QSAR Studies on N-aryl Derivative Activity Towards Alzheimer’s Disease

Abstract
:1. Introduction
2. Results and Discussion

| Descriptor | Type | Significance |
|---|---|---|
| ALOGP98 | Thermodynamic descriptor | Logarithm of partition coefficient |
| WIENER | Graph–theoretical descriptor | Sum of chemical bonds between atoms |
| CHI-V-1-3 | Topological descriptor | Molecular connectivity indices |
| CHI-1 | ||
| KAPPA-1-AM | Topological descriptor | Molecular Shape Kappa indices |
| DIPOLE-MAG | Electronic descriptor | Dipole moment |
| PHI | Topological descriptor | Molecular flexibility indices |
| LogZ | Topological descriptor | Logarithm of Hosoya index |
| HBOND DONOR | Structural descriptor | Number of hydrogen bond donors groups |

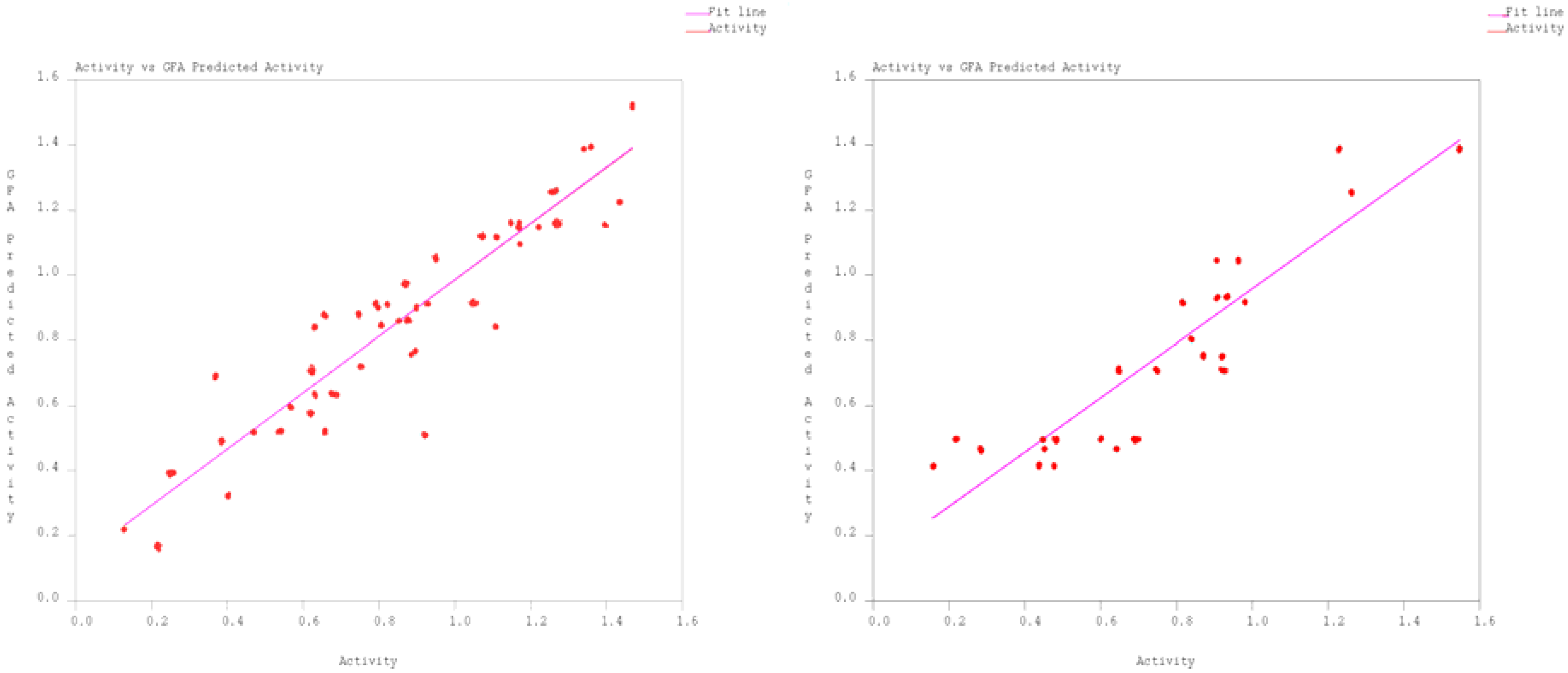{kind=link}
| Compd. No. | Substituents | Experimental Activity | GFA Predicted Activity | GFA Residuals Activity | ||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | ||||
| 54A | NH2 | H | H | 0.285 | 0.467 | -0.182 |
| 55A | H | CO | H | 0.437 | 0.416 | 0.020 |
| 56A | H | H | NH2 | 0.642 | 0.467 | 0.174 |
| 57A | H | H | OH | 0.748 | 0.709 | 0.038 |
| 58A | H | OCH3 | H | 0.818 | 0.918 | -0.100 |
| 59A | H | H | H | 1.23 | 1.389 | -0.159 |
| 60A | OH | H | H | 0.926 | 0.709 | 0.216 |
| 61A | CO2H | H | H | 0.91 | 0.933 | -0.023 |
| 62B | NO2 | H | H | 1.549 | 1.389 | 0.159 |
| 63B | CO2H | H | H | 0.158 | 0.416 | -0.258 |
| 64B | H | CO2H | H | 0.477 | 0.416 | 0.060 |
| 65B | Cl | H | H | 0.217 | 0.496 | -0.279 |
| 66B | H | Cl | H | 0.484 | 0.496 | -0.012 |
| 67B | H | H | Cl | 0.448 | 0.496 | -0.048 |
| 68B | H | OH | H | 0.65 | 0.709 | -0.059 |
| 69B | H | H | NH2 | 0.453 | 0.467 | -0.014 |
| 70A | H | H | OCH3 | 0.981 | 0.918 | 0.062 |
| 71A | F | H | H | 0.935 | 0.933 | 0.001 |
| 72C | H | H | OH | 0.906 | 1.045 | -0.139 |
| 73C | H | CO2H | H | 0.921 | 0.752 | 0.168 |
| 74D | H | CO2H | H | 0.872 | 0.752 | 0.119 |
| 75D | H | H | OCH3 | 1.262 | 1.254 | 0.007 |
| 76D | H | OH | H | 0.965 | 1.045 | -0.080 |
| 77D | H | NH2 | H | 0.84 | 0.803 | 0.036 |
| 78D | NH2 | H | CH | 0.6 | 0.496 | 0.103 |
| 79D | H | H | Cl | 0.692 | 0.496 | 0.095 |
| Compd. No. | Substituents | Experimental Activity | GFA Predicted Activity | GFA Residuals Activity | ||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | ||||
| 60A | OH | H | H | 1.176 | 1.22 | -0.044 |
| 20B | H | NH2 | H | 0.448 | 0.469 | -0.021 |
| 19C | H | H | H | 1.813 | 1.885 | -0.072 |
| 21C | H | NO2 | H | 1.247 | 1.114 | 0.131 |
| 72C | H | H | OH | 1.752 | 1.567 | 0.184 |
| 26C | OH | H | H | 1.648 | 1.771 | -0.123 |
| 35C | H | H | F | 1.942 | 1.901 | 0.04 |
| 37D | H | NO2 | H | 1.181 | 1.115 | 0.065 |
| 74D | H | CO2 | H | 0.986 | 1.115 | -0.129 |
| 76D | H | OH | H | 1.531 | 1.422 | 0.108 |
| 50D | H | NH2 | H | 1.424 | 1.369 | 0.054 |
| 77D | H | H | NH2 | 1.136 | 1.088 | 0.047 |
| 43D | H | NO2 | H | 1.139 | 1.234 | -0.095 |
| 7B | NO2 | H | H | 0.991 | 0.923 | 0.067 |
| 62B | H | Cl | H | 1.363 | 1.285 | 0.077 |
| 86C | H | OCH3 | H | 1.77 | 1.755 | 0.014 |
| 45D | H | CI | H | 1.477 | 1.522 | -0.045 |
| 39D | H | NO | H | 1.26 | 1.425 | -0.165 |
| 59A | H | H | NO2 | 0.561 | 0.636 | -0.075 |
| 2A | H | OH | H | 0.791 | 0.803 | -0.012 |

3. Experimental
3.1. Data screening and Molecular Modeling
3.2. Generation of QSAR models using GFA technique
4. Conclusions
References
- Lahiri, D.K.; Farlow, M.R.; Greig, N.H.; Sambamurti, K. Current Drug Targets for Alzheimer’s disease Treatment. Drug Develop. Res. 2004, 56, 267–281. [Google Scholar]
- Cummings, J.L. Alzheimer’s disease. Drug Ther. 2004, 351, 56–67. [Google Scholar]
- Campiani, G.; Kozikowski, A.P.; Wang, S.M.; Liu, M.; Nacci, V.; Saxena, A.; Doctor, B.P. Synthesis and Anti cholinesterase Activity of Huperzine A Analogues Containing Phenol and Catechol Replacements for the Pyridone ring. Bioorg. Med. Chem. 1998, 8, 1413–1418. [Google Scholar] [CrossRef]
- Guevara-Salazar, J.A.; Espinoza-Fonseca, M.; Beltran, H.I.; Correa-Basurto, J.; Trujillo-Ferrara, D.Q.Z.J.G. The Electronic Influence on the Active Site-Directed Inhibition of Acetylcholinesterase by N-aryl substituted Succinimides. J. Mex. Chem. Soc. 2007; 51, 173–177. [Google Scholar]
- Trujillo-Ferrara, J.; Correa-Basurto, J.; Espinosa, J.; Garcia, J. Solvent-free Synthesis of Arylamides and Arylimides, Analogues of Acetylcholine. Syn. Commu. 2005, 35, 1–7. [Google Scholar] [CrossRef]
- Duardo-Sanchez, A.; Patlewicz, G.; Lopez-Diaz, A. Current topics on software use in medicinal chemistry: Intellectual property, taxes, and regulatory issues. Curr. Top. Med. Chem. 2008, 8, 1666–1675. [Google Scholar] [CrossRef]
- Gonzalez-Diaz, H.; Vilar, S.; Santana, L.; Uriarte, E. Medicinal chemistry and bioinformatics – current trends in drugs discovery with networks topological indices. Curr. Top. Med. Chem. 2007, 7, 1015–1029. [Google Scholar] [CrossRef]
- Gonzalez-Diaz, H.; Prado-Prafo, F.; Uberia, F.M. Predicting antimicrobial drugs and targets with the MARCH-INSIDE approach. Curr. Top. Med. Chem. 2008, 8, 1676–1690. [Google Scholar] [CrossRef]
- Caballero, J.; Fernandez, M. Artificial neural networks from MATLAB in medicinal chemistry. Bayesian-regularized generic neural networks (BRGNN): Application to the prediction of the anatagonistic actitivity against human platelet thrombin receptor (PAR-1). Curr. Top. Med. Chem. 2008; 8, 1580–1605. [Google Scholar]
- Gonzalez, M.P.; Teran, C.; Saiz-Urra, L.; Teijeria, M. Variable selection methods in QSAR: An overview. Curr. Top. Med. Chem. 2008, 8, 1606–1627. [Google Scholar] [CrossRef]
- Helguera, A.M.; Combes, R.D.; Gonzalez, M.P.; Cordeiro, M.N. Applications of 2D Descriptors in drug design: a DRAGON tale. Curr. Top. Med. Chem. 2008, 8, 1628–1655. [Google Scholar]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Liu, A.; Gunag, H.; Zhu, L.; Du, G. 3D-QSAR Analysis of a Newtype of Acetylcholinesterase inhibitors. Sci. China Life Sci. 2007, 50, 726–730. [Google Scholar] [CrossRef]
- Recanatini, M.; Cavalli, A.; Hansch, C.A. Comparative QSAR analysis of Acetylcholinesterase Inhibitors currently studied for the Treatment of Alzheimer’s disease. Chem. Biol. Interact. 1997, 105, 199–228. [Google Scholar] [CrossRef]
- Shen, L.L.; Liu, G.X.; Tang, Y. Molecular docking & 3D-QSAR studies of 2-substituted 1-indanone derivatives as Acetylcholinesterase inhibitors. Acta Pharmacol. Sin. 2007, 28, 2053–2063. [Google Scholar] [CrossRef]
- Castilho, C.; Guido, R.V. Andricopulo. Classical and Hologram QSAR Studies on a Series of Tacrine Derivatives as Butyrylcholinesterase Inhibitors. Lett. Drug Des. Discov. 2007, 4, 106–113. [Google Scholar] [CrossRef]
- Correa-Basurto, J.; Flores-Sandoval, C.; Marvin-Cruz, J.; Rojo-Dominguez, A.; Espinoza-Fonseca, L.M.; Trujillo-Ferrara, J.G. Docking and Quantum Mechanic Studies on Cholinesterase and their Inhibitors. Eur. J. Med. Chem. 2007, 42, 10–19. [Google Scholar] [CrossRef]
- Hahn, M.; Rogers, D. Receptor surface models. 2 application to quantitative structure-activity relationships studies. J. Med. Chem. 1995, 38, 2091–2102. [Google Scholar] [CrossRef]
- Hopfinger, A.J.; Kawayami, Y. QSAR analysis of a set of benzothiopyranoindazole anti-cancer analogs based upon their DNA intercalation properties as determined by molecular dynamics simulation. Anticancer Drug Des. 1992, 7, 203–217. [Google Scholar]
- Kawakami, Y.; Inoue, A.; Kawai, T.; Wakita, M.; Sugimoto, H.; Hopfinger, A.J. The rationale for E2020 as a potent acetylcholinesterase inhibitor. Bioorgan. Med. Chem. 1996, 4, 1429–1449. [Google Scholar] [CrossRef]
- Jung, M.; Tak, J.; Lee, Y.; Jung, Y. Quantitative structure–activity relationship (QSAR) of tacrine derivatives against acetylcholinesterase (AChE) activity using variable selections. Bioorg. Med. Chem. Lett. 2007, 17, 1082–1090. [Google Scholar] [CrossRef]
- Anand, V.R.; Vithal, M.K. 3D-QSAR of Cyclooxgenase-2 Inhibitors by Genetic Function Approximation. Internet Electron. J. Mol. Des. 2003, 2, 242–261. [Google Scholar]
- Cho, D.H.; Lee, S.K.; Kim, B.T.; No, K.T. Quantitative Structure-Activity Relationship (QSAR) Study of New Fluorovinyloxyacetamides. Bull Korean Chem. Soc. 2001, 22(No. 4.). [Google Scholar]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Solomon, K.A.; Sundararajan, S.; Abirami, V. QSAR Studies on N-aryl Derivative Activity Towards Alzheimer’s Disease. Molecules 2009, 14, 1448-1455. https://doi.org/10.3390/molecules14041448
Solomon KA, Sundararajan S, Abirami V. QSAR Studies on N-aryl Derivative Activity Towards Alzheimer’s Disease. Molecules. 2009; 14(4):1448-1455. https://doi.org/10.3390/molecules14041448
Chicago/Turabian StyleSolomon, Kamalakaran Anand, Srinivasan Sundararajan, and Veluchamy Abirami. 2009. "QSAR Studies on N-aryl Derivative Activity Towards Alzheimer’s Disease" Molecules 14, no. 4: 1448-1455. https://doi.org/10.3390/molecules14041448
APA StyleSolomon, K. A., Sundararajan, S., & Abirami, V. (2009). QSAR Studies on N-aryl Derivative Activity Towards Alzheimer’s Disease. Molecules, 14(4), 1448-1455. https://doi.org/10.3390/molecules14041448