Introduction
P. microphyllus (Rutaceae) is originally from the Amazon region in Brazil where it is known as
jaborandi [
1]. This plant is a rich source of imidazole alkaloids, of which pilocarpine is the best known and the only one being economically exploited. Pilocarpine has been well studied as it is used not only for the treatment of glaucoma [
2] but also as a stimulant of sweat and lachrymal glands [
3,
4].
The possibility of production of pilocarpine by callus cell lines in bioreactors has been evaluated, with the objective of protecting the
jaborandi plant from uncontrolled exploitation [
5]. Studies using electrospray ionization mass spectrometry (ESI-MS) to investigate the regulation of pilocarpine biosynthesis in
P. microphyllus callus showed that pilosine (another imidazole alkaloid) was also produced depending on the cell line and treatment [
6]. Due to the lack of standards of other imidazole alkaloids produced in
jaborandi, posterior studies were conducted using electrospray ionization mass spectrometry (ESI-MS). These studies resulted in the characterization, by direct insertion tandem mass spectrometry (ESI-MS/MS and ESI-MS/MS/MS), of five known imidazole alkaloids, as well as three new alkaloids, suggesting the existence of three different biosynthetic pathways for imidazole alkaloids in the
jaborandi plant [
7]. Although the presence of pilocarpine has been reported in 10 species of the
Pilocarpus genus [
1], there is no information as to the presence and concentration of the other imidazole alkaloids, whose pharmacological properties are still unknown.
Diverse chromatographic methods exist for the analysis of pilocarpine, mainly for the purpose of quality control of eye drops. Most of the analytical methods applied to the quantification of pilocarpine employ HPLC with either octadecyl [
8,
9], phenyl [
10,
11] or cyclodextrin [
12] columns and UV detection. More recently, monolithic HPLC columns have also been tested [
13]. As pilocarpine and related compounds lack chromophoric groups, UV detection must be carried out at low wavelengths (210-214 nm) where several other compounds may interfere [
14], particularly when plant extracts are analyzed. Lower detection limits and richer structural information could be achieved by HPLC-ESI-MS analysis. However triethylamine, which is commonly used in buffer solutions to prevent tailing [
13] is incompatible with ESI-MS detection. Merbel
et al. [
14] tested several stationary phases and eluents, determining that a combination of an Inertsil octadecyl column with an eluent using an ammonium acetate buffer and acetonitrile organic modifier was compatible for the LC-APCI-MS separation of pilocarpine, isopilocarpine and their respective acids extracted from plasma. It has also been our experience also that not all brands of octadecyl columns are capable of adequately separating imidazole alkaloids because of tailing.
Herein we describe a HPLC-ESI-MS/MS method developed for the analysis of imidazole alkaloids in the extracts of P. microphyllus samples. The method used is based on concepts used in previous papers, but has been modified and improved, in order to separate over a dozen imidazole alkaloids in a single 22 minute chromatographic run. The soft ionization technique used (ESI) permits the observation of protonated ([M + H] +) molecules, rather than fragment ions. Therefore analysis in the full scan mode (TIC) instead of selected ion mode (SIM), permits the detection and quantification of both known and novel alkaloids in the sample. Two different types of samples were analyzed: P. microphyllus leaves and a paste that is left over after the industrial extraction of pilocarpine. The composition of the two samples could be qualitatively compared based on the results of the analysis. A calibration curve based on pilocarpine reference standard was built and used to compare the concentration of imidazole alkaloids in the samples.
Results and Discussion
In previous studies of the imidazole alkaloids in
jaborandi, ESI-MS fingerprinting was used as a fast and simple technique for the comparison of samples [
6,
7]. However, using direct infusion ESI-MS it was not possible to distinguish between isomers in the plant extracts. The present method was developed and applied in order to chromatographically separate and quantify the alkaloids in the
jaborandi samples, including the isomers. This method has modified and substantially improved previous methods, such as that proposed by Merbel
et al. [
14], which was isocratic, and separated only four alkaloids in 20 minutes. By using a gradient, we achieved the separation of 13 compounds in 22 minutes. Several of these compounds have only been reported once before in a previous study by our group [
7].
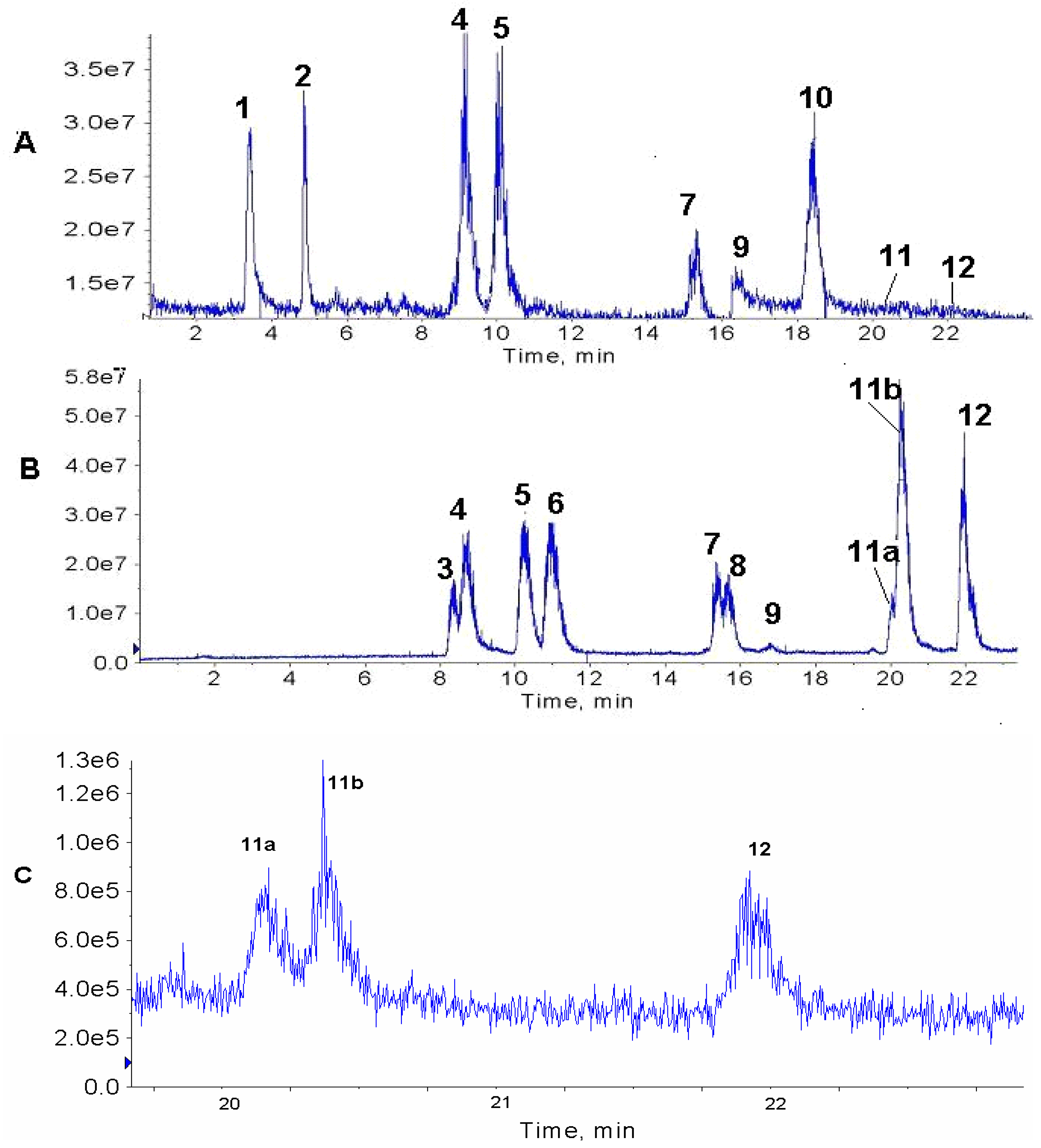
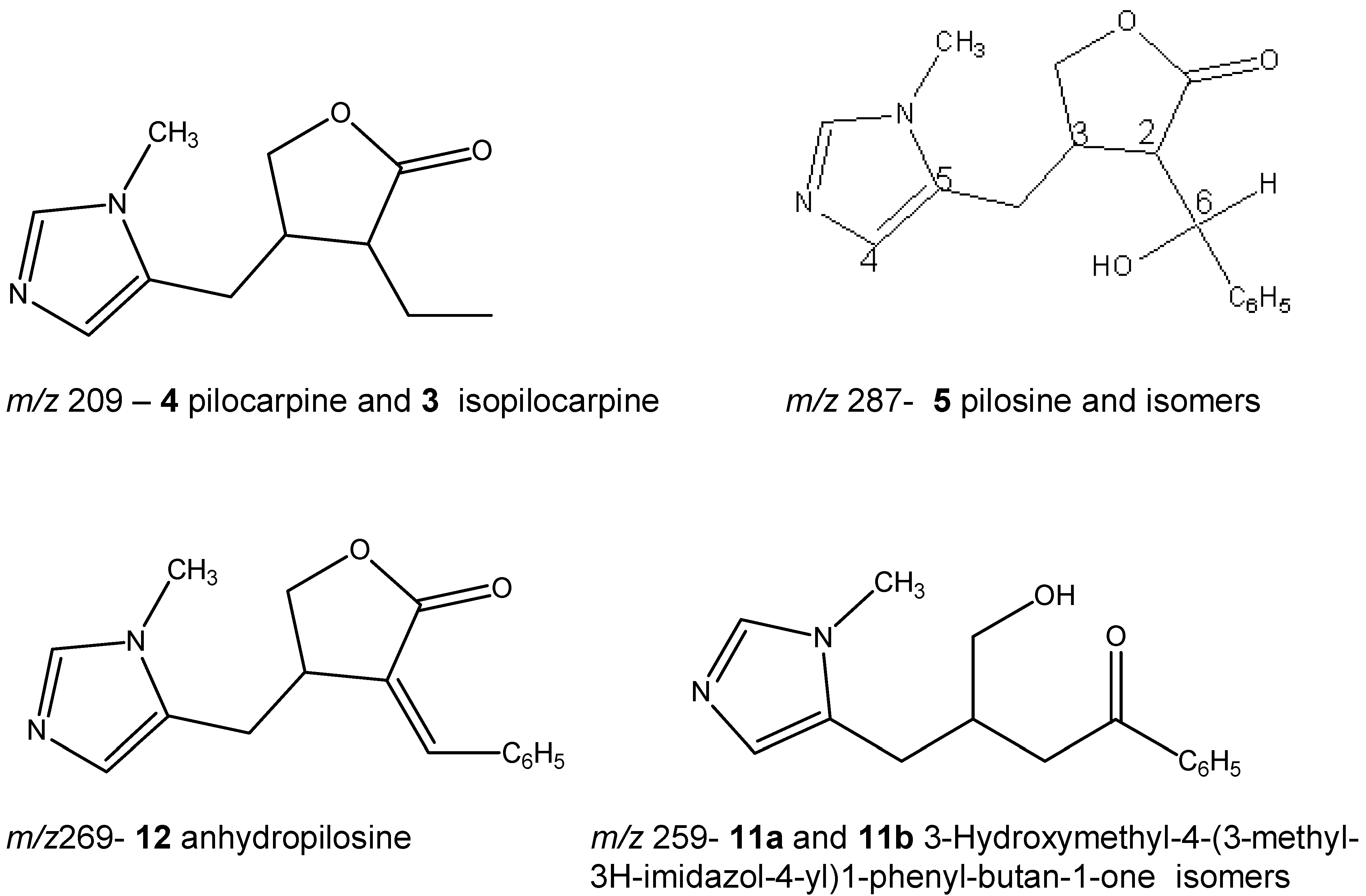
Figure 1 compares the chromatogram of a leaf extract (A) with that of the paste (B) that is left over after the industrial extraction of pilocarpine. The presence of the epimeric isopilocarpine (
3) and pilocarpine (
4) are clearly observed in the paste (
Figure 1B), but in the leaf extract only pilocarpine (
4) is detected. The ESI-MS/MS of the protonated molecules of these isomers of
m/z 209 are very similar (
Figures 2A and B); therefore, they can only be distinguished by their retention times. Both samples were spiked with standard pilocarpine, peak
4 increased in both samples and no new peak appeared in the leaf sample. This confirmed that peak
4 in both chromatograms was pilocarpine. This is in accordance with the results of Merbel
et al. [
14], who reported that isopilocarpine eluted slightly before its isomer, pilocarpine. No other isomers of this molecule have been reported.
A series of peaks corresponding to ions of
m/z 287 elute (
5, 6, 7, 8 and
9) in sequence in the paste (
Figure 1B), whereas in the leaf extract only
5,
7 and
9 were observed (
Figure 1 A). These compounds have slightly different ESI-MS/MS allowing them to be distinguished, and are presented for the first time in literature in
Figure 2 (F, G, H, I and J). We have found references to five isomers of molecular formula C
16H
18N
2O
3 found in species
P. microphyllus and
P. jaborandi: pilosine (2
R,3
R,6
S), isopilosine (2
S,3
R,6
R) and epiisopilosine (2
S,3
R,6
S) [
15], as well as piloturin and epiisopiloturin [
16] in which the imidazole ring is linked at the C
4 (rather than at the C
5 as in pilosine). At the present moment we are able to identify just one of these isomers, that is, pilosine (
5). Both extracts were spiked with the standard of pilosine to confirm that peak
5 is in fact pilosine, in spite of slight differences in the retention time between samples. Further analysis using a semi-preparative HPLC column should allow us to isolate and identify the other isomers. Isomers were not present in leaves in the same proportion as they were found in the industrial paste. This may be the result of the industrial extraction procedure, as it is known that a long time in the presence of acids (normally used in the extraction of alkaloids) can cause the opening and closing of the lactone ring and formation of isomers.
Figure 1.
HPLC-MS TIC chromatograms of (A) jaborandi leaf extract and (B) paste extract. Numbered peaks correspond to: 1 m/z 202, 2 m/z 248, 3 isopilocarpine m/z 209, 4 pilocarpine m/z 209, 5, pilosine m/z 287, 6 m/z 287, 7 m/z 287, 8 m/z 287, 9 m/z 287, 10 m/z 316, 11 3-hydroxymethyl-4-(3methyl-3H-imidazol-4-yl)1-phenyl-butan-1-one m/z 259, and 12 anhydropilosine m/z 269. (C) XIC of ions m/z 259 (11a and 11b) and m/z 269 (12) from the leaf extract chromatogram.
Figure 1.
HPLC-MS TIC chromatograms of (A) jaborandi leaf extract and (B) paste extract. Numbered peaks correspond to: 1 m/z 202, 2 m/z 248, 3 isopilocarpine m/z 209, 4 pilocarpine m/z 209, 5, pilosine m/z 287, 6 m/z 287, 7 m/z 287, 8 m/z 287, 9 m/z 287, 10 m/z 316, 11 3-hydroxymethyl-4-(3methyl-3H-imidazol-4-yl)1-phenyl-butan-1-one m/z 259, and 12 anhydropilosine m/z 269. (C) XIC of ions m/z 259 (11a and 11b) and m/z 269 (12) from the leaf extract chromatogram.
Compounds corresponding to peaks
1 (
m/z 202 ),
2 (
m/z 248) and
10 (
m/z 316) in the leaf extract (
Figure 1A) have not been identified yet. These ions but have been observed previously in fingerprints of extracts of leaves and callus of
P. microphyllus [
6]. Peaks
11a and
11b (
m/z 259) correspond to a protonated compound identified for the first time in leaves of
P. microphyllus in a previous study [
7], whose suggested structure corresponds to 3-hydroxymethyl-4-(3-methyl-3
H-imidazol-4-yl)1-phenyl-butan-1-one, based on its ESI-MS/MS. The chromatograms suggest that two isomers are present (
Figures 1B and C), although the ESI-MS/MS for both were identical (
Figures 2C and D). The observation that the compound of
m/z 259 is, in fact, a mixture of two isomers, is presented for the first time in this study as a result of the chromatographic method used. Peak
12 (
m/z 269) corresponds to protonated anhydropilosine, by comparison of its ESI-MS/MS (
Figure 2 E) to the compound identified in a previous study [
7].
In the present study, chromatograms were obtained using full scan mode (TIC) instead of the selective ion mode (SIM), as the purpose of this study was to observe whichever compounds were present in the extracts rather than observe low concentrations of specific compounds. By using TIC, in a single chromatographic run, several new alkaloids from
Pilocarpus microphyllus, as well as some previously identified alkaloids could be detected (
Figures 1A and B). However, extracting specific ions (XIC) from the chromatogram allows us to observe low concentrations that cannot be seen in the TIC, as exemplified here for
m/z 259 and 269 in the leaf extract (
Figure 1C). Therefore, for the exploratory analysis of samples of alkaloids from
jaborandi, the TIC mode is the most adequate. However, for the routine analysis of these extracts, using the same chromatographic method in SIM could permit a manifold gain in sensitivity, if such were the need.
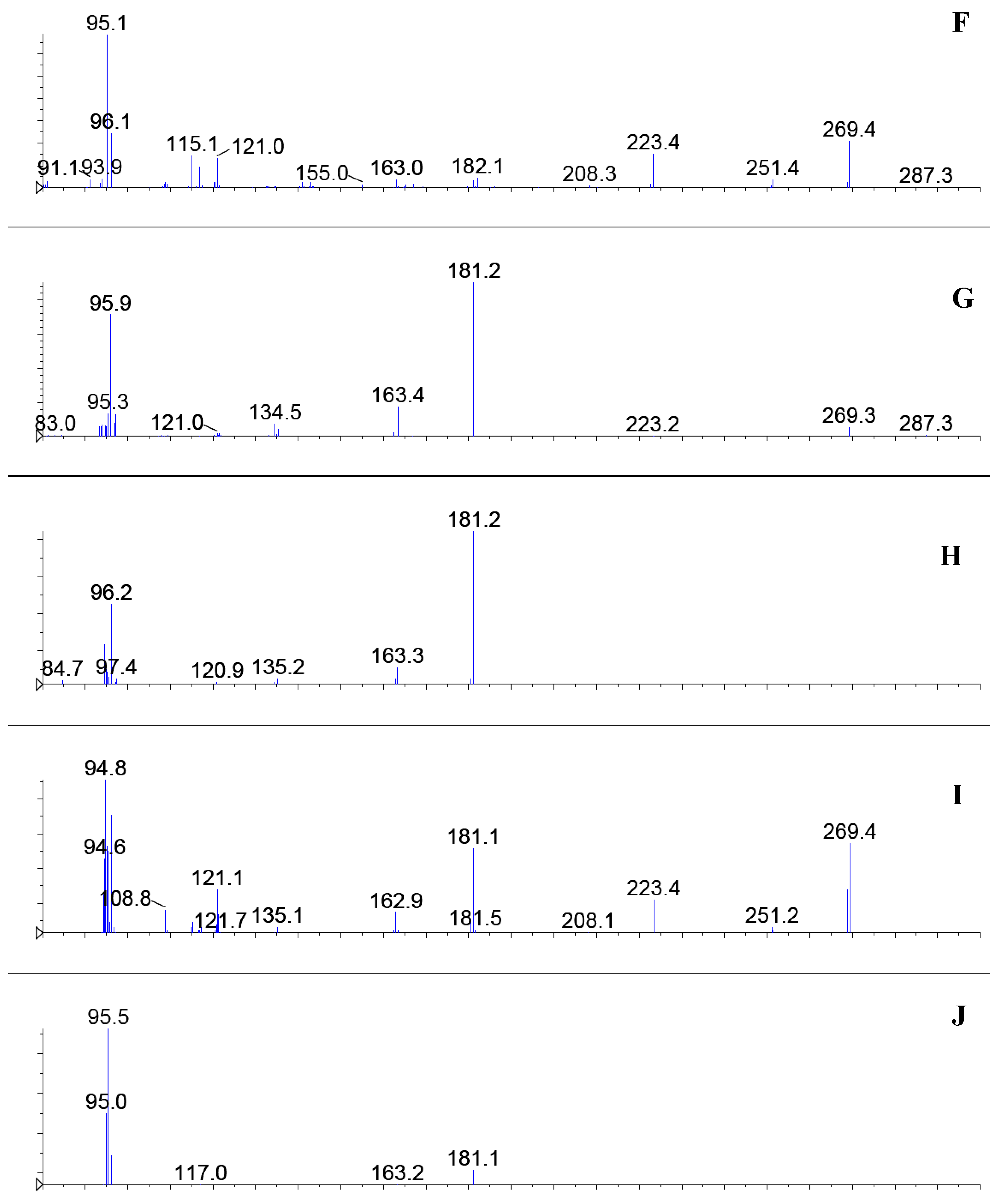
Figure 2.
ESI-MS/MS of selected ions from the chromatograms presented in
Figure 1: A)
m/z 209, peak
3, isopilocarpine; B)
m/z 209, peak
4, pilocarpine; C)
m/z 259, peak
11a, D)
m/z 259, peak
11b; E)
m/z 269, peak
12, anhydropilosine; F)
m/z 287, peak
5, pilosine, G)
m/z 287, peak
6, H)
m/z 287, peak
7; I)
m/z 287, peak
8 and J)
m/z 287, peak
9
Figure 2.
ESI-MS/MS of selected ions from the chromatograms presented in
Figure 1: A)
m/z 209, peak
3, isopilocarpine; B)
m/z 209, peak
4, pilocarpine; C)
m/z 259, peak
11a, D)
m/z 259, peak
11b; E)
m/z 269, peak
12, anhydropilosine; F)
m/z 287, peak
5, pilosine, G)
m/z 287, peak
6, H)
m/z 287, peak
7; I)
m/z 287, peak
8 and J)
m/z 287, peak
9
The ionization method used by Merbel
et al. [
14] was APCI, which is more energic and causes in-source fragmentation, therefore neither pilocarpic nor isopilocarpic acid could be quantified as parent ions (
m/z 227), but only as their fragments (
m/z 209). The use of a much softer ionization (ESI) permitted the observation of all the compounds based on the mass of the protoned [M + H]
+ molecules, not of fragment ions, which is essential for bio-prospection of new compounds.
A calibration curve with a linear regression coefficient of 0.9949 for values between 0.5 and 20 μg/mL was built using pilocarpine, a value comparable to that obtained by Merbel
et al. [
14]. This calibration curve enabled us to calculate the concentrations of pilocarpine and isopilocarpine in both extracts (
Table 1). It was also possible to obtain an estimate of the concentrations of the other alkaloids based on the area of their peaks in the chromatogram as their areas and concentration fell within the range of the calibration curve, except for compounds
11a, 11b and
12 in the leaf extract whose concentrations fell below these values.
Table 1.
HPLC-ESI-MS analysis of leaf and paste extract (10 μL injections). Areas of peaks and quantification are based on a calibration curve using a pure pilocarpine standard. Compounds are numbered as in
Figure 1.
Table 1.
HPLC-ESI-MS analysis of leaf and paste extract (10 μL injections). Areas of peaks and quantification are based on a calibration curve using a pure pilocarpine standard. Compounds are numbered as in Figure 1.
| Compound as [M + H]+ (m/z) | Leaves concentration (µg/ mL) | Paste concentration (µg/ mL) |
|---|
| 1 (202) | 1.6 | |
| 2 (248) | 0.9 | |
| 3 (209) | | 1.5 |
| 4 (209) | 3.0 | 3.9 |
| 5 (287) | 2.8 | 4.1 |
| 6 (287) | | 4.6 |
| 7 (287) | 1.2 | 1.9 |
| 8 (287) | | 1.9 |
| 9 (287) | 0.7 | 0.2 |
| 10 (316) | 2.1 | |
| 11a (259) | | 1.2 |
| 11b (259) | | 7.6 |
| 12 (269) | | 4.6 |
It is noteworthy that the concentration of pilocarpine seems higher in the paste than in the leaf extract. However, one must bear in mind that the paste is enriched of alkaloids since the industrial process removed other leaf components. The relation between the concentrations of pilocarpine in the leaf extract in comparison to the other peaks (24%) is higher than in the paste (12%). Pilosine makes up 22% of the leaf alkaloids but only 14% of the paste. A series of compounds which are found in the paste, but not in the leaf extract, could be degradation products. For example, compounds of
m/z 259 and 269, whose concentration was below the quantification limit in the leaf extract, made up 42% of the paste. Compounds of
m/z 202, 248 and 316, observed in the leaf extract, were absent in the paste. These variables could also be linked to the time of the year in which the samples were obtained, as the production of each of these imidazole alkaloids varies, depending on the season [
7]. Further studies will be necessary to clarify all these points. However, the estimated concentration obtained using the HPLC-ESI-MS method proposed is sufficient to compare samples from different parts of the plant, of different plants, of plants or callus submitted to different treatments in order to study the production of imidazole alkaloids in this species.






{kind=link}
{kind=link}
{kind=link}
{kind=link}