Application of Prodrugs to Inflammatory Diseases of the Gut

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction
Inflammatory Bowel Disease
Pathogenesis of IBD
LPS and TNF production and IBD
NF-κB and IBD
Reactive Oxygen Species (ROS)
The Prodrug Approach
Anti-Inflammatory Prodrugs:
5-Aminosalicylic acid

Acid-regulation and short chain fatty acids
Short chain fatty acids
Carnitine (β-hydroxy-γ-trimethylaminobutyrate)
Immunomodulators and Immunosuppressors
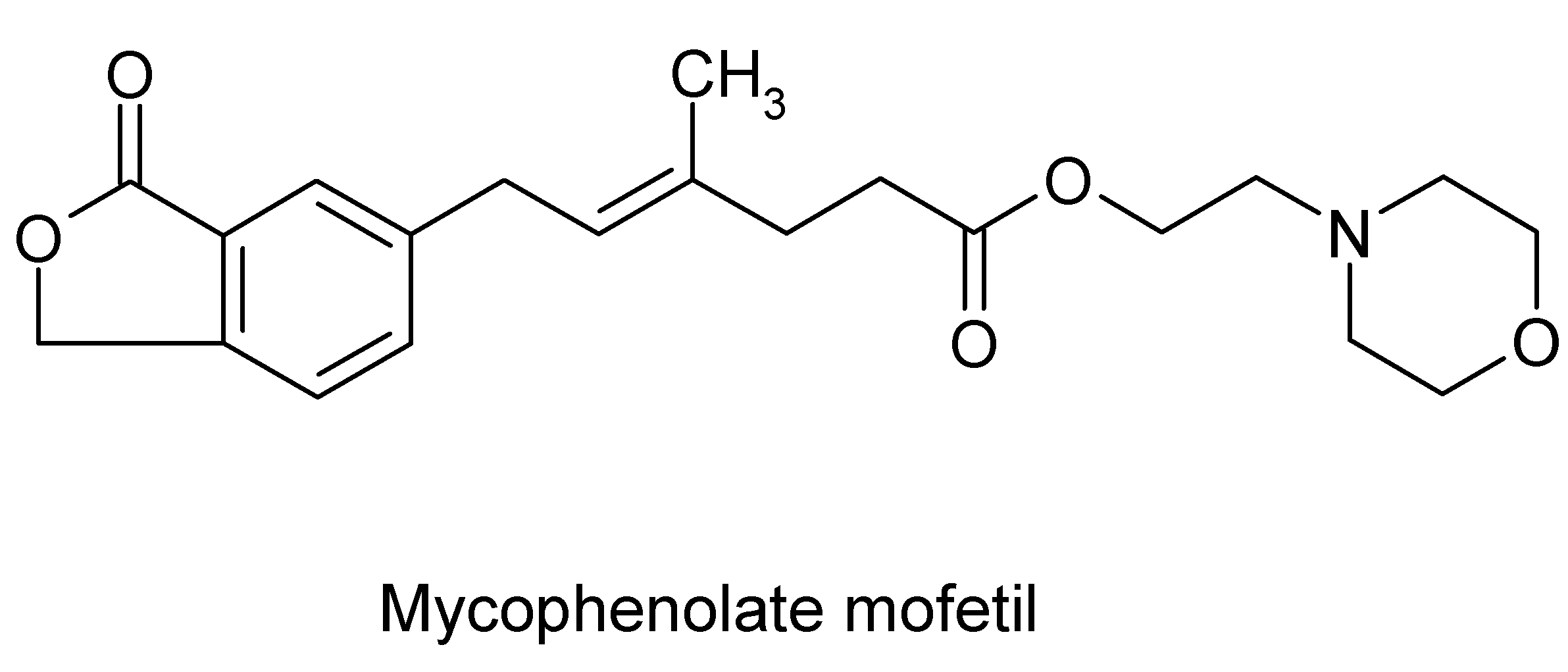
Mycophenolate mofetil

Azathioprine
Glucocorticosteroids
Antioxidants
Glutathione (GSH)
Cysteine Prodrugs:
Cysteine and glutathione prodrugs



S-adenosylmethionine (SAMe, Adomet) Metabolism and Gut

Conclusions
Acknowledgements
References
- Tursi, A.; Brandimarte, G.; Giorgetti, G.M.; Nasi, G. Assessment of orocaecal transit time in different localization of Crohn's disease and its possible influence on clinical response to therapy. Eur. J. Gastroenterol. Hepatol. 2003, 15, 69–74. [Google Scholar] [CrossRef]
- Schwab, M.; Klotz, U. Pharmacokinetic considerations in the treatment of inflammatory bowel disease. Clin. Pharmacokinet. 2001, 40, 723–751. [Google Scholar] [CrossRef]
- Dressman, J.B.; Vertzoni, M.; Goumas, K.; Reppas, C. Estimating drug solubility in the gastrointestinal tract. Adv. Drug Deliv. Rev. 2007, 59, 591–602. [Google Scholar] [CrossRef]
- Albert, A. Chemical aspects of selective toxicity. Nature 1958, 182, 421–422. [Google Scholar] [CrossRef]
- Stella, V.J.; Charman, W.N.A.; Naringrekar, V.H. Prodrugs. Do they have advantages in clinical practice? Drugs 1985, 29, 455–473. [Google Scholar] [CrossRef]
- Bundgaard, H. Design and application of prodrugs. In A Textbook of Drug Design and Development; Krogsgaard-Larsen, P., Bundgaard, H., Eds.; Harwood: Reading, UK, 1991; pp. 113–191. [Google Scholar]
- Ettmayer, P.; Amidon, G.L.; Clement, B.; Testa, B. Lessons learned from marketed and investigational prodrugs. J. Med. Chem. 2004, 47, 2393–2404. [Google Scholar] [CrossRef]
- Han, H.K.; Amidon, G.L. Targeted prodrug design to optimize drug delivery. AAPS Pharm. Sci. 2000, 2, E6. [Google Scholar]
- Rubinstein, A. Approaches and opportunities in colon-specific drug delivery. Crit. Rev. Ther. Drug Carr. Syst. 1995, 12, 101–149. [Google Scholar] [CrossRef]
- Hovgaard, L.; Brondsted, H. Current applications of polysaccharides in colon targeting. Crit. Rev. Ther. Drug Carr. Syst. 1996, 13, 185–223. [Google Scholar] [CrossRef]
- Fang, L.; Battisti, R.F.; Cheng, H.; Reigan, P.; Xin, Y.; Shen, J; Ross, D.; Chan, K.K.; Martin, J.; Wang, P.G.; Sun, D. Enzyme specific activation of benzoquinone ansamycin prodrugs using HuCC49DeltaCH2-beta-Galactosidase conjugates. J. Med. Chem. 2006, 49, 6290–6297. [Google Scholar] [CrossRef]
- Curini, M.; Epifano, F.; Genovese, S. Synthesis of a novel prodrug of 3-(4′-geranyloxy-3′-methoxyphenyl)-2-trans-propenoic acid for colon delivery. Bioorg. Med. Chem. Lett. 2005, 15, 5049–5052. [Google Scholar] [CrossRef]
- Podolskiy, D.K. Inflammatory bowel disease. N. Engl. J. Med. 2002, 347, 417–429. [Google Scholar] [CrossRef]
- Hutfless, S.M.; Weng, X.; Liu, L.; Allison, J.; Herrinton, L.J. Mortality by Medication use among patients with inflammatory bowel disease, 1996-2003. Gastroenterology 2007, 133, 1779–1786. [Google Scholar] [CrossRef]
- Cohen, R.D.; Woseth, D.M.; Thistel, R.A.; Hanauer, S.B. A meta-analysis and overview of the literature on treatment options for left-sided ulcerative colitis and ulcerative proctitis. Am. J. Gastroenterol. 2000, 95, 1263–1276. [Google Scholar] [CrossRef]
- Li, M.C.; He, S.H. IL-10 and its related cytokines for treatment of inflammatory bowel disease. World J. Gastroenterol. 2004, 10, 620–625. [Google Scholar]
- Neurath, M.F.; Pettersson, S.; Strober, W. Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-kB abrogates established experimental colitis in mice. Nature Med. 1996, 2, 998–1004. [Google Scholar] [CrossRef]
- Neuman, M.G. Immune dysfunction in inflammatory bowel disease. Transl. Res. 2007, 149, 173–186. [Google Scholar] [CrossRef]
- Kozuch, P.L.; Hanauer, S.B. General principles and pharmacology of biologics in inflammatory bowel disease. Gastroenterol. Clin. North Am. 2006, 35, 757–773. [Google Scholar] [CrossRef]
- Kozuch, P.L.; Hanauer, S.B. Treatment of inflammatory bowel disease: A review of medical therapy. World J. Gastroenterol. 2008, 4, 354–377. [Google Scholar] [CrossRef]
- Sartor, R.B. Enteric microflora in IBD: pathogens or commensals? Inflamm. Bowel Dis. 1997, 3, 230–235. [Google Scholar] [CrossRef]
- Robertson, D.J; Sandler, R.S. Measles virus and Crohn’s disease: a critical appraisal of the current literature. Inflamm. Bowel Dis. 2001, 7, 51–57. [Google Scholar] [CrossRef]
- Sartor, RB. Microbial factors in the pathogenesis of Crohn’s disease, ulcerative colitis, and experimental intestinal inflammation. In InInflammatory Bowel Disease; Kirsner, J.B., Shorter, R.G., Eds.; Williams and Wilkens: Baltimore, 1995; pp. 96–124. [Google Scholar]
- Elson, C.O.; Sartor, R.B.; Tennyson, G.S.; Riddell, R.H. Insights into the pathogenesis of IBD provided by new rodent models of colitis. Inflam. Bowel Dis. 1995, 1, 64–75. [Google Scholar]
- Sadlack, B.; Merz, H.; Schorle, H.; Schimpl, A.; Feller, A.C.; Horak, I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell 1993, 75, 255–261. [Google Scholar]
- Sellon, R.K.; Tonkonogy, S.; Schultz, M.; Dieleman, L.A.; Grenther, W.; Balish, E.; Rennick, D.M.; Sartor, R.B. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in IL-10-deficient mice. Infect. Immun. 1998, 66, 5224–5231. [Google Scholar]
- MacDermott, R.P.; Stenson, W.F. Inflammatory Bowel Disease. In Immunology and Immunopathology of the liver and gastrointestinal tract; Targan, S.R., Shanahan, F., Eds.; Igaku-Shoin: New York, 1990; pp. 459–486. [Google Scholar]
- Gaetke, L.M.; Frederich, R.C.; Oz, H.S.; McClain, C.J. Zinc deficiency induced changes in plasma leptin, metabolic rate, and physical activity in rats. J. Nutr. Biochem. 2002, 13, 237–244. [Google Scholar] [CrossRef]
- Grave, G. Antioxidant nutrients in inflammatory bowel disease. In Progress in inflammatory bowel disease, Trends in IBD Care; CCFA: New York, 1994; pp. 12–14. [Google Scholar]
- Hatoum, O.A.; Binion, D.G.; Gutterman, D.D. Paradox of simultaneous intestinal ischaemia and hyperaemia in inflammatory bowel disease. Eur. J. Clin. Invest. 2005, 35, 599–609. [Google Scholar] [CrossRef]
- Berg, R.D. The indigenous gastrointestinal microflora. Trends Microbiol. 1996, 4, 430–435. [Google Scholar] [CrossRef]
- Shanahan, F. Probiotics in inflammatory bowel disease – therapeutic rationale and role. Drug Deliv. Rev. 2004, 56, 809–818. [Google Scholar] [CrossRef]
- Wang, J.H.; Redmond, H.P.; Watson, R.W.; Bouchei-Hayes, D. Role of lipopolysaccharide and tumor necrosis factor-alpha in induction of hepatocyte necrosis. Am. J. Physiol. 1995, 269, G297–G304. [Google Scholar]
- Beutler, B.; Kruys, V. Lipopolysaccharide signal transduction, regulation of tumor necrosis factor biosynthesis, and signaling by tumor necrosis factor itself. J. Cardiovasc. Pharmacol. 1995, 25 (suppl. 2), S1–S8. [Google Scholar] [CrossRef]
- Pastor Rojo, O.; López San Román, A.; Albéniz Arbizu, E.; de la Hera Martínez, A.; Ripoll Sevillano, E.; Albillos Martínez, A. Serum lipopolysaccharide-binding protein in endotoxemic patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 269–277. [Google Scholar] [CrossRef]
- Gaetke, L.; Oz, H.S.; Frederich, R.; McClain, C. Anti-TNF-α Antibody Normalizes Serum Leptin in IL-2 Deficient Mice. J. Amer. Coll. Nutri. 2003, 22, 415–420. [Google Scholar] [CrossRef]
- Conner, E.M.; Brand, S.; Davis, J.M.; Laroux, F.S.; Palombella, V.J.; Fuseler, J.W.; Kang, D.Y.; Wolf, R.E.; Grisham, M.B. Proteasome inhibition attenuates nitric oxide synthase expression, VCAM-1 transcription and the development of chronic colitis. J. Pharmacol. Exp. Ther. 1997, 282, 1615–1622. [Google Scholar]
- Scheinman, R.I.; Cogswell, P.C.; Lofquist, A.K.; Baldwin, A.S. Role of transcriptional activation of IkB-α in mediation of immunosuppression by glucocorticoids. Science 1995, 270, 283–286. [Google Scholar]
- Auphan, N.; DiDonato, J.A.; Rosette, C.; Helmberg, A.; Karin, M. Immunosuppression by glucocorticoids: inhibition of NF-ĸB activity through induction of IkB synthesis. Science 1995, 270, 286–290. [Google Scholar]
- Wahl, C.; Liptay, S.; Adler, G.; Schmid, R.M. Sulfasalazine: a potent and specific inhibitor of nuclear factor kappa B. J. Clin. Invest. 1998, 101, 1163–1174. [Google Scholar] [CrossRef]
- Yang, F.; de Villiers, W.J.S.; McClain, C.J.; Varilek, G.W. Mesalamine (5-ASA) modulates NF-kB/ IkB-α activity In Caco-2 cells. Gastroenterology 1997, 112, A1125. [Google Scholar]
- Meyer, S.; Kohler, N.G.; Joly, A. Cyclosporine A is an uncompetitive inhibitor of proteasome activity and prevents NF-κB activation. FEBS Lett. 1997, 413, 354–358. [Google Scholar] [CrossRef]
- Grisham, M.B.; Granger, D.N. Neutrophil-mediated mucosal injury. Role of reactive oxygen metabolites. Dig. Dis. Sci. 1988, 33, 6S–15S. [Google Scholar] [CrossRef]
- Beckman, K.B.; Ames, B.N. Oxidative decay of DNA. J. Biol. Chem. 1997, 272, 19633–19636. [Google Scholar] [CrossRef] [Green Version]
- Babbs, C.F. Oxygen radicals in ulcerative colitis. Free Radic. Biol. Med. 1992, 13, 169–181. [Google Scholar] [CrossRef]
- Suematsu, M.; Suzuki, M.; Kitahora, T.; Miura, S.; Suzuki, K.; Hibi, T.; Watanabe, M.; Nagata, H.; Asakura, H.; Tsuchiya, M. Increased respiratory burst of leukocytes in inflammatory bowel diseases–the analysis of free radical generation by using chemiluminescence probe. J. Clin. Lab. Immunol. 1987, 24, 125–128. [Google Scholar]
- Simmonds, N.J.; Allen, R.E.; Stevens, T.R.; Van Someren, R.N.; Blake, D.R.; Rampton, D.S. Chemiluminescence assay of mucosal reactive oxygen metabolites in inflammatory bowel disease. Gastroenterology 1992, 103, 186–196. [Google Scholar]
- Shiratora, Y.; Aoki, S.; Takada, H.; Kiriyama, H.; Ohto, K.; Hai, K.; Teraoka, H.; Matano, S.; Matsumoto, K.; Kamii, K. Oxygen-derived free radical generating capacity of polymorphonuclear cells in patients with ulcerative colitis. Digestion 1989, 44, 163–171. [Google Scholar] [CrossRef]
- Stein, R.B.; Hanauer, S.B. Comparative tolerability of treatments for inflammatory bowel disease. Drug Saf. 2000, 23, 429–448. [Google Scholar] [CrossRef]
- Dubuquoy, L.; Rousseaux, C.; Thuru, X.; Peyrin-Biroulet, L.; Romano, O.; Chavatte, P.; Chamaillard, M.; Desreumaux, P. PPARγ as a new therapeutic target in inflammatory bowel disease. Gut 2006, 55, 1341–1349. [Google Scholar] [CrossRef]
- Sands, B.E. Therapy of inflammatory bowel disease. Gastroenterology 2000, 118, S68–S82. [Google Scholar] [CrossRef]
- Kinget, R.; Kalala, W.; Vervoort, L.; Mooter, G.U. Colonic drug targeting. J. Drug. Target. 1998, 6, 129–149. [Google Scholar] [CrossRef]
- Scheline, R. Metabolism of foreign compounds by gastrointestinal microorganisms. Pharmacol. Rev. 1973, 25, 451–523. [Google Scholar]
- Hanauer, S.B. Review article: high-dose aminosalicylates to induce and maintain remissions in ulcerative colitis. Aliment. Pharmacol. Ther. 2006, Suppl 3, 37–40. [Google Scholar] [CrossRef]
- Staerk Laursen, L.; Stokholm, M.; Bukhave, K.; Rask-Madsen, J.; Lauritsen, K. Disposition of 5-aminosalicylic acid by olsalazine and three mesalazine preparations in patients with ulcerative colitis: comparison of intraluminal colonic concentrations, serum values, and urinary excretion. Gut 1990, 31, 1271–1276. [Google Scholar] [CrossRef]
- Dhaneshwar, S.S.; Gairola, N.; Kandpal, M.; Vadnerkar, G.; Bhatt, L. Colon-specific, mutual azo prodrug of 5-aminosalicylic acid with L-tryptophan: synthesis, kinetic studies and evaluation of its mitigating effect in trinitrobenzenesulfonic acid- induced colitis in rats. Bioorg. Med. Chem. 2007, 15, 4903–4909. [Google Scholar] [CrossRef]
- Bishop, J.; Furman, M.; Thomson, M. Omeprazole for gastroesophageal reflux disease in the first 2 years of life: a dose-finding study with dual-channel pH monitoring. J. Pediatr. Gastroenterol. Nutr. 2007, 45, 50–55. [Google Scholar] [CrossRef]
- Oosterhuis, B.; Jonkman, J.H. Omeprazole: pharmacology, pharmacokinetics and interactions. Digestion 1989, 44 suppl, 9–17. [Google Scholar] [CrossRef]
- Topping, D.L.; Clifton, P.M. Short chain fatty acids and human colonic function: roles of resistant starch and non-starch polysaccharides. Physiol. Rev. 2001, 81, 1031–1064. [Google Scholar]
- Bergman, E.N. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 1990, 70, 567–590. [Google Scholar]
- Brouns, F.; Kettlitz, B.; Arrigoni, E. Resistant starch and "butyrate revolution". Trends Food Sci. Technol. 2002, 13, 251–261. [Google Scholar] [CrossRef]
- Caderni, G.; Luceri, C.; Lancioni, L.; Tessitore, L.; Dolara, P. Slow-release pellets of sodium butyrate increase apoptosis in the colon of rats treated with azoxymethane, without affecting aberrant crypt foci and colonic proliferation. Nutr. Cancer 1998, 30, 175–181. [Google Scholar] [CrossRef]
- Scheppach, W.; Sommer, H.; Kirchner, T.; Paganelli, G.M.; Bartram, P.; Christl, S.; Richter, F.; Dusel, G.; Kasper, H. Effect of butyrate enema on the colonic mucosa in distal ulcerative colitis. Gasteroentrology 1992, 103, 51–56. [Google Scholar]
- Illman, R.J.; Topping, D.L.; McIntosh, G.H.; Trimble, R.P.; Storer, G.B.; Taylor, M.N. The hypocholesterolaemic effects of dietary propionate: studies in whole animals and perfused rat liver. Ann. Nutr. Metab. 1988, 32, 97–107. [Google Scholar] [CrossRef]
- Annison, G.; Illman, R.J.; Topping, D.L. Acetylated propionylated or butyrylated starches raise large bowel short-chain fatty acids preferentially when fed to rats. Nutrition 2003, 133, 3523–3528. [Google Scholar]
- Bennett, M.J.; Rinaldo, P.; Strauss, A.W. Inborn errors of mitochondrial fatty acid oxidation. Crit. Rev. Clin. Lab. Sci. 2000, 37, 1–44. [Google Scholar] [CrossRef]
- Rinaldo, P. Fatty acid transport and mitochondrial oxidation disorders. Semin. Liver Dis. 2001, 21, 489–500. [Google Scholar] [CrossRef]
- Rinaldo, P.; Matern, D.; Bennett, M.J. Fatty acid oxidation disorders. Annu. Rev. Physiol. 2002, 64, 477–502. [Google Scholar] [CrossRef]
- Srinivas, S.R.; Prasad, P.D.; Umapathy, N.D.; Ganapathy, V.; Shekhawat, P.S. Transport of butyryl-L-carnitine, a potential prodrug, via the carnitine transporter OCTN2 and the amino acid transporter ATB0,+. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G1046–G1053. [Google Scholar]
- Lipsky, J.J. Mycophenolate mofetil. Lancet 1996, 348, 1357–1359. [Google Scholar] [CrossRef]
- Gosio, B. Riceche bacteriologiche e chimiche sulle alterazioni del mais. Riv. Igiene. Sanita Pubblica 1896, 7, 825–868. [Google Scholar]
- Hupe, D.J.; Azzolina, B.A.; Behrens, N.D. IMP dehydrogenase from the intracellular parasitic protozoan Eimeria Tenella and its inhibition by mycophenolic acid. J. Biol. Chem. 1986, 261, 8363–8369. [Google Scholar]
- Oz, H.S.; Hughes, W. Novel anti-Pneumocystis carinii effects of the immunosuppressant mycophenolate mofetil in contrast to provocative effects of tacrolimus, sirolimus and dexamethasone. J. Infect. Dis. 1997, 175, 901–904. [Google Scholar] [CrossRef]
- Oz, H.S.; Hughes, W.T.; Varilek, G. Provocative effects of immunosuppressants rapamycin, tacrolimus and dexamethasone on P. carinii pneumonitis in contrast to anti-PCP effects of the mycophenolate mofetil. Transplant 2001, 72, 1464–1465. [Google Scholar] [CrossRef]
- Azevedo, L.S.; Castro, M.C.R.; Paula, F.J.; Ianhez, L.E.; David-Neto, E. Mycophenolate mofetil may protect against Pneumocystis carinii pneumonia in renal patients. Rev. Inst. Med. Trop. S. Paulo 2005, 47, 143–145. [Google Scholar] [CrossRef]
- Zeeh, J.M.; Zorlu, Riley, N.E.; Hoffmann, P.; Ruwe, M.; Goebell, H.; Gerken, G.; Dignass, A.U. Mycophenolate mofetil reduces tissue damage and inflammation in an experimental model of colitis in rats. Scand. J. Gastroenterol. 2001, 36, 66–70. [Google Scholar]
- Neurath, M.F.; Wanitschke, R.; Peters, M.; Krummenauer, F.; Meyer, zum Buschenfelde, K.H.; Schlaak, J.F. Randomised trial of mycophenolate mofetil versus azathioprine for treatment of chronic active Crohn’s disease [comment]. Gut 1999, 44, 625–628. [Google Scholar] [CrossRef]
- Orth, T.; Peters, M.; Schlaak, J.F.; Krummenauer, F.; Wanitschke, R.; Mayet, W.J.; Galle, P.R.; Neurath, M.F. Mycophenolate mofetil versus azathioprine in patients with chronic active ulcerative colitis: a 12-month pilot study. Am. J. Gastroenterol. 2000, 95, 1201–1207. [Google Scholar] [CrossRef]
- Florin, T.H.; Roberts, R.K.; Watson, M.R.; Radford-Smith, G.L. Treatment of steroid refractory inflammatory bowel disease (IBD) with mycophenolate mofetil (MMF). Aust. N. Z. J. Med. 1998, 28, 344–345. [Google Scholar] [CrossRef]
- Miehsler, W.; Reinisch, W.; Moser, G.; Gangl, A.; Vogelsang, H. Is mycophenolate mofetil an effective alternative in azathioprine-intolerant patients with chronic active Crohn’s disease. Am. J. Gastroenterol. 2001, 96, 782–787. [Google Scholar] [CrossRef]
- Golconda, M.S.; Valente, J.F.; Bejarano, P.; Gilinsky, N.; First, MR. Mycophenolate mofetil induced colonic ulceration in renal transplant recipients. Transplant. Proc. 1999, 31, 272–273. [Google Scholar] [CrossRef]
- Shaw, L.M.; Nowak, I. Mycophenolic acid: measurement and relationship to pharmacologic effects. Ther. Drug Monit. 1995, 17, 685–689. [Google Scholar] [CrossRef]
- Loftus, C.G.; Egan, L.J.; Sandborn, W.J. Cyclosporine, tacrolimus, and mycophenolate mofetil in the treatment of inflammatory bowel disease. Gastroenterol. Clin. N. Am. 2004, 33, 141–169. [Google Scholar] [CrossRef]
- Breen, D.P.; Marinaki, A.M.; Arenas, M.; Hayes, P.C. Pharmacogenetic association with adverse drug reactions to azathioprine immunosuppressive therapy following liver transplantation. Liver Transpl. 2005, 11, 826–833. [Google Scholar] [CrossRef]
- Marín-Jimenez, I.; Pena, A.S. Budesonide for ulcerative colitis. Rev. Esp. Enferm. Dig. 2006, 98, 362–373. [Google Scholar]
- Parks, D.A.; Bulkley, G.B.; Granger, D.N. Role of oxygen-derived free radicals in digestive tract diseases. Surgery 1983, 94, 415–422. [Google Scholar]
- Tsuchiya., M. Free radicals in digestive diseases. In Proceedings of the 1st International Symposium on Free Radicals in Digestive Diseases; Excerpta Medica: New York, 1988; p. 244. [Google Scholar]
- Baker, S.S.; Campbell, C.L. Enterocyte injury by O2-dependent processes. Gastroenterology 1991, 101, 716–720. [Google Scholar]
- Mrtensson, J.; Jain, A.; Meister, A. Glutathione is required for intestinal function. Proc. Nat. Acad. Sci. 1990, 87, 1715–1719. [Google Scholar] [CrossRef] [Green Version]
- Uhlig, S.; Wendel, A. The physiological consequences of glutathione variations. Life. Sci. 1992, 51, 1083–1094. [Google Scholar] [CrossRef]
- Mercier, S.; Breuille, D.; Mosoni, L.; Obled, C.; Patureau Mirand, P. Chronic Inflammation Alters Protein Metabolism in Several Organs of Adult Rats. J. Nutr. 2002, 132, 1921–1928. [Google Scholar]
- Oz, H.S.; Chen, T.; McClain, C.J.; deVilliers, W. Antioxidants a novel therapy in a murine model of colitis. J. Nutri. Biochem. 2005, 16, 297–304. [Google Scholar] [CrossRef]
- Dieleman, L.A.; Palmen, M. J.; Akol, H.; Bloemena, E.; Pena, A.S.; Meuwissen, S.G.; et al. Chronic experimental colitis induced by dextran sulfate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin. Exp. Immunol. 1998, 114, 385–391. [Google Scholar] [CrossRef]
- Oz, H.S.; Chen, T.; de Villiers, W.; McClain, C. Metallothionein overexpression does not protect against inflammatory bowel disease in a DSS murine colitis model. Med. Sci. Monit. 2005, 11, BR69–73. [Google Scholar]
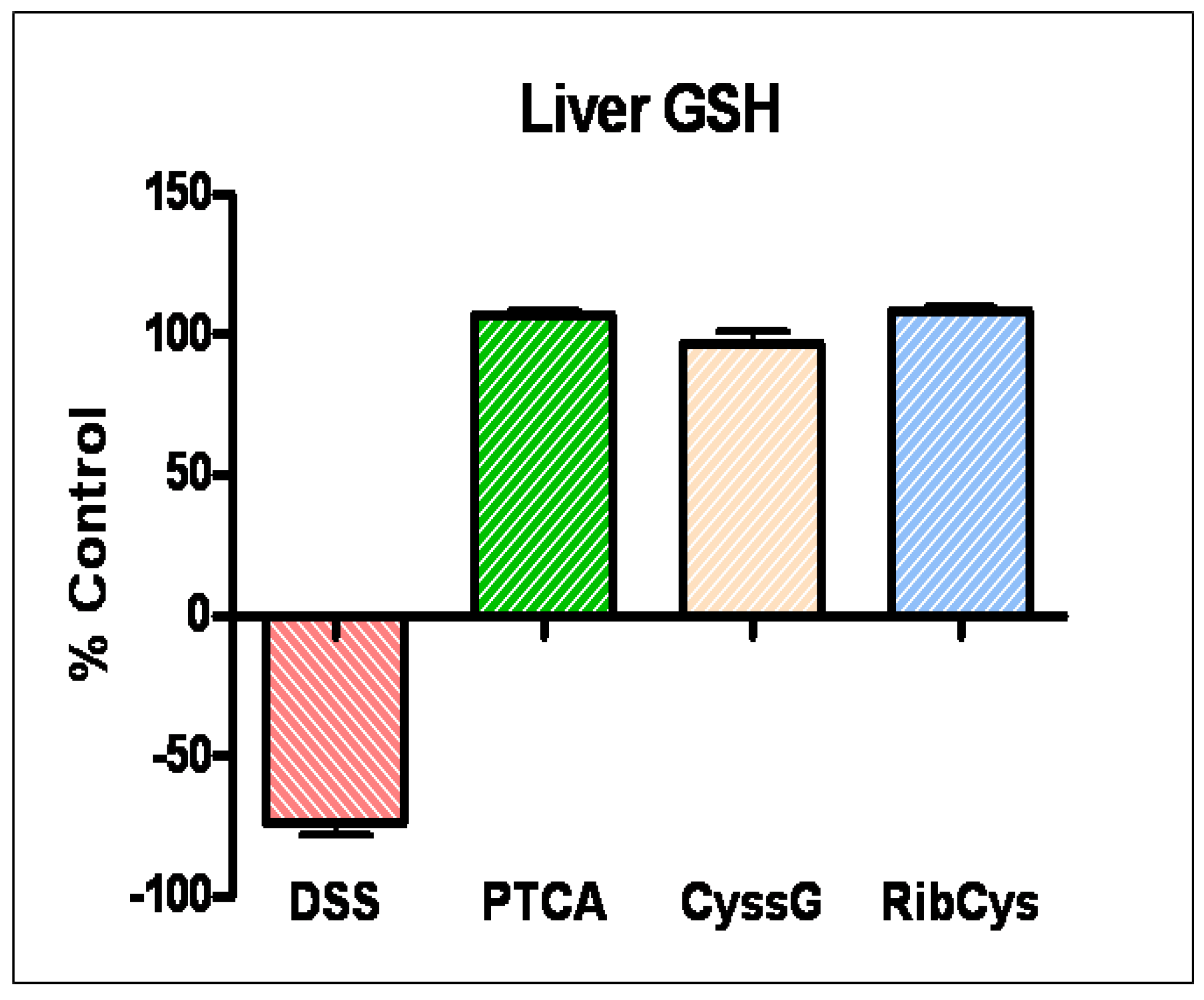
- Sido, B.; Hack, V.; Hochlehnert, A.; Lipps, H.; Herfarth, C.; Dröge, W. Impairment of intestinal glutathione synthesis in patients with inflammatory bowel disease. Gut 1998, 42, 485–492. [Google Scholar] [CrossRef]
- Chen, T.S.; Richie, J.P., Jr; Lang, C.A. Life span profiles of glutathione and acetaminophen detoxification. Drug Metab. Disp. 1990, 18, 882–887. [Google Scholar]
- Oz, H.S.; McClain, C.; Nagasawa, H.; Ray, M.; Chen, T. Diverse Antioxidants protect Against Acetaminophen Hepatotoxicity. J. Biochem. Mol. Tox. 2004, 18, 361–368. [Google Scholar]
- Robinson, M.K.; Rounds, J.D; Hong, R.W.; Jacobs, D.O.; Wilmore, D.W. Glutathione deficiency increases organ dysfunction after hemorrhagic shock. Surgery 1992, 112, 140–7, 148–149. [Google Scholar]
- Koch, T.R.; Yuan, L.X.; Fink, J.G.; Petro, A.; Opara, E.C. Induction of enlarged intestinal lymphoid aggregates during acute glutathione depletion in a murine model. Dig. Dis. Sci. 2000, 45, 2115–2121. [Google Scholar] [CrossRef]
- Tian, J.; Washizawa, N.; Gu, L.H.; Levin, M.S.; Wang, L.; Rubin, D.C.; Mwangi, S.; Srinivasan, S.; Gao, Y.; Jones, D.P.; Ziegler, T.R. Stimulation of colonic mucosal growth associated with oxidized redox status in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1081–R1091. [Google Scholar]
- Roberts, J.C.; Nagasawa, H.T.; Zera, R.T.; Fricke, R.F.; Goon, D.J. Prodrugs of L-cysteine as protective agents against acetaminophen-induced hepatotoxicity. 2-(Polyhydroxyalkyl)- and 2-(polyacetoxyalkyl)-thiazolidine-4(R)-carboxylic acids. J. Med. Chem. 1987, 30, 1891–1896. [Google Scholar] [CrossRef]
- Kleinman, W.A.; Richie Jr, J.P. Status of glutathione and other thiols and disulfides in human plasma. Biochem. Pharmacol. 2000, 60, 19–29. [Google Scholar] [CrossRef]
- Baker, D.H.; Czarnecki-Maulden, G.L. Pharmacologic role of cysteine in ameliorating or exacerbating mineral toxicities. J. Nutr. 1987, 117, 1003–1010. [Google Scholar]
- Ardite, E.; Sans, M.; Panes, J.; Romero, F.; Pique, J.M.; Fernandez-Checa, J. Replenishment of glutathione levels improves mucosal function in experimental acute colitis. Lab. Invest. 2000, 80, 735–744. [Google Scholar] [CrossRef]
- Kim, H.S.; Berstad, A. Experimental colitis in animal model. Scand. J. Gastroentrol. 1992, 27, 529–537. [Google Scholar] [CrossRef]
- Iwai, A.; Iwashita, E. Changes in colonic inflammation induced by dextran sulfate sodium (DSS) during short- and long-term administration of rebamipide. Dig. Dis. Sci. 1998, 43, 143S–147S. [Google Scholar]
- Nagasawa, H.T.; Goon, D.J.; Muldoon, W.P.; Zera, R.T. 2-Substituted thiazolidine-4(R)-carboxylic acids as prodrugs of L-cysteine, protection of mice against acetaminophen hepatotoxicity. J. Med. Chem. 1984, 27, 591–596. [Google Scholar] [CrossRef]
- Rathbun, W.B.; Nagasawa, H.T.; Killen, C.E. Prevention of naphthalene-induced cataract and hepatic glutathione loss by the L-cysteine prodrugs, MTCA and PTCA. Exp. Eye Res. 1996, 62, 433–441. [Google Scholar] [CrossRef]
- Butler, J.D.B.; Spielberg, S.P. Accumulation of cystine from glutathione-cysteine mixed disulfide in cystinotic fibroblasts; blockade by an inhibitor of γ-glutamyl transpeptidase. Life Sci. 1982, 31, 2563–2570. [Google Scholar] [CrossRef]
- Berkeley, L.I.; Cohen, J.F.; Crankshaw, D.L.; Shirota, F.; Nagasawa, H.T. Hepatoprotection by L-cysteine-glutathione mixed disulfide, a sulfhydryl-modified prodrug of glutathione. J. Biochem. Mol. Toxicol. 2003, 17, 95–97. [Google Scholar] [CrossRef]
- Eriksson, S.A.; Mannervik, B. The reduction of the L-cysteine-glutathione mixed disulfide in rat liver. Involvement of an enzyme catalyzing thiol-disulfide interchange. FEBS Lett. 1970, 7, 26–28. [Google Scholar] [CrossRef]
- Oz, H.S.; Ray, M.; Chen, T.S.; McClain, C.J. Efficacy of a TGF-β2 Containing Nutritional Support Formula in a Murine Model of IBD. J. Amer. Coll. Nutri. 2004, 23, 220–226. [Google Scholar] [CrossRef]
- Zhong, J.; Eckhardt, E.R.; Oz, H.S.; Bruemmer, D.; de Villiers, W.J. Osteopontin-deficiency protects mice from dextran sodium sulfate-induced colitis. Inflam. Bowel. Dis. 2006, 12, 1–7. [Google Scholar]
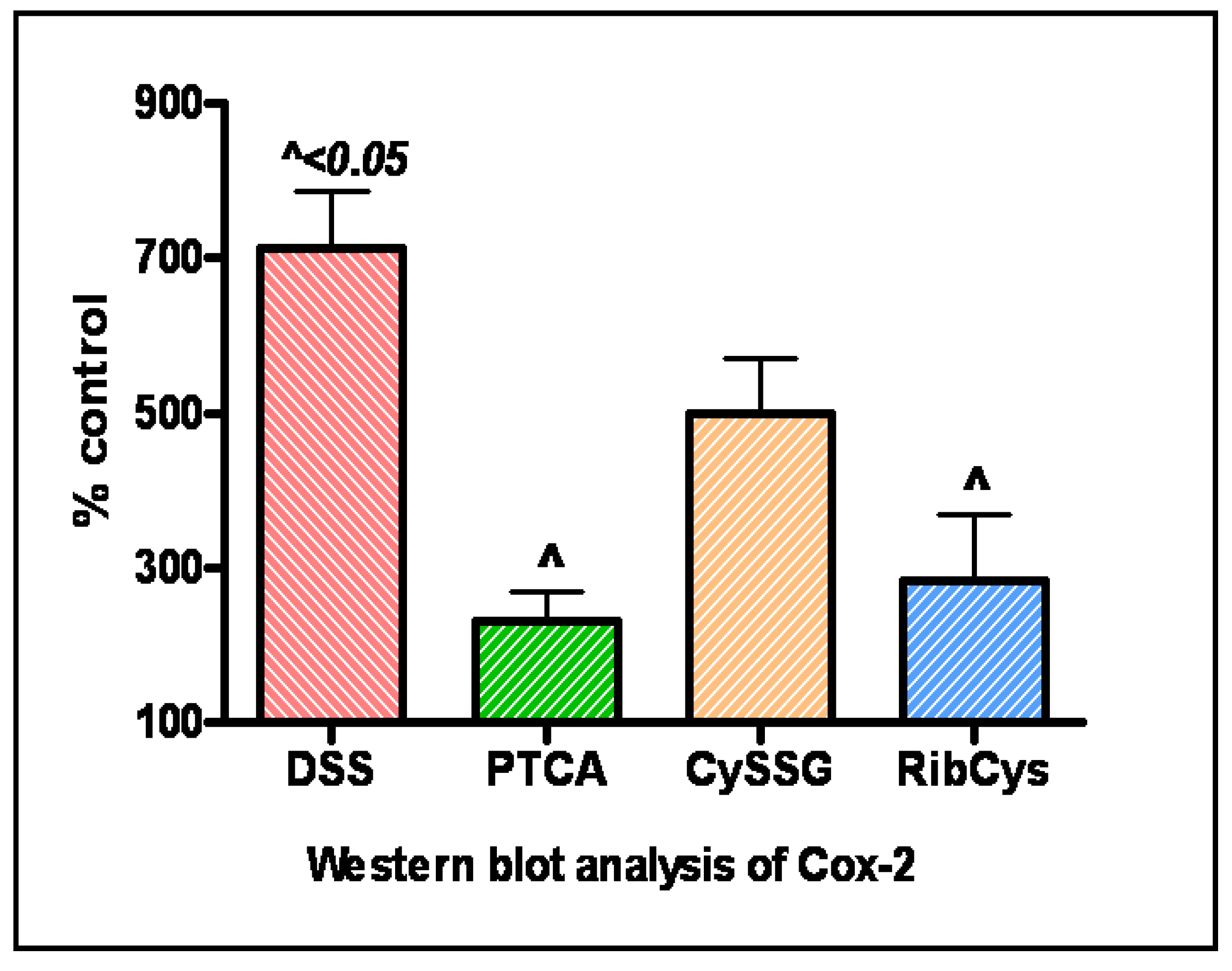
- Oz, H.S.; Chen, T.; Nagasawa, H. Comparative Efficacies of Two Cysteine Prodrugs and a Glutathione Delivery Agent in a Colitis Model. Translat. Res. 2007, 150, 122–129. [Google Scholar]
- Srinivasan, C.; Williams, W.M.; Ray, M.B.; Chen, T.S. Prevention of acetaminophen-induced liver toxicity by 2(R,S)-n-propylthiazolidine-4(R)-carboxylic acid in mice. Biochem. Pharmacol. 2001, 61, 245–252. [Google Scholar] [CrossRef]
- Srinivasan, C.; Williams, W.M.; Nagasawa, H.T.; Chen, T.S. Effects of 2(RS)-n-propylthiazolidine-4(R)-carboxylic acid on extrahepatic sulfhydryl levels in mice treated with acetaminophen. Biochem. Pharmacol. 2001, 61, 925–931. [Google Scholar] [CrossRef]
- Oz, H.S.; Im, H.J.; Chen, T.S.; de Villiers, W.J.; McClain, C.J. Glutathione enhancing agents protect against Steatohepatitis in a model. J. Biochem. Mol. Tox. 2006, 20, 39–47. [Google Scholar]
- Holleschau, A.M.; Rathbun, W.B.; Nagasawa, H.T. An HPLC radiotracer method for assessing the ability of L-Cysteine prodrugs to maintain glutathione levels in the cultured rat lens. Curr. Eye Res. 1996, 15, 510. [Google Scholar]
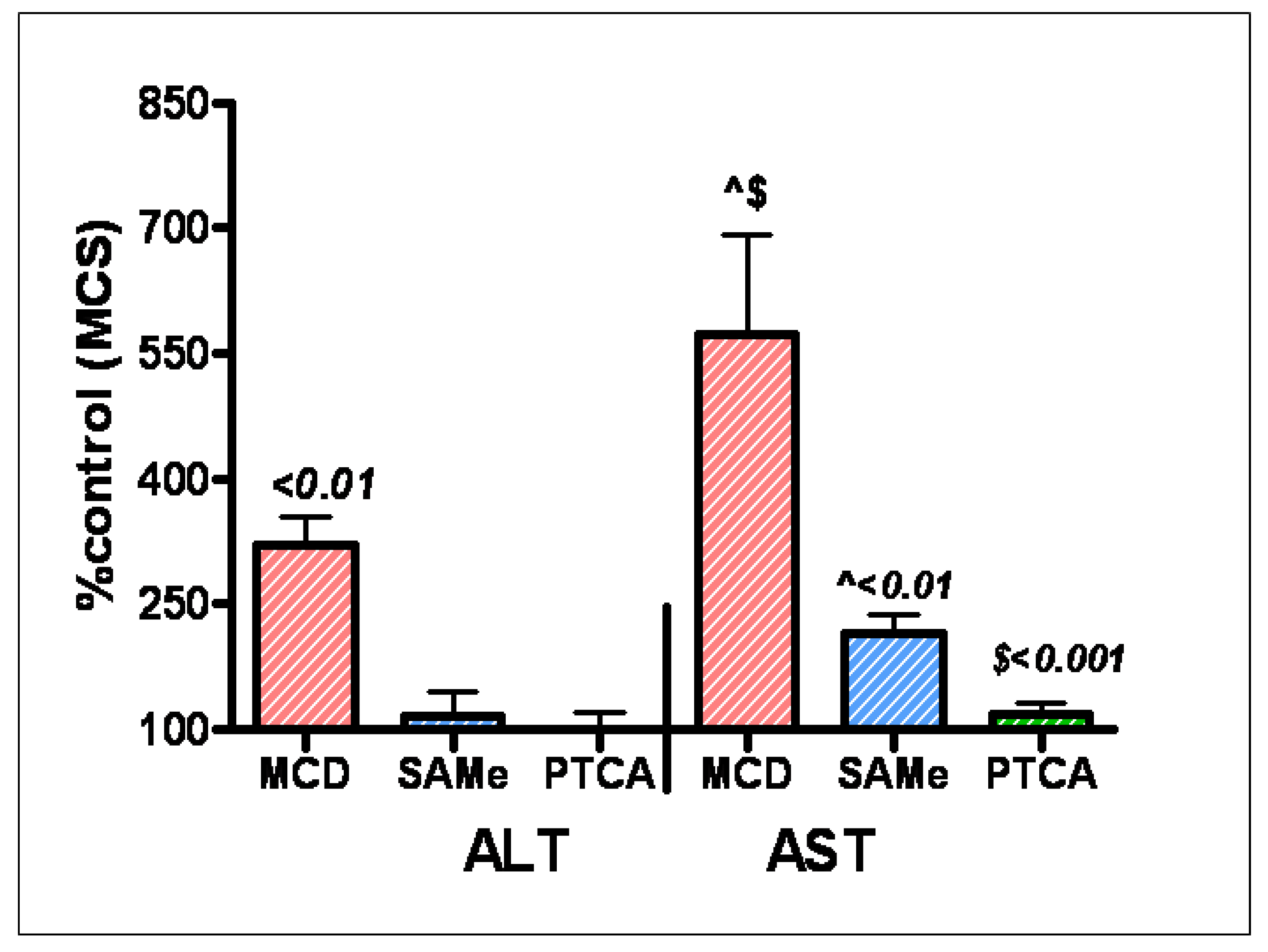
- Mato, J.M.; Alvarez, L.; Ortiz, P.; Pajares, M.A. S-adenosylmethionine synthesis: molecular mechanisms and clinical implications. Pharmacol. Ther. 1997, 73, 265–280. [Google Scholar] [CrossRef]
- Lu, S.C. S-Adenosylmethionine. Int. J. Biochem. Cell. Biol. 2000, 32, 391–395. [Google Scholar] [CrossRef]
- Giulidori, P.; Galli-Kienle, M.; Catto, E.; Stramentinoli, E. Transmethylation, transsulfuration, and aminopropylation reactions of S-adenosyl-L-methionine in vivo. J. Biol. Chem. 1984, 259, 4205–4211. [Google Scholar]
- Roediger, W.E.; Babidge, W.; Millard, S. Methionine derivatives diminish sulphide damage to colonocytes--implications for ulcerative colitis. Gut 1996, 39, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Roediger, W.E.; Babidge, W.J. Thiol methyltransferase activity in inflammatory bowel disease. Gut 2000, 47, 206–210. [Google Scholar] [CrossRef]
- Moore, J.W.; Babidge, W.J.; Millard, S.H.; Roediger, W.E. Thiolmethyltransferase activity in the human colonic mucosa: implications for ulcerative colitis. J. Gastroenterol. Hepatol. 1997, 12, 678–684. [Google Scholar] [CrossRef]
- Obayashi, M.; Matsui-Yuasa, I.; Matsumoto, T.; Kitano, A.; Kobayashi, K.; Otani, S. Polyamine metabolism in colonic mucosa from patients with ulcerative colitis. Am. J. Gastroenterol. 1992, 87, 736–740. [Google Scholar]
- Oz, H.S.; Chen, T.; Neuman, M. Methionine deficiency and liver injury in a dietary NASH model. Dig. Dis. Sci. 2007. [Google Scholar] [CrossRef] [Green Version]
- Sample Availability: Contact the authors.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Oz, H.S.; Ebersole, J.L. Application of Prodrugs to Inflammatory Diseases of the Gut. Molecules 2008, 13, 452-474. https://doi.org/10.3390/molecules13020452
Oz HS, Ebersole JL. Application of Prodrugs to Inflammatory Diseases of the Gut. Molecules. 2008; 13(2):452-474. https://doi.org/10.3390/molecules13020452
Chicago/Turabian StyleOz, Helieh S., and Jeffrey L. Ebersole. 2008. "Application of Prodrugs to Inflammatory Diseases of the Gut" Molecules 13, no. 2: 452-474. https://doi.org/10.3390/molecules13020452
APA StyleOz, H. S., & Ebersole, J. L. (2008). Application of Prodrugs to Inflammatory Diseases of the Gut. Molecules, 13(2), 452-474. https://doi.org/10.3390/molecules13020452