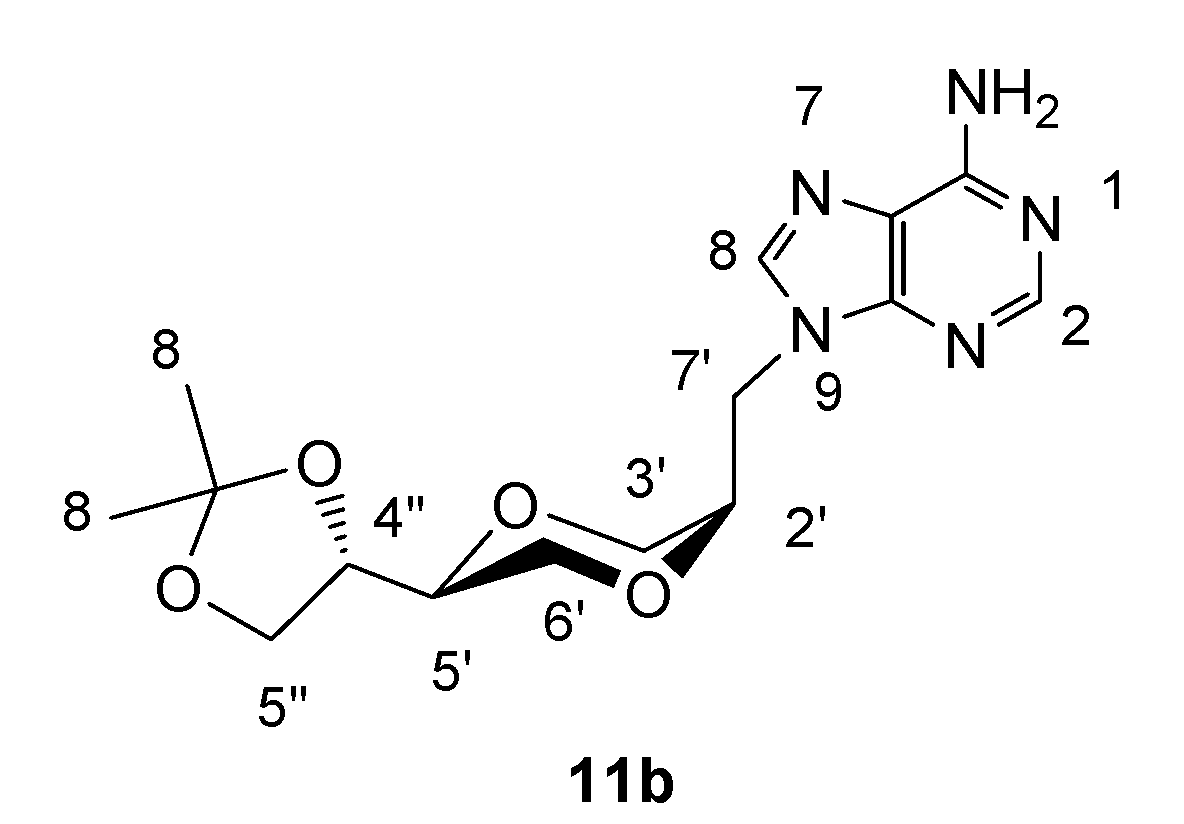
(2R,5S)-5-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]-2-(adenin-9-yl-methyl)-1,4-dioxane (11b)
Synthesis of 11b was carried out as described for 11a. The product exhibited the following spectroscopic properties: 1H-NMR (CDCl3, 400 MHz): δ= 8.35 (s, 1H, H-2), 7.87 (s, 1H, H-8), 6.14 (s, 2H, NH2), 4.54 (dd, 1H, J= 14.8 Hz, 8.8 Hz, H-7’), 4.32-4.38 (m, 2H, H-4”, H-7’), 4.06- 4.11 (m, 1H, H-2’), 4.05 (dd, J= 8.4 Hz, 6.6 Hz, H-5”), 3.80-3.90 (m, 3H, H-3’, H-6’), 3.77 (dd, J= 8.4 Hz, 6.8 Hz, H-5”), 3.61-3.67 (m, 2H, H-5’, H-6’), 1.46 (s, 1H, H-10), 1.39 (s, 1H, H-10) ppm; 13C-NMR (CDCl3, 100 MHz): δ= 155.7, 153.1, 150.1, 140.9, 119.4, 109.9, 74.6, 73.8, 71.1, 65.6, 65.1, 63.0, 42.8, 26.5, 25.4 ppm; IR (neat): 3276, 3134, 2984, 2935, 2870, 1676, 1600, 1575, 1126, 1066 cm-1; HRMS (ESI) m/z: for C15H22N5O4 M+, Calcd. 336.1671, Found 336.1675.
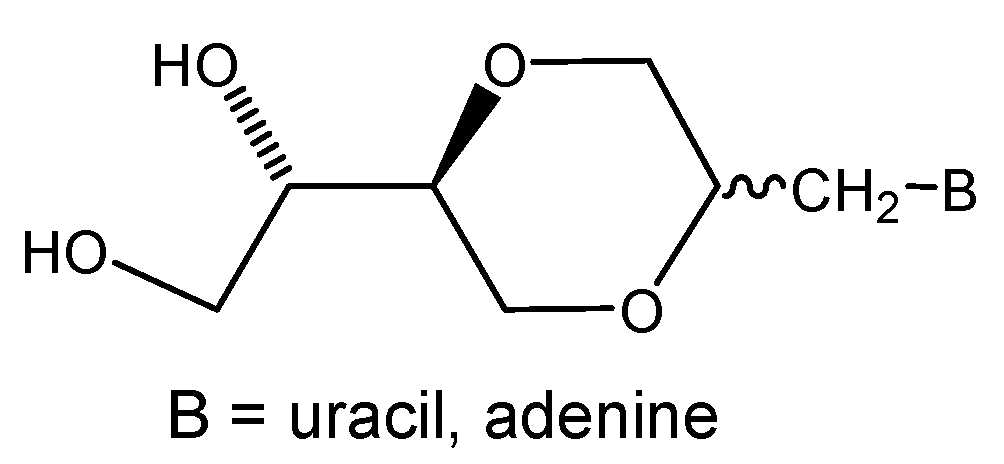
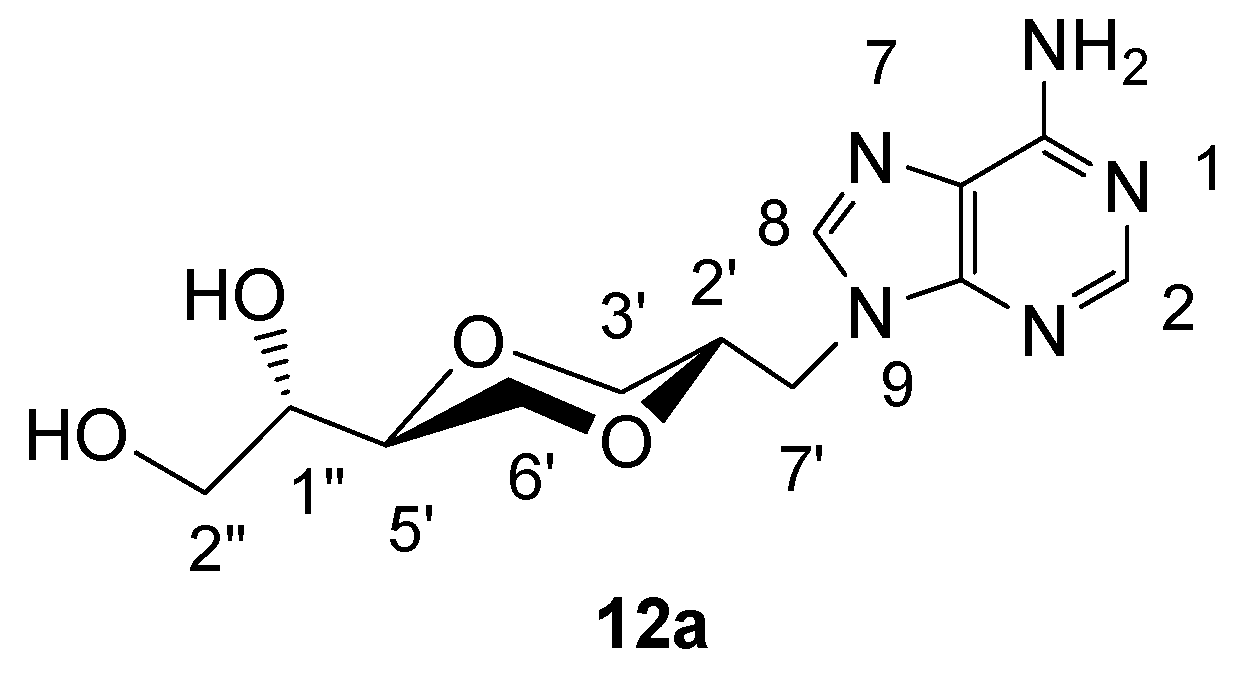
(2S,5S)-5-[(1S)-1,2-dihydroxyethyl]-2-(adenin-9-yl-methyl)-1,4-dioxane (12a)
Compound 11a (108 mg, 0.32 mmol) was dissolved in methanol (10 mL), Amberlyst 15 (45 mg) was added and the mixture was refluxed until TLC showed that all 11a was consumed. The solution was filtered and the solvent was evaporated. The product 12a (81 mg, 85 %) exhibited the following spectroscopic properties: 1H-NMR (DMSO-d6, 400 MHz): δ= 8.17 (s, 1H, H-2), 8.08 (s, 1H, H-8), 7.35 (brs, 2H, NH2), 4.3-4.8 (brs, 2H, OH), 4.21 (dd, 1H, J= 14.4 Hz, 4.2 Hz, H-7’), 4.13 (dd, 1H, J= 14.4 Hz, 6.8 Hz, H-7’), 3.83-3.88 (m, 2H, H-2’, H-3’), 3.67-3.73 (m, 1H, H-6’), 3.17-3.48 (m, 6H, H-6’, H-5’, H-3’, H-1”, H-2”) ppm; 13C-NMR (DMSO-d6, 100 MHz): δ= 155.5, 151.9, 149.6, 141.6, 118.4, 75.2, 72.6, 70.8, 68.1, 67.5, 61.9, 43.7 ppm; IR (neat): 3271, 3117, 2918, 2881, 1668, 1604, 1120, 1106, 1066 cm-1; HRMS (ESI) m/z: for C12H17N5O4 [M+1]+, Calcd. 296.1358, Found 296.1356.
Figure 11.
Structure 12a.
Figure 11.
Structure 12a.
(2R,5S)-5-[(1S)-1,2-dihydroxyethyl]-2-(adenin-9-yl-methyl)-1,4-dioxane (12b)
Compound 12b was prepared using the same method as described for the synthesis of 12a. Thus, 11b (79 mg, 0.24 mmol) in methanol (10 mL) containing added Amberlyst-15 (36 mg) was refluxed until TLC showed that all 11b was consumed. The solution was filtered and the solvent was evaporated. The product 12b (49 mg, 70%) exhibited the following spectroscopic properties: 1H-NMR (DMSO-d6, 400 MHz): δ= 8.16 (s, 1H, H-2 or H-8), 8.15 (s, 1H, H-8 or H-2), 7.30 (brs, 2H, NH2), 4.69 (dd, 1H, J= 14.4 Hz, 9.6 Hz, H-7’), 4.22 (dd, 1H, J= 14.4 Hz, 4.2 Hz, H-7’), 4.00-4.05 (m, 1H, H-2’), 3.92-3.98 (m, 1H, H-2” or H-6’), 3.78 (dd, 1H, J = 12.2 Hz, 2.2 Hz, H-3’), 3.69 (dd, 1H, J= 12.2 Hz, 3.2 Hz, H-3’), 3.34-3.56 (m, 5H, remining protons) ppm; 13C-NMR (DMSO-d6, 100MHz): δ= 155.7, 152.0, 149.6, 141.3, 118.5, 75.5, 70.4, 69.6, 65.6, 62.2, 61.4, 40.6 ppm; IR (neat): 3271, 3125, 2883, 1674, 1604, 1119, 1065 cm-1; HRMS (ESI) m/z: for C12H17N5O4 [M+1]+, Calcd. 296.1358, Found 296.1355.
Figure 10.
Structure 12b.
Figure 10.
Structure 12b.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}















