Synthesis and Antimalarial Evaluation of Cyclic β-Amino Acid-Containing Dipeptides

Abstract
:Introduction
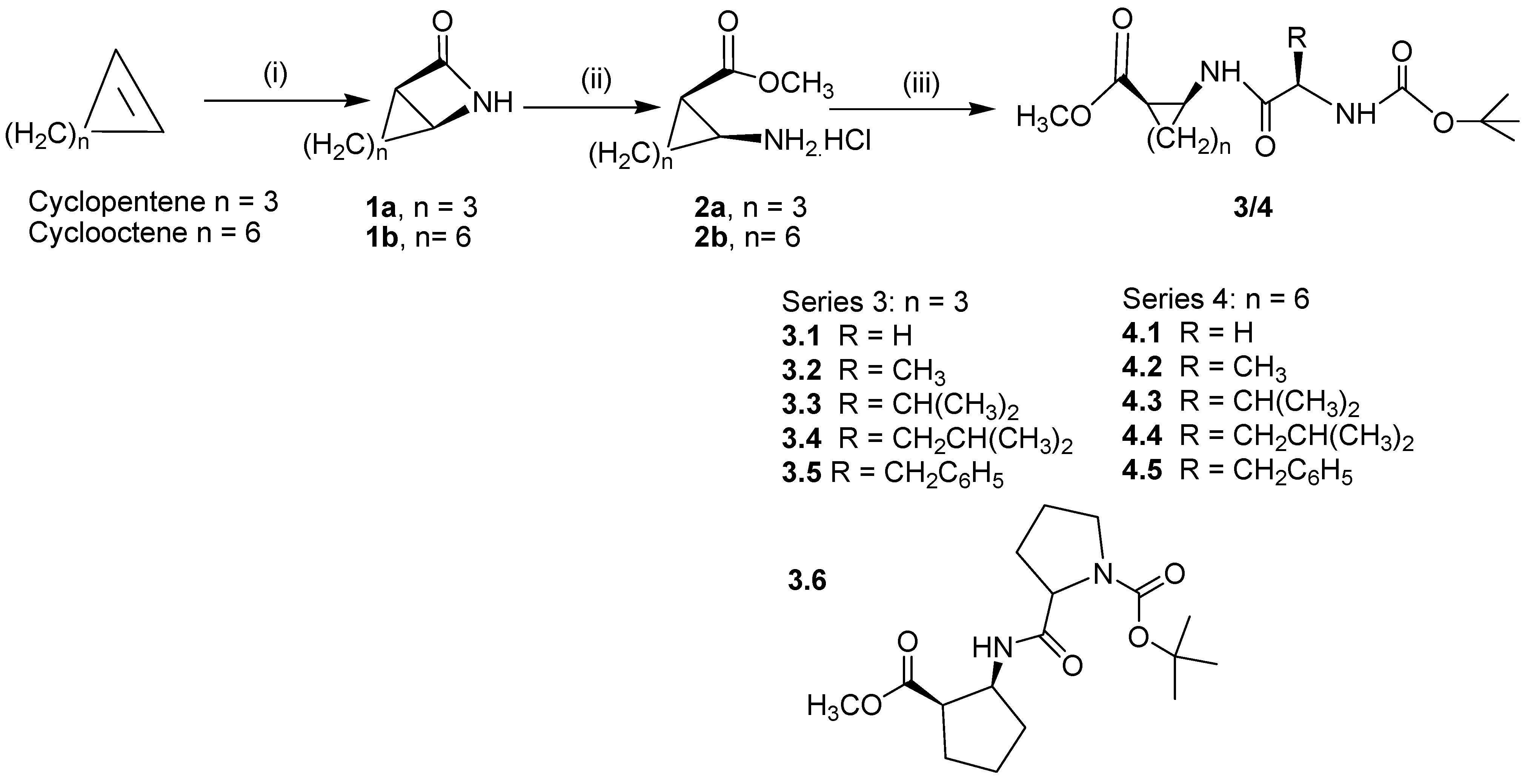
Result and Discussion

In vitro and in vivo evaluation of antimalarial activity

{kind=link}
| Compound | IC50 | IC90 | IC99 | |||
|---|---|---|---|---|---|---|
| CS | CR | CS | CR | CS | CR | |
| 3.1 | 18.44 | 19.87 | 25.64 | 29.87 | 31.45 | 36.36 |
| 3.2 | 17.14 | 21.87 | 24.36 | 31.45 | 32.14 | 36.31 |
| 3.3 | 12.14 | 14.52 | 13.52 | 17.14 | 36.89 | 39.87 |
| 3.4 | 9.41 | 9.21 | 9.12 | 10.52 | 11.42 | 12.36 |
| 3.5 | 8.96 | 9.67 | 11.36 | 14.31 | 12.89 | 15.87 |
| 3.6 | 14.56 | 16.31 | 14.87 | 15.83 | 16.21 | 17.33 |
| 4.1 | 12.11 | 11.21 | 4.11 | 4.52 | 6.11 | 6.88 |
| 4.2 | 9.17 | 8.14 | 7.21 | 8.31 | 7.98 | 8.47 |
| 4.3 | 6.14 | 7.39 | 10.88 | 9.13 | 11.43 | 10.19 |
| 4.4 | 3.87 | 3.53 | 3.42 | 6.21 | 4.54 | 7.41 |
| 4.5 | 3.64 | 3.54 | 19.55 | 21.69 | 31.48 | 39.78 |
| Chloroquine | 0.0241 | 0.038 | ||||
| Compound | Dose (mg/kg/day) | Percent suppressiona on day 4 | Mice alive on day 28 |
|---|---|---|---|
| 3.4 | 100 | 36.22 | 0/5 |
| 50 | Nil | 0/5 | |
| 3.5 | 100 | 21.43 | 0/5 |
| 50 | Nil | 0/5 | |
| 4.2 | 100 | 24.31 | 0/5 |
| 50 | 6.21 | 0/5 | |
| 4.3 | 100 | 32.65 | 1/5 |
| 50 | 12.24 | 0/5 | |
| 4.4 | 100 | 58.28 | 2/5 |
| 50 | 21.22 | 1/5 | |
| 4.5 | 100 | 85.29 | 0/5 |
| 50 | 48.68 | 2/5 | |
| Chloroquine | 8 | 100 | 5/5 |
Conclusions
Experimental
General
General procedure for the synthesis of β-lactams 1: 9-aza-bicyclo[6.2.0]decan-10-one (1b, n = 6)
General procedure for the synthesis of 2-aminocyclooctanecarboxylic acid methyl ester hydrochloride, (2b, n = 6)
Typical experimental procedure for the prepartion of compounds 3.1-3.6: 2-(2-tert-butoxycarbonyl-aminoacetylamino)-cyclopentanecarboxylic acid methyl ester (3.1)
Typical experimental procedure for the prepartion of compounds 4.1-4.5: 2-(2-tert-butoxycarbonyl-aminoacetylamino)-cyclooctanecarboxylic acid methyl ester (4.1)
In vitro antimalarial evaluation
In vivo antimalarial evaluation
Acknowledgements
References and Notes
- Wyler, D.J. Malaria: overview and update. Clin. Infect. Dis. 1993, 16, 449–456. [Google Scholar] Kano, S. Global malaria control into the 21st century. Prog. Med 2001, 21, 319–324. [Google Scholar] Greenwood, B.; Mutabingwa, T. Malaria in 2002. Nature 2002, 415, 670–672. [Google Scholar]
- Ridley, R.G. Enhanced: planting the seeds of new antimalarial drugs. Science 1999, 285, 1502–1503. [Google Scholar]
- Murray, M.C.; Perkin, M.E. The synthesis and testing of arenearylpyrimidylmethanes as antimalarial agents. Annu. Rep. Med. Chem. 1996, 31, 141–150. [Google Scholar]
- Hyde, J.E. Mechanisms of resistance of Plasmodium falciparum to antimalarial drugs. Microb. Inf. 2002, 4, 165–174. [Google Scholar]
- Alisky, J.M.; Chertkova, E.L.; Iczkowski, K.A. Drug interactions and pharmacogenetic reactions are the basis for chloroquine and mefloquine-induced psychosis. Med. Hypoth. 2006, 67, 1090–1094. [Google Scholar]
- Cook, G.C. Fatal Stevens-Johnson syndrome associated with fansidar and chloroquine. Lancet 1985, 14, 92. [Google Scholar] [CrossRef] Dua, V.K.; Sarin, R.; Sharma, V.P. Sulphadoxine concentrations in plasma, red blood cells and whole blood in healthy and Plasmodium falciparum malaria cases after treatment with Fansidar using high-performance liquid chromatography. J. Pharm. Bio. Med. Anal. 1994, 12, 1317–1323. [Google Scholar] Rombo, L.; Stenbeck, J.; Lobel, H.O.; Campbell, C.; Papaionou, M.; Miller, K. Does chloroquine contribute to the risk of serious adverse reactions to fansidar? Lancet 1985, 326, 1298–1299. [Google Scholar]
- Werbovetz, K.A. Target-based drug discovery for malaria, leishmaniasis, and trypanosomiasis. Curr. Med. Chem. 2000, 7, 835–860. [Google Scholar] [CrossRef] [PubMed]
- Berry, C. Synthesis of plasmepsin II inhibitors–potential antimalarial agents. Curr. Opin. Drug Discovery Dev. 2003, 3, 624–629. [Google Scholar]
- Blackman, M.J. Proteases involved in erythrocyte invasion by the malaria parasite function and potential as chemotherapeutic targets. Curr. Drug Targets 2000, 1, 59–83. [Google Scholar] Francis, S. E.; Sullivan, D.J., Jr.; Goldberg, D.E. Hemoglobin metabolism in the malaria parasite plasmodium falciparum. Annu. Rev. Microbiol 1997, 51, 97–123. [Google Scholar]
- Haque, T.S.; Skillman, A.G.; Lee, T.S.; Habashita, H.; Gluzman, I.Y.; Ewing, T.J.A.; Goldberg, D.E.; Kuntz, I.D.; Ellman, J.A. Potent, low–molecular weight non–peptide inhibitors of malarial aspartyl protease plasmepsin II. J. Med. Chem. 1999, 42, 1428–1440. [Google Scholar] [CrossRef] [PubMed]
- Weiesner, J.; Ortmann, R.; Jomaa, H..; Schlitzer, M. New antimalarial drugs. Angew. Chem. Int Ed. 2003, 42, 5274–5293. [Google Scholar] Singh, S.B.; Zink, D.L.; Polishook, J.D.; Dombrowski, A. W.; Darkin-Rattray, S.J.; Schmatz, D.M.; Goetz, M.A. Apicidins: Novel cyclic tetrapeptides as coccidiostats and antimalarial agents from Fusarium pallidoroseum. Tetrahedron Lett. 1996, 37, 8077–8080. [Google Scholar]
- Thongtan, J.; Saenboonrueng, J.; Rachtawee, P.; Isaka, M. An Antimalarial Tetrapeptide from the Entomopathogenic Fungus Hirsutella sp. BCC 1528. J. Nat. Prod. 2006, 69, 713–714. [Google Scholar]
- Fulop, F. The chemistry of 2-aminocycloalkanecarboxylic acids. Chem. Rev. 2001, 101, 2181–2204. [Google Scholar] Fulop, F.; Martinek, T.A.; Toth, G.K. Application of alicyclic β–amino acids in peptide chemistry. Chem. Soc. Rev. 2006, 35, 323–334. [Google Scholar]
- Knapp, S. Synthesis of complex nucleoside antibiotics. Chem. Rev. 1995, 95, 1859–1876. [Google Scholar] [CrossRef]
- Cativiela, C.; Diaz-de-Villegas, M.D. Asymmetric synthesis of α-amino acids using polymer-supported Cinchona alkaloid-derived ammonium salts as chiral phase-transfer catalysts. Tetrahedron Asymmetry 2000, 11, 645–732. [Google Scholar] Lambert, J.N.; Mitchell, J.P.; Roberts, K. The synthesis of cyclic peptides. J. Chem. Soc., Perkin Trans. 1 2001, 471–484. [Google Scholar]
- Parsons, P.J.; Camp, N.P.; Underwood, J.M.; Harvey, M.D. Tandem reactions of anions: A short and efficient route to ± Anatoxin-a. Tetrahedron 1996, 52, 11637–11642. [Google Scholar] Szakonyi, Z.; Fulo, F.; Bernath, G.; Evanics, F.; Riddell, G. Synthesis and ring–chain tautomerism of angularly substituted cycloalkane–fused tetrahydro–1,3–oxazines. Tetrahedron 1998, 54, 1013–1020. [Google Scholar] Szakonyi, Z.; Martinek, T.; Sillanpaeae, R.; Fulop, F. Regio- and stereoselective synthesis of the enantiomers of monoterpene-based beta-amino acid derivatives. Tetrahedron Asymmetry 2007, 18, 2442–2447. [Google Scholar] Sathe, M.; Ghorpade, R.; Kaushik, M.P. Highly efficient methanolysis of bicyclic β-lactams to β-amino ester using silica chloride. Chem. Lett. 2006, 35, 1004–1005. [Google Scholar]
- Trager, W.; Jensen, J.B. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Contact the authors.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Sathe, M.; Thavaselvam, D.; Srivastava, A.K.; Kaushik, M.P. Synthesis and Antimalarial Evaluation of Cyclic β-Amino Acid-Containing Dipeptides. Molecules 2008, 13, 432-443. https://doi.org/10.3390/molecules13020432
Sathe M, Thavaselvam D, Srivastava AK, Kaushik MP. Synthesis and Antimalarial Evaluation of Cyclic β-Amino Acid-Containing Dipeptides. Molecules. 2008; 13(2):432-443. https://doi.org/10.3390/molecules13020432
Chicago/Turabian StyleSathe, Manisha, D. Thavaselvam, A. K. Srivastava, and M. P. Kaushik. 2008. "Synthesis and Antimalarial Evaluation of Cyclic β-Amino Acid-Containing Dipeptides" Molecules 13, no. 2: 432-443. https://doi.org/10.3390/molecules13020432
APA StyleSathe, M., Thavaselvam, D., Srivastava, A. K., & Kaushik, M. P. (2008). Synthesis and Antimalarial Evaluation of Cyclic β-Amino Acid-Containing Dipeptides. Molecules, 13(2), 432-443. https://doi.org/10.3390/molecules13020432