2-Amido-3-(1H-Indol-3-yl)-N-Substitued-Propanamides as a New Class of Falcipain-2 Inhibitors. 1. Design, Synthesis, Biological Evaluation and Binding Model Studies

Abstract
:1. Introduction
2. Results and Discussion
2.1. Identification of Binders (Hits) by Virtual Screening

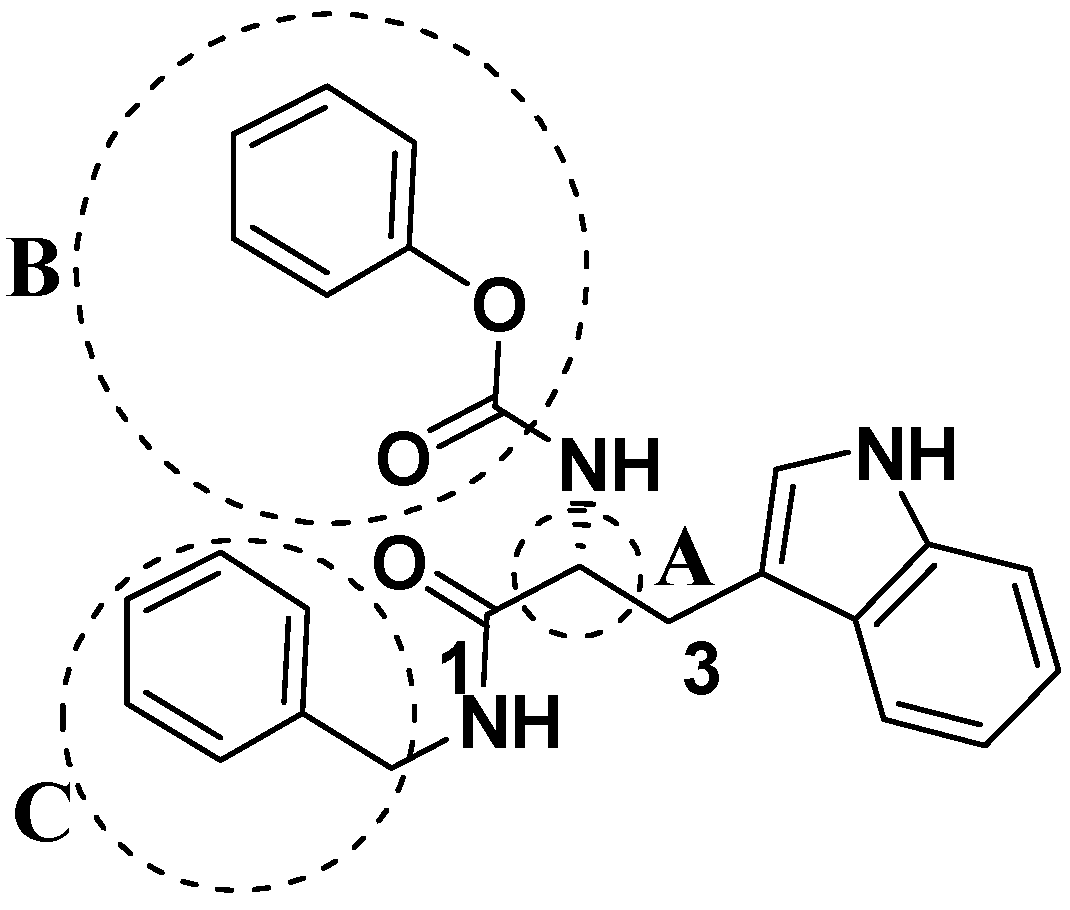
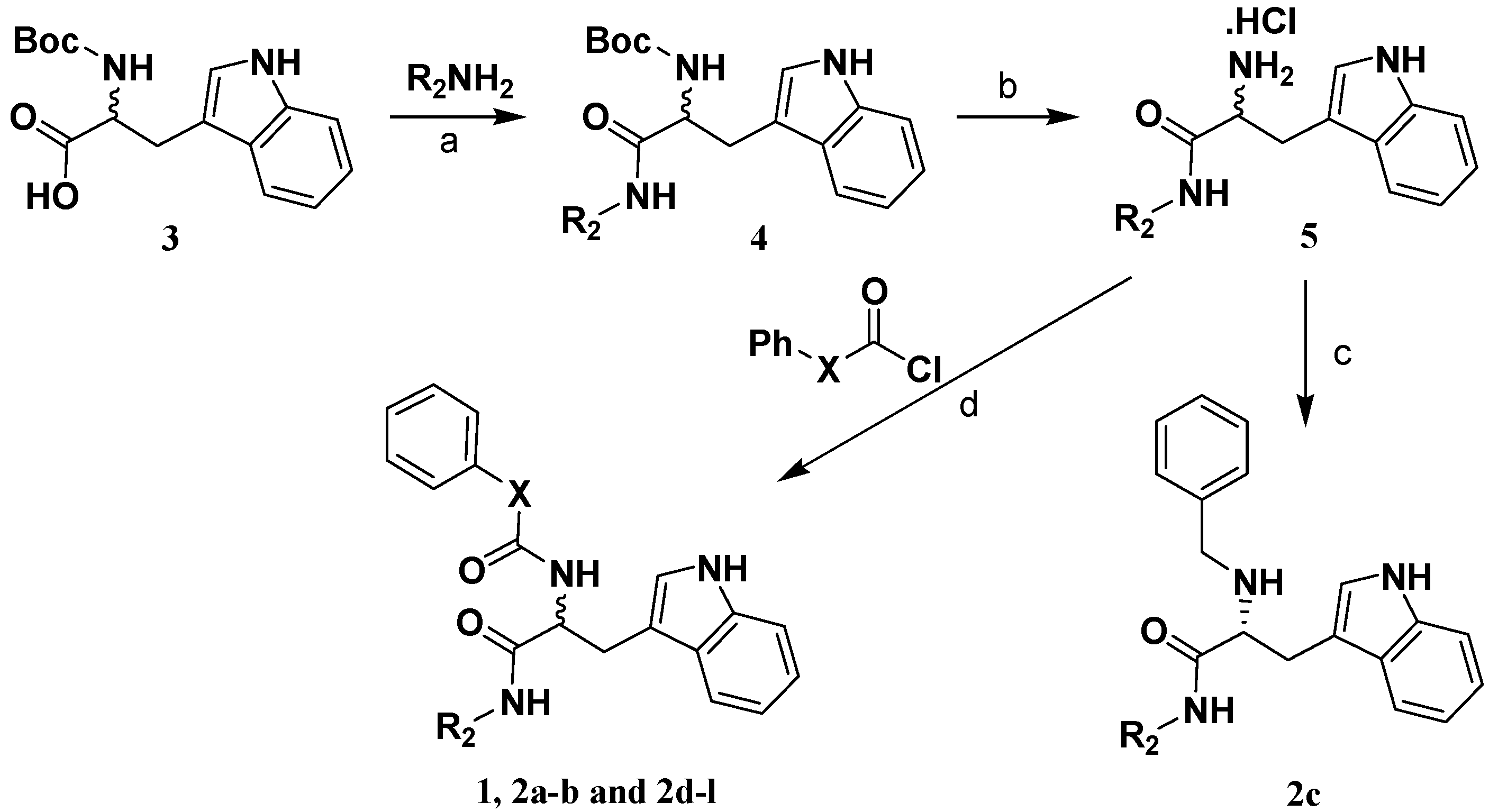
2.2. Analog Design and Synthesis




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | C2 configuration | R1 | R2 | Inhibition rate at 10 μM (%) | IC50 (μM) |
|---|---|---|---|---|---|
| 1 | R |  |  | 29.4 | 29.7 |
| 2a | S | | | 14.3 | - |
| 2b | R |  | | 25.7 | 39.4 |
| 2c | R | | | 13.0 | - |
| 2d | R | |  | 11.8 | - |
| 2e | R | |  | 3.1 | - |
| 2f | R | |  | 13.6 | - |
| 2g | R | |  | 3.1 | - |
| 2h | R | |  | - | - |
| 2i | R | |  | 2.2 | - |
| 2j | R | |  | 6.6 | - |
| 2k | R | |  | 49.5 | 10.0 |
| 2l | R | |  | 32.9 | 13.3 |
2.3. Enzyme Inhibition Assay
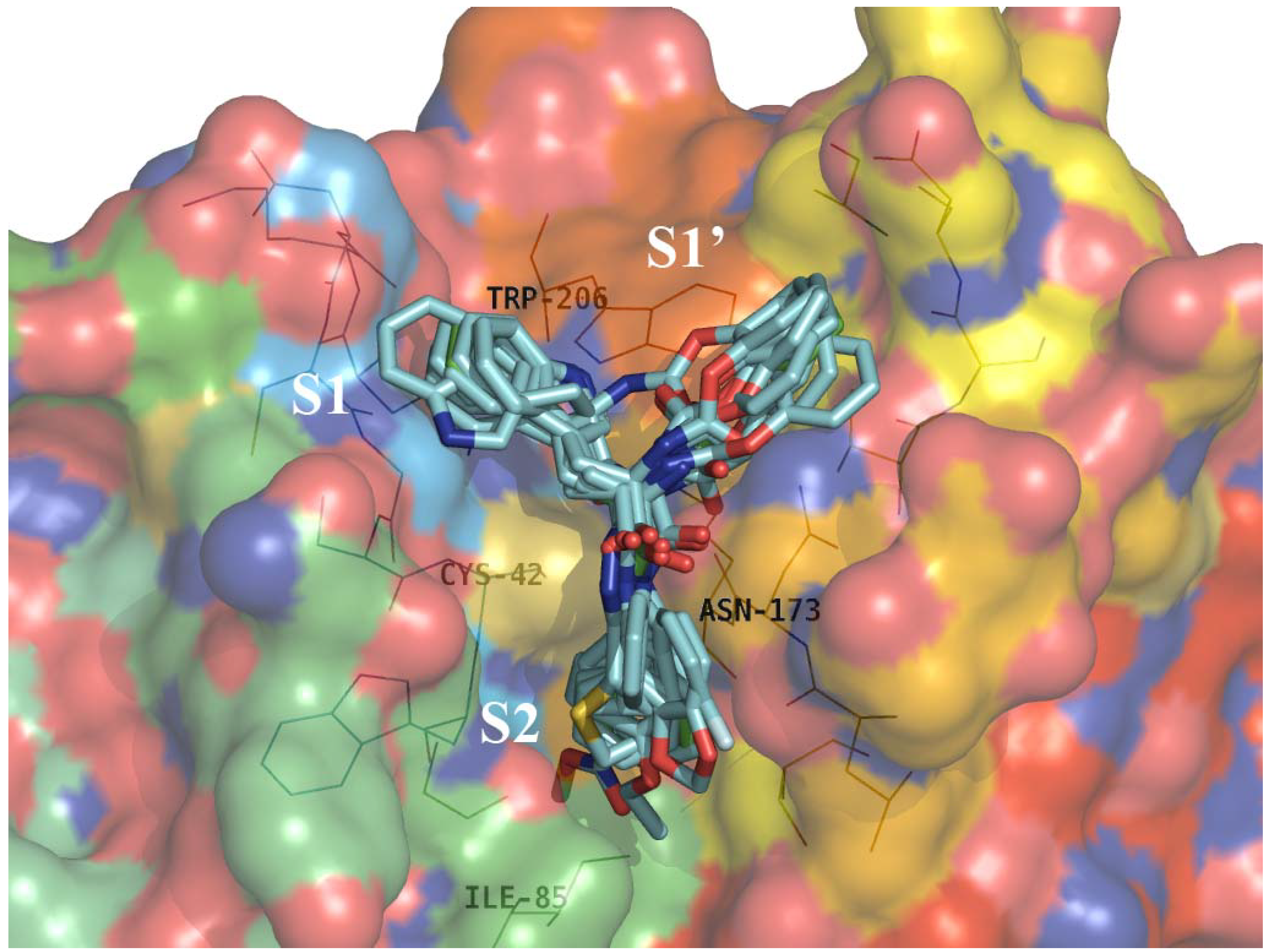
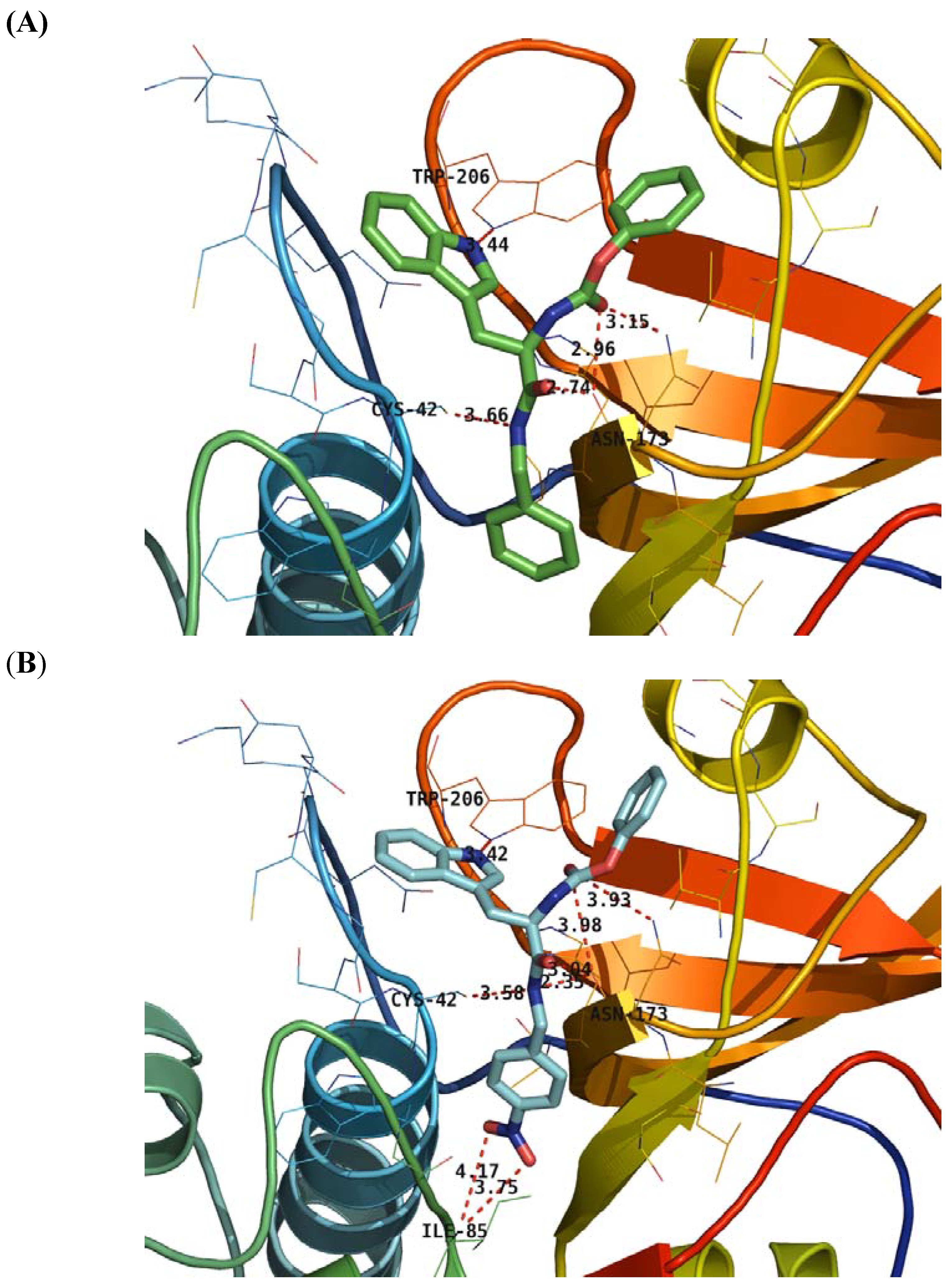
2.4. Structure Activity Relationship Correcting with the Binding Models


3. Experimental
3.1. Virtual Screening by Molecular Docking
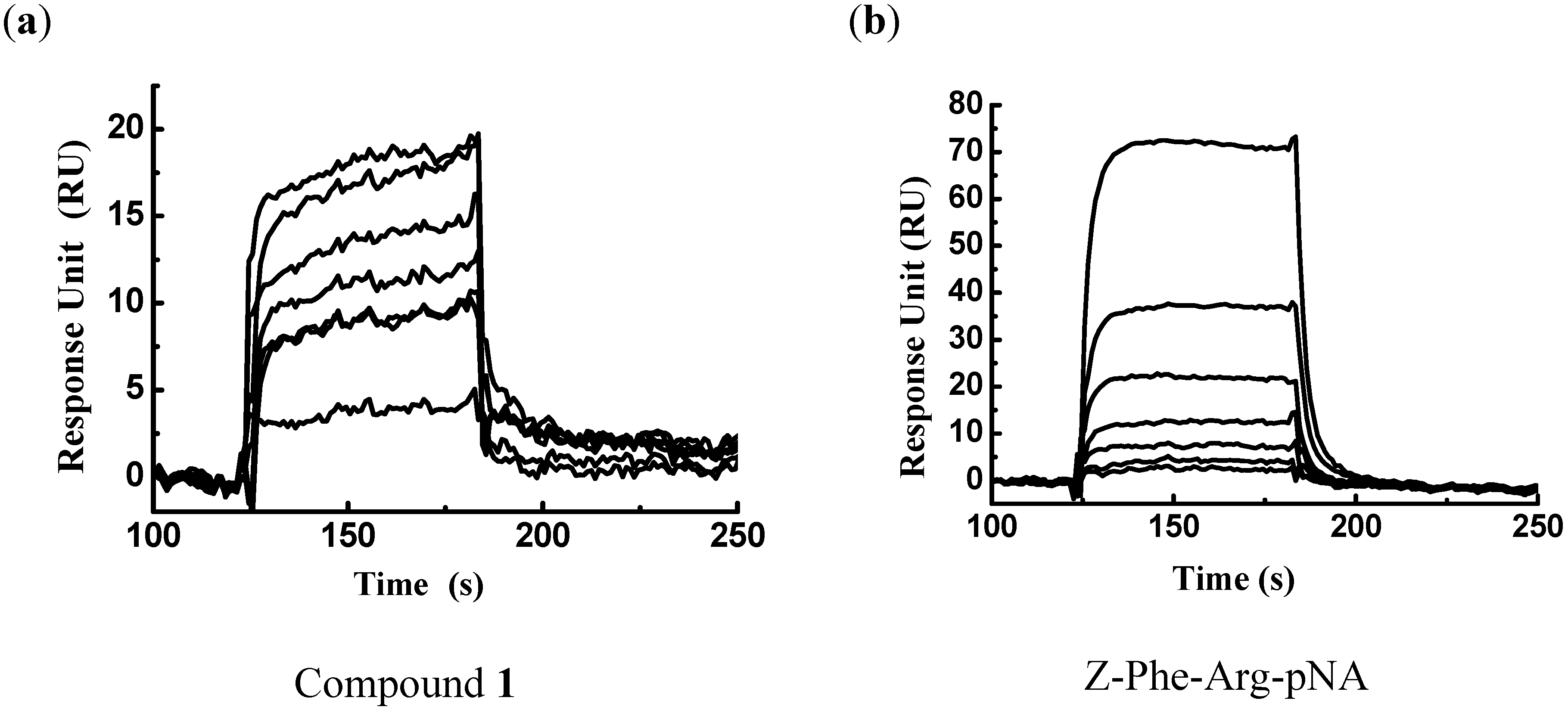
3.2. Surface Plasmon Resonance-based FP-2/Ligand Binding Assay
3.3. Enzyme Inhibition Assay
3.4. Preparation of (R)-2-phenoxycarboxamido-3-(1H-indol-3-yl)-N-benzylpropanamide (1).
3.5 Preparation of 2-amide-3-(1H-indol-3-yl)-N-substitued-propanamide compounds 2a,b and 2d-l
4. Conclusions
Acknowledgements
References and Notes
- Vangapandu, S.; Jain, M.; Kaur, K.; Patil, P.; Patel, S.R.; Jain, R. Recent advances in antimalarial drug development. Med. Res. Rev. 2007, 27, 65–107. [Google Scholar] [CrossRef]
- Fidock, D.A.; Rosenthal, P.J.; Croft, S.L.; Brun, R.; Nwaka, S. Antimalarial drug discovery: Efficacy models for compound screening. Nat. Rev. Drug Discov. 2004, 3, 509–520. [Google Scholar] [CrossRef]
- Wellems, T.E.; Plowe, C.V. Chloroquine-resistant malaria. J. Infect. Dis. 2001, 184, 770–776. [Google Scholar] [CrossRef]
- Trape, J.F. The public health impact of chloroquine resistance in Africa. Am. J. Trop. Med. Hyg. 2001, 64, 12–17. [Google Scholar]
- Plowe, C.V. Monitoring antimalarial drug resistance: making the most of the tools at hand. J. Exp. Biol. 2003, 206, 3745–3752. [Google Scholar] [CrossRef]
- Shenai, B.R.; Sijwali, P.S.; Singh, A.; Rosenthal, P.J. Characterization of native and recombinant falcipain-2, a principal trophozoite cysteine protease and essential hemoglobinase of Plasmodium falciparum. J. Biol. Chem. 2000, 275, 29000–29010. [Google Scholar] [CrossRef]
- Dua, M.; Raphael, P.; Sijwali, P.S.; Rosenthal, P.J.; Hanspal, M. Recombinant falcipain-2 cleaves erythrocyte membrane ankyrin and protein 4.1. Mol. Biochem. Parasitol. 2001, 116, 95–99. [Google Scholar] [CrossRef]
- Rosenthal, P.J.; Olson, J.E.; Lee, G.K.; Palmer, J.T.; Klaus, J.L.; Rasnick, D. Antimalarial effects of vinyl sulfone cysteine proteinase inhibitors. Antimicrob. Agents Chemother. 1996, 40, 1600–1603. [Google Scholar]
- Shenai, B.R; Lee, B.J.; Alvarez-Hernandez, A.; Chong, P.Y.; Emal, C.D.; Neitz, R.J.; Roush, W.R.; Rosenthal, P.J. Structure-activity relationships for inhibition of cysteine protease activity and development of Plasmodium falciparum by peptidyl vinyl sulfones. Antimicrob. Agents Chemother. 2003, 47, 154–160. [Google Scholar] [CrossRef]
- Lee, B.J.; Singh, A.; Chiang, P.; Kemp, S.J.; Goldman, E.A.; Weinhouse, M.I.; Vlasuk, G.P.; Rosenthal, P.J. Antimalarial activities of novel synthetic cysteine protease inhibitors. Antimicrob. Agents Chemother. 2003, 47, 3810–3814. [Google Scholar] [CrossRef]
- Domínguez, J.N.; López, S.; Charris, J.; Iarruso, L.; Lobo, G.; Semenov, A.; Olson, J.E.; Rosenthal, P.J. Synthesis and antimalarial effects of phenothiazine inhibitors of a Plasmodium falciparum cysteine protease. J. Med. Chem. 1997, 40, 2726–2732. [Google Scholar] [CrossRef]
- Huang, L.; Lee, A.; Ellman, J.A. Identification of potent and selective mechanism-based inhibitors of the cysteine protease cruzain using solid-phase parallel synthesis. J. Med. Chem. 2002, 45, 676–684. [Google Scholar] [CrossRef]
- Greenbaum, D.C.; Mackey, Z.; Hansell, E.; Doyle, P.; Gut, J.; Caffrey, C.R.; Lehrman, J.; Rosenthal, P.J.; McKerrow, J.H.; Chibale, K. Synthesis and structure-activity relationships of parasiticidal thiosemicarbazone cysteine protease inhibitors against Plasmodium falciparum, Trypanosoma brucei, and Trypanosoma cruzi. J. Med. Chem. 2004, 47, 3212–3219. [Google Scholar] [CrossRef]
- Desai, P.V.; Patny, A.; Sabnis, Y.; Tekwani, B.; Gut, J.; Rosenthal, P.; Srivastava, A.; Avery, M. Identification of novel parasitic cysteine protease inhibitors using virtual screening. 1. The ChemBridge database. J. Med. Chem. 2004, 47, 6609–6615. [Google Scholar] [CrossRef]
- Domínguez, J.N.; León, C.; Rodrigues, J.; Gamboa de Domínguez, N.; Gut, J.; Rosenthal, P.J. Synthesis and evaluation of new antimalarial phenylurenyl chalcone derivatives. J. Med. Chem. 2005, 48, 3654–3658. [Google Scholar] [CrossRef]
- Desai, P.V.; Patny, A.; Gut, J.; Rosenthal, P.J.; Tekwani, B.; Srivastava, A.; Avery, M. Identification of novel parasitic cysteine protease inhibitors by use of virtual screening. 2. The available chemical directory. J. Med. Chem. 2006, 49, 1576–1584. [Google Scholar] [CrossRef]
- Batra, S.; Sabnis, Y.A.; Rosenthal, P.J.; Avery, M.A. Structure-based approach to falcipain-2 inhibitors: Synthesis and biological evaluation of 1,6,7-trisubstituted dihydroisoquinolines and isoquinolines. Bioorg. Med. Chem. 2003, 11, 2293–2299. [Google Scholar] [CrossRef]
- Chiyanzu, I.; Hansell, E.; Gut, J.; Rosenthal, P.J.; Mckerrow, J.H.; Chibale, K. Synthesis and evaluation of isatins and thiosemicarbazone derivatives against cruzain, falcipain-2 and rhodesain. Bioorg. Med. Chem. Lett. 2003, 13, 3527–3530. [Google Scholar] [CrossRef]
- Chipeleme, A.; Gut, J.; Rosenthal, P.J.; Chibale, K. Synthesis and biological evaluation of phenolic Mannich bases of benzaldehyde and (thio)semicarbazone derivatives against the cysteine protease falcipain-2 and a chloroquine resistant strain of Plasmodium falciparum. Bioorg. Med. Chem. 2007, 15, 273–282. [Google Scholar] [CrossRef]
- Rosenthal, P.J.; Wollish, W.S.; Palmer, J.T.; Rasnick, D. Antimalarial effects of peptide inhibitors of a Plasmodium falciparum cysteine proteinase. J. Clin. Invest. 1991, 88, 1467–1472. [Google Scholar] [CrossRef]
- Rosenthal, P.J.; Lee, G.K.; Smith, R.E. Inhibition of a Plasmodium vinckei cysteine proteinase cures murine malaria. J. Clin. Invest. 1993, 91, 1052–1056. [Google Scholar] [CrossRef]
- Sajid, M.; McKerrow, J. Cysteine proteases of parasitic organisms. Mol. Biochem. Parasitol. 2002, 120, 1–21. [Google Scholar] [CrossRef]
- Jorgensen, W.L. The many roles of computation in drug discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef]
- Sabnis, Y.; Rosenthal, P.J.; Desai, P.; Avery, M.A. Homology modeling of falcipain-2: Validation, de novo ligand design and synthesis of novel inhibitors. J. Biomol. Struct. Dyn. 2002, 19, 765–774. [Google Scholar] [CrossRef]
- Goh, L.L.; Sim, T.S. Homology modeling and mutagenesis analyses of Plasmodium falciparum falcipain 2A: Implications for rational drug design. Biochem. Biophys. Res. Commun. 2004, 323, 565–572. [Google Scholar] [CrossRef]
- Hogg, T.; Nagarajan, K.; Herzberg, S.; Chen, L.; Shen, X.; Jiang, H.; Wecke, M.; Blohmke, C.; Hilgenfeld, R.; Schmidt, C.L. Structural and functional characterization of Falcipain-2, a hemoglobinase from the malarial parasite Plasmodium falciparum. J. Biol. Chem. 2006, 281, 25425–25437. [Google Scholar] [CrossRef]
- Wang, S.X.; Pandey, K.C.; Somoza, J.R.; Sijwali, P.S.; Kortemme, T.; Brinen, L.S.; Fletterick, R.J.; Rosenthal, P.J.; Mckerrow, J.H. Structural basis for unique mechanisms of folding and hemoglobin binding by a malarial protease. Proc. Natl. Acad. Sci. USA 2006, 103, 11503–11508. [Google Scholar] [CrossRef]
- Li, H.; Chen, L.; Huang, J.; Zhu, J.; Shen, X.; Li, J.; Hilgenfeld, R.; Jiang, H. Identification of novel Falcipain-2 inhibitors as potential antimalarial agents through structure-based virtual screening. Submitted to J. Med. Chem.
- Peters, W. Drug resistance in Plasmodium berghei Vincke and Lips, 1948. I. Chloroquine resistance. Expl. Parasite 1965, 17, 80–89. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar]
- Rosenthal, P.J.; McKerrow, J.H.; Rasnick, D.; Leech, J.H. Plasmodium falciparum: Inhibitors of lysosomal cysteine proteinases inhibit a trophozoite proteinase and block parasite development. Mol. Biochem. Parasitol. 1989, 35, 177–183. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds reported in this paper are available from authors.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhu, J.; Chen, T.; Chen, L.; Lu, W.; Che, P.; Huang, J.; Li, H.; Li, J.; Jiang, H. 2-Amido-3-(1H-Indol-3-yl)-N-Substitued-Propanamides as a New Class of Falcipain-2 Inhibitors. 1. Design, Synthesis, Biological Evaluation and Binding Model Studies. Molecules 2009, 14, 494-508. https://doi.org/10.3390/molecules14010494
Zhu J, Chen T, Chen L, Lu W, Che P, Huang J, Li H, Li J, Jiang H. 2-Amido-3-(1H-Indol-3-yl)-N-Substitued-Propanamides as a New Class of Falcipain-2 Inhibitors. 1. Design, Synthesis, Biological Evaluation and Binding Model Studies. Molecules. 2009; 14(1):494-508. https://doi.org/10.3390/molecules14010494
Chicago/Turabian StyleZhu, Jin, Tong Chen, Lili Chen, Weiqiang Lu, Peng Che, Jin Huang, Honglin Li, Jian Li, and Hualiang Jiang. 2009. "2-Amido-3-(1H-Indol-3-yl)-N-Substitued-Propanamides as a New Class of Falcipain-2 Inhibitors. 1. Design, Synthesis, Biological Evaluation and Binding Model Studies" Molecules 14, no. 1: 494-508. https://doi.org/10.3390/molecules14010494
APA StyleZhu, J., Chen, T., Chen, L., Lu, W., Che, P., Huang, J., Li, H., Li, J., & Jiang, H. (2009). 2-Amido-3-(1H-Indol-3-yl)-N-Substitued-Propanamides as a New Class of Falcipain-2 Inhibitors. 1. Design, Synthesis, Biological Evaluation and Binding Model Studies. Molecules, 14(1), 494-508. https://doi.org/10.3390/molecules14010494