Regioselectivity and Tautomerism of Novel Five-Membered Ring Nitrogen Heterocycles Formed via Cyclocondensation of Acylthiosemicarbazides

Abstract
:Introduction
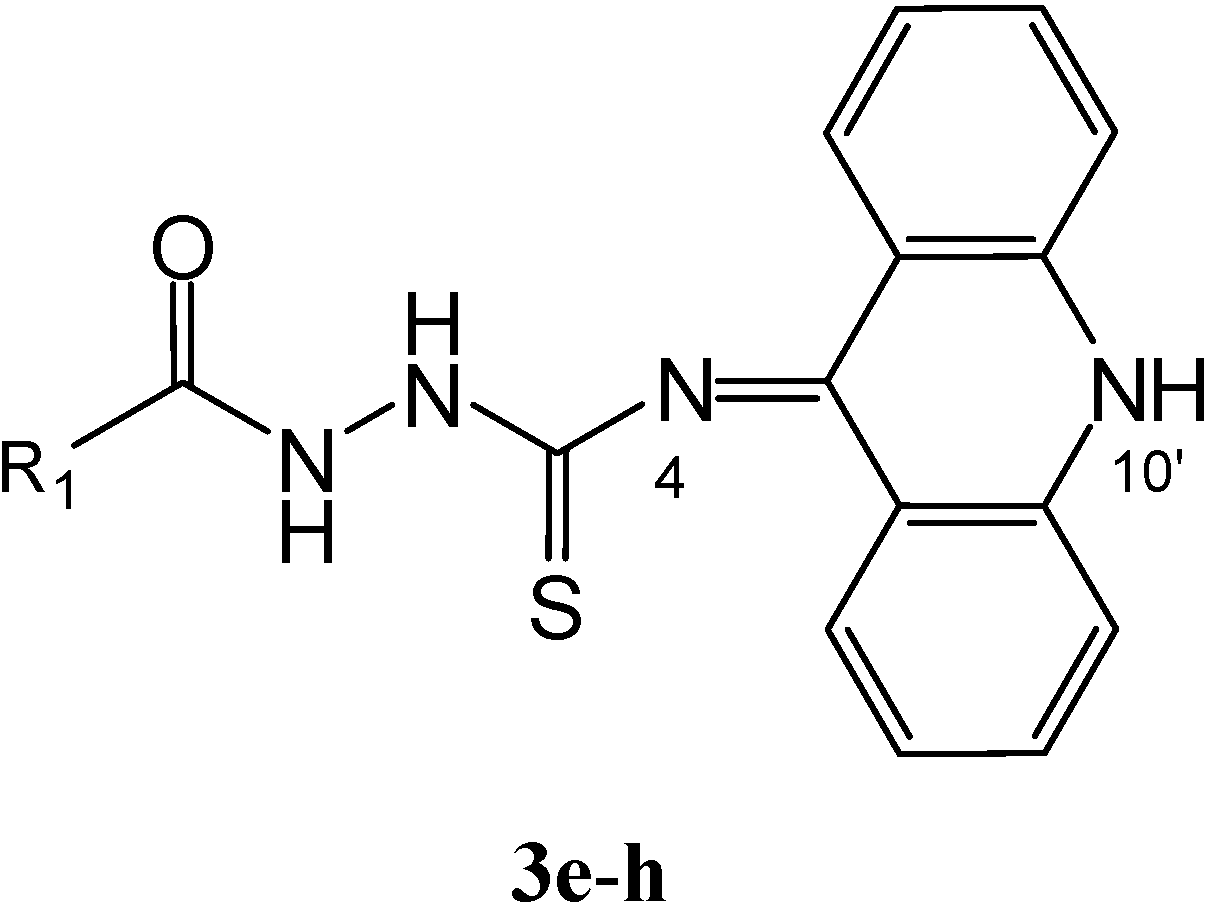
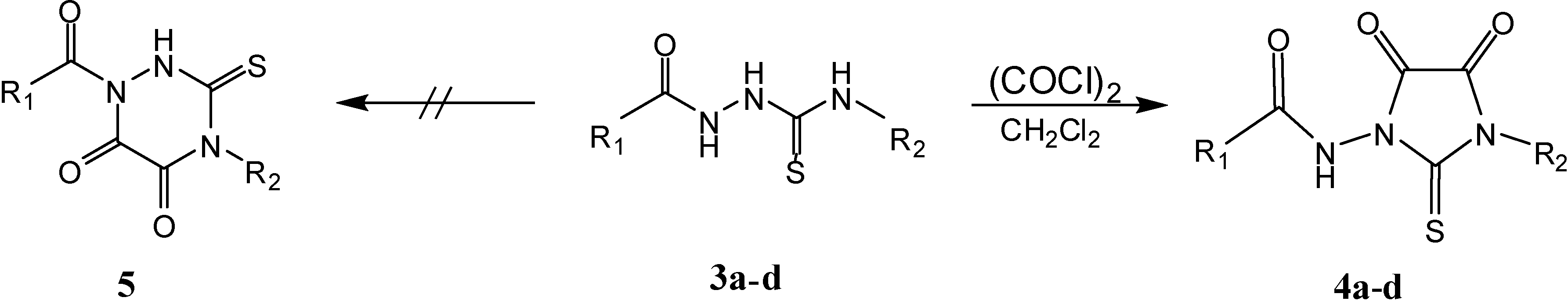
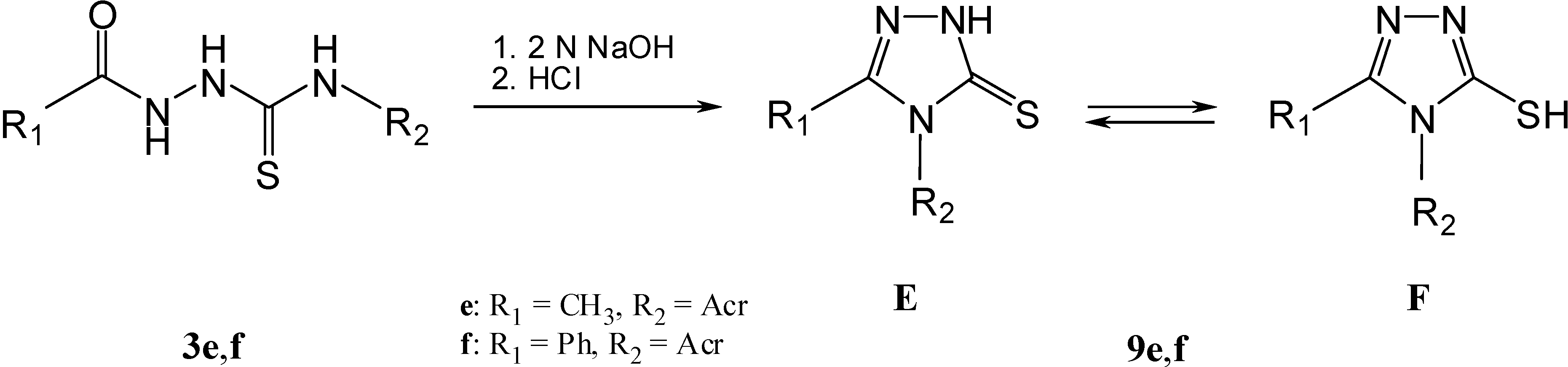
Results and Discussion

| R1 | CH3 | Ph | 3-Py | 4-Py | CH3 | Ph | 3-Py | 4-Py |
| R2 | Ph | Ph | Ph | Ph | Acr | Acr | Acr | Acr |






{kind=link}
{kind=link}
{kind=link}
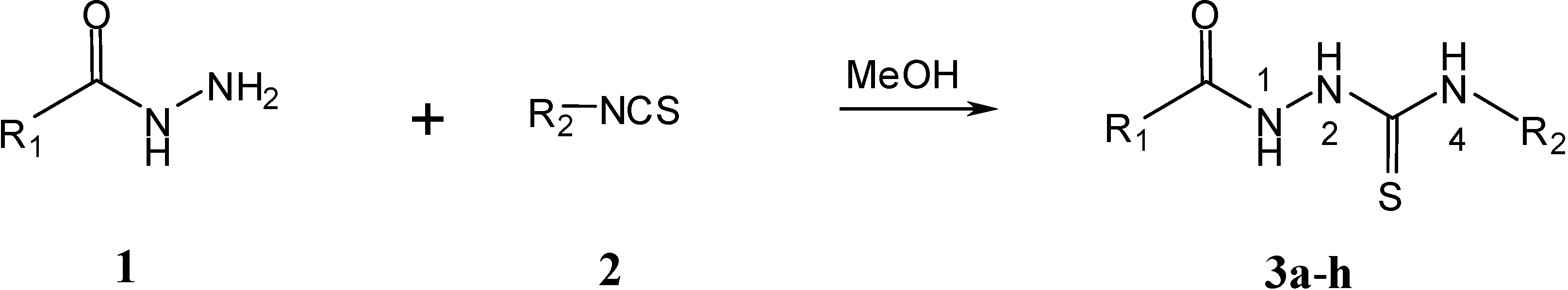{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 9a | 9b | 9c | 9d | 9e | 9f | |
|---|---|---|---|---|---|---|
| δ 1H (NH) [ppm]1 | 13.66
8.79 | 14.12
9.15 | 14.23
9.19 | 14.34
9.21 | 14.13
8.96 | 14.65
9.30 |
| δ 13C (C=S) [ppm]1 | 167.3
180.7 | 168.5
180.9 | 168.8
180.9 | 169.1
181.3 | 168.4
181.3 | 169.2
181.6 |
| ΔEthione/thiol [kcal mol−1] | 13.05 | 12.17 | 11.42 | 12.00 | 12.59 | 11.64 |


Biological activity
| Compound | Conc. [× 10−5 M] | Cytotoxicity [%] | Conc. [× 10−6 M] | Cytotoxicity [%] |
| 3f | 1 | 0 | 1 | 0 |
| 3g | 1 | 24 | 1 | 18 |
| 3h | not tested | – | 1 | 15 |
| 3h∙2HCl | 7 | 100 | 1 | 8 |
| 7b | 5 | 46 | 1 | 0 |
| 7d | 8 | 82 | 1 | 0 |
| 10 | 1 | 35 | 1 | 31 |
Conclusions
Experimental
General
General procedure for the syntheses of 1,4-disubstituted acylthiosemicarbazides 3e–h
General procedure for the syntheses of 1,3-disubstituted imidazolidine-4,5-diones 4a–d
General procedure for the syntheses of 1,3-thiazolan-5-yliden acetates 7a–d and 8a,e–h
General procedure for the syntheses of 1,2,4-triazoles 9e,f
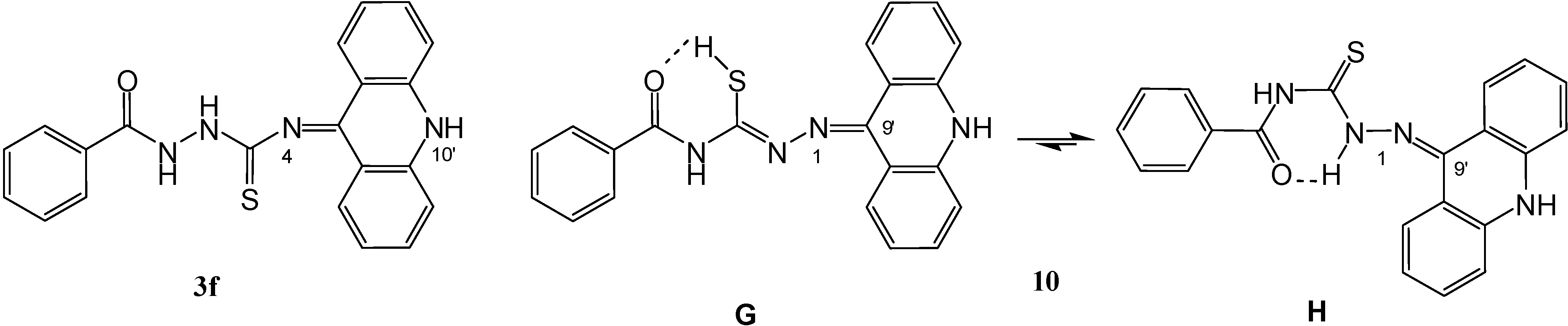
1-(9',10'-Dihydroacridin-9'-ylidene)-4-(benzoyl)-thiosemicarbazide (10)
Biological activity tests
Acknowledgements
References
- Smicius, R.; Jakubkiene, V.; Burbuliene, M. M.; Mikalainyte, A.; Vainilavicius, P. Synthesis of 1-(6-methyl-2,4-dioxo-1,2,3,4-tetrahydro-3-pyrimidinyl)acetyl-4-alkyl(aryl)thiosemicarbazides and their heterocyclisation to 1,2,4-triazoles and 1,3,4-thiadiazoles. J. Chem. Res.-S 2002, 170–172. [Google Scholar]
- Gülerman, N. N.; Doğan, H. N.; Rollas, S.; Johansson, C.; Çelik, C. Synthesis and structure elucidation of some new thioether derivatives of 1,2,4-triazoline-3-thiones and their antimicrobial activities. Farmaco 2001, 56, 953–958. [Google Scholar] [CrossRef]
- Singh, S. P.; Parmar, S. S.; Raman, K.; Stenberg, V. I. Chemistry and biological activity of thiazolidinones. Chem. Rev. 1981, 81, 175–203. [Google Scholar] [CrossRef]
- Argentieri, T. M.; Butera, J. A. An overview of potassium channel activators for the treatment of overactive bladder: a survey of new structures 2000–2005. Expert Opin. Ther. Patents 2006, 16, 573–585. [Google Scholar] [CrossRef]
- Colanceska-Ragenovic, K.; Dimova, V.; Kakurinov, V.; Molnar, D. G.; Buzarovska, A. Synthesis, antibacterial and antifungal activity of 4-substituted-5-aryl-1,2,4-triazoles. Molecules 2001, 6, 815–824. [Google Scholar] [CrossRef]
- Zamani, K.; Faghihi, K.; Tofighi, T.; Shariatzadeh, M. R. Synthesis and antimicrobial activity of some pyridyl and naphthyl substituted 1,2,4-triazole and 1,3,4-thiadiazole derivatives. Turk. J. Chem. 2004, 28, 95–100. [Google Scholar]
- Gažová, Z.; Bellová, A.; Daxnerová, Z.; Imrich, J.; Kristian, P.; Tomaščiková, J.; Bagelová, J.; Fedunová, D.; Antalík, M. Acridine derivatives inhibit lysozyme aggregation. Eur. Biophys. J. 2008, in press. [Google Scholar]
- Ergenç, N.; Çapan, G. Synthesis and anticonvulsant activity of new 4-thiazolidone and 4-thiazoline derivatives. Farmaco 1994, 49, 133–135. [Google Scholar]
- Kelarev, V. I.; Silin, M. A.; Kotova, I. G.; Kobrakov, K. I.; Rybina, I. I.; Korolev, V. K. Synthesis of N-substituted 3-aminothiazolidin-4-ones containing heteroaryl fragments. Chem. Heterocycl. Comp. 2003, 39, 213–222. [Google Scholar] [CrossRef]
- Ulrich, H.; Sayigh, A. A. R. The reaction of oxalyl chloride with substituted ureas and thioureas. J. Org. Chem. 1965, 30, 2781–2783. [Google Scholar] [CrossRef]
- Özkýrýmlý, S.; Hamalý, O. Synthesis and hypnotic activity of some 2-thioxo-4,5-imidazolidinedione derivatives. Farmaco 1995, 50, 65–67. [Google Scholar]
- Tomaščiková, J.; Imrich, J.; Danihel, I.; Böhm, S.; Kristian, P. Heterocyclization of (acridin-9-yl)thiosemicarbazides with dimethyl acetylenedicarboxylate. Coll. Czech. Chem. Commun. 2007, 72, 347–362. [Google Scholar] [CrossRef]
- Demeunynck, M.; Charmantray, F.; Martelli, A. Interest of acridine derivatives in the anticancer chemotherapy. Curr. Pharm. Des. 2001, 7, 1703–1724. [Google Scholar] [CrossRef]
- Baxendale, Ian R.; Ley, S. V.; Martinelli, M. The rapid preparation of 2-aminosulfonamide-1,3,4-oxadiazoles using polymer-supported reagents and microwave heating. Tetrahedron 2005, 61, 5323–5349. [Google Scholar] [CrossRef]
- Foks, H.; Janowiec, M.; Zwolska, Z.; Augustynowicz-Kopec, E. Synthesis and tuberculostatic activity of some piperazinylmethyl derivatives of 1,2,4-triazole-3-thiones. Phosphorus Sulfur Silicon Relat. Elem. 2005, 180, 537–543. [Google Scholar] [CrossRef]
- Kalyoncuoglu, N.; Rollas, S.; Sür-Altmer, D.; Yegenoglu, Y.; Ang, Ö. 1-[p-(Benzoylamino)benzoyl]-4-substituted thiosemicarbazides: synthesis and antibacterial and antifungal activities. Pharmazie 1992, 47, 796–797. [Google Scholar]
- Klika, K. D.; Balentová, E.; Bernát, J.; Imrich, J.; Vavrušová, M.; Kleinpeter, E.; Pihlaja, K.; Koch, A. Tautomerism, regioisomerism, and cyclization reactions of acridinyl thiosemicarbazides. J. Heterocycl. Chem. 2006, 43, 633–643. [Google Scholar] [CrossRef]
- Balentová, E.; Imrich, J.; Bernát, J.; Suchá, L.; Vilková, M.; Prónayová, N.; Kristian, P.; Pihlaja, K.; Klika, K. D. Stereochemistry, tautomerism, and reactions of acridinyl thiosemicarbazides in the synthesis of 1,3-thiazolidines. J. Heterocycl. Chem. 2006, 43, 645–656. [Google Scholar] [CrossRef]
- Klika, K. D.; Balentová, E.; Bernát, J.; Imrich, J.; Vavrušová, M.; Pihlaja, K.; Koch, A.; Kleinpeter, E.; Kelling, A.; Schilde, U. Structural revision of products resulting from the reaction of methylhydrazine with acridin-9-yl isothiocyanate due to unexpected acridinyl migration and further reactions. ARKIVOC 2006, (xvi), 93–108. [Google Scholar]
- Bernát, J.; Balentová, E.; Kristian, P.; Imrich, J.; Sedlák, E.; Danihel, I.; Böhm, S.; Prónayová, N.; Pihlaja, K.; Klika, K. D. Methylation of acridin-9-ylthioureas. Structure, fluorescence and biological properties of products. Coll. Czech. Chem. Commun. 2004, 69, 833–849. [Google Scholar] [CrossRef]
- Klika, K. D.; Imrich, J.; Vilková, M.; Bernát, J.; Pihlaja, K. Unusual structures derived from N-acridin-9-yl methyl N´-acridin-9-yl thiourea based on the propensity of N-10 to retain H. J. Heterocycl. Chem. 2006, 43, 739–743. [Google Scholar]
- Klika, K. D.; Bernát, J.; Imrich, J.; Chomča, I.; Sillanpää, R.; Pihlaja, K. Unexpected formation of a spiro acridine and fused ring system from the reaction between an N-acridinylmethyl substituted thiourea and bromoacetonitrile under basic conditions. J. Org. Chem. 2001, 66, 4416–4418. [Google Scholar] [CrossRef]
- Kristian, P.; Balentová, E.; Bernát, J.; Imrich, J.; Sedlák, E.; Danihel, I.; Böhm, S.; Prónayová, N.; Klika, K. D.; Pihlaja, K.; Baranová, J. Fluorescence and structure of methylated acridin-9-ylthioureas. Chem. Pap. 2004, 58, 268–275. [Google Scholar]
- Adcock, B. Aminoacridines . In Acridines; Acheson, R. M., Ed.; (from The Chemistry of Heterocyclic Compounds, Weissberger, A.; Taylor, E. C., Eds.); J. Wiley: New York, 1973; p. 109. [Google Scholar]
- Géci, I.; Valtamo, P.; Imrich, J.; Kivelä, H.; Kristian, P.; Pihlaja, K. Regiospecific synthesis, structure and electron ionization mass spectra of 1,3-thiazolidin-4-ones containing the acridine skeleton. J. Heterocycl. Chem. 2005, 42, 907–918. [Google Scholar] [CrossRef]
- Kristian, P. Isothiocyanates and their synthetic formers V. Synthesis of substituted 9-isothiocyanatoacridines and study of the relation between their physicochemical properties and structure. Chem. Pap. 1969, 23, 371–376. [Google Scholar]
- Rostamizadeh, S.; Mollahoseini, K.; Moghadasi, S. A One-pot synthesis of 4,5-disubstituted-1,2,4-triazole-3-thiones on solid support under microwave irradiation. Phosphorus Sulfur Silicon Relat. Elem. 2006, 181, 1839–1845. [Google Scholar] [CrossRef]
- Hazzaa, A. A. B.; Ashour, F.; Shafik, R. M. Synthesis of 1,2,4-triazolylthiopropanol-amines as potential cardiovascular agents. Pharmazie 1980, 35, 324–325. [Google Scholar]
- Vailikhit, V.; Treesuwan, W.; Hannongbua, S. A combined MD-ONIOM2 approach for 1H NMR chemical shift calculations including a polar solvent. Theochem-J. Mol. Struct. 2007, 806, 99–104. [Google Scholar] [CrossRef]
- Hritzová, O.; Černák, J.; Šafař, P.; Fröhlichová, Z.; Csöregh, I. Furan derivatives of substituted phenylthiourea: spectral studies, semi-empirical quantum-chemical calculations and X-ray structure analyses. J. Mol. Struct. 2005, 743, 29–48. [Google Scholar] [CrossRef]
- Becke, A. D. Density-functional thermochemistry III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A., Jr.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hagesawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J.B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzales, C.; Pople, J. A. Gaussian 03, Revision B.05; Gaussian, Inc.: Pittsburgh PA, 2003. [Google Scholar]
- Wolinski, K.; Hinton, J. F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. 1990, 112, 8251–8260. [Google Scholar]
- Kristian, P. Isothiocyanates VII. Synthesis of 9-isothiocyanatoacridine and some of its derivatives. Chem. Pap. 1961, 15, 641–646. [Google Scholar]
- Albert, A.; Ritchie, B. 9-Aminoacridine. Org. Synth. 1942, 22, 5–8. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunolog. Meth. 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Sabol, M.; Kutschy, P.; Siegfried, L.; Miroššay, A.; Suchý, M.; Hrbková, H.; Dzurilla, M.; Marušková, R.; Starková, J.; Paulíková, E. Cytotoxic effect of cruciferous phytoalexins against murine L1210 leukemia and B16 melanoma. Biologia 2000, 55, 701–707. [Google Scholar]
- Sample Availability: Samples of compounds 4a, 7a–d, 8a,e–h 9e and 10 are available from the authors.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Tomaščiková, J.; Imrich, J.; Danihel, I.; Böhm, S.; Kristian, P.; Pisarčíková, J.; Sabol, M.; Klika, K.D. Regioselectivity and Tautomerism of Novel Five-Membered Ring Nitrogen Heterocycles Formed via Cyclocondensation of Acylthiosemicarbazides. Molecules 2008, 13, 501-518. https://doi.org/10.3390/molecules13030501
Tomaščiková J, Imrich J, Danihel I, Böhm S, Kristian P, Pisarčíková J, Sabol M, Klika KD. Regioselectivity and Tautomerism of Novel Five-Membered Ring Nitrogen Heterocycles Formed via Cyclocondensation of Acylthiosemicarbazides. Molecules. 2008; 13(3):501-518. https://doi.org/10.3390/molecules13030501
Chicago/Turabian StyleTomaščiková, Jana, Ján Imrich, Ivan Danihel, Stanislav Böhm, Pavol Kristian, Jana Pisarčíková, Marián Sabol, and Karel D. Klika. 2008. "Regioselectivity and Tautomerism of Novel Five-Membered Ring Nitrogen Heterocycles Formed via Cyclocondensation of Acylthiosemicarbazides" Molecules 13, no. 3: 501-518. https://doi.org/10.3390/molecules13030501
APA StyleTomaščiková, J., Imrich, J., Danihel, I., Böhm, S., Kristian, P., Pisarčíková, J., Sabol, M., & Klika, K. D. (2008). Regioselectivity and Tautomerism of Novel Five-Membered Ring Nitrogen Heterocycles Formed via Cyclocondensation of Acylthiosemicarbazides. Molecules, 13(3), 501-518. https://doi.org/10.3390/molecules13030501