General
Unless otherwise stated, all chemical reagents purchased (Merck or Aldrich) were of the highest commercially available purity and were used without previous purification. Melting points were measured (in triplicate) on a Stuart-Scientific SMP3 apparatus and are uncorrected. IR spectra were recorded as thin films in a Nicolet Impact 420 spectrometer and frequencies are reported in cm-1. Optical rotations were measured with a sodium lamp (λ=589 nm, D line) on a Perkin Elmer 241 digital polarimeter equipped with 1 dm cells at the temperature indicated in each case. Low resolution mass spectra were recorded on a Shimadzu QP-2000 spectrometer at 70eV ionising voltage and are given as m/z (% rel. int.) 1H-, 13C- (DEPT 135 and DEPT 90), sel. 1D 1H NOESY, sel. 1D 1H TOCSY, 2D HSQC and 2D HMBC spectra were recorded in CDCl3 solutions and are referenced to the residual peaks of CHCl3 at δ 7.26 ppm and δ 77.0 ppm for 1H and 13C, respectively, on a Bruker Avance 400 Digital NMR spectrometer, operating at 400.1MHz for 1H and 100.6MHz for 13C. Chemical shifts are reported in δ ppm and coupling constants (J) are given in Hz. Silica gel (Merck 200-300 mesh) was used for C.C. and silica gel plates HF-254 for TLC. TLC spots were detected by heating after spraying with 25% H2SO4 in H2O.
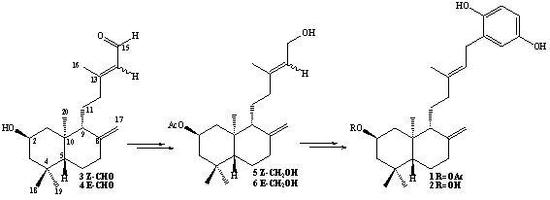
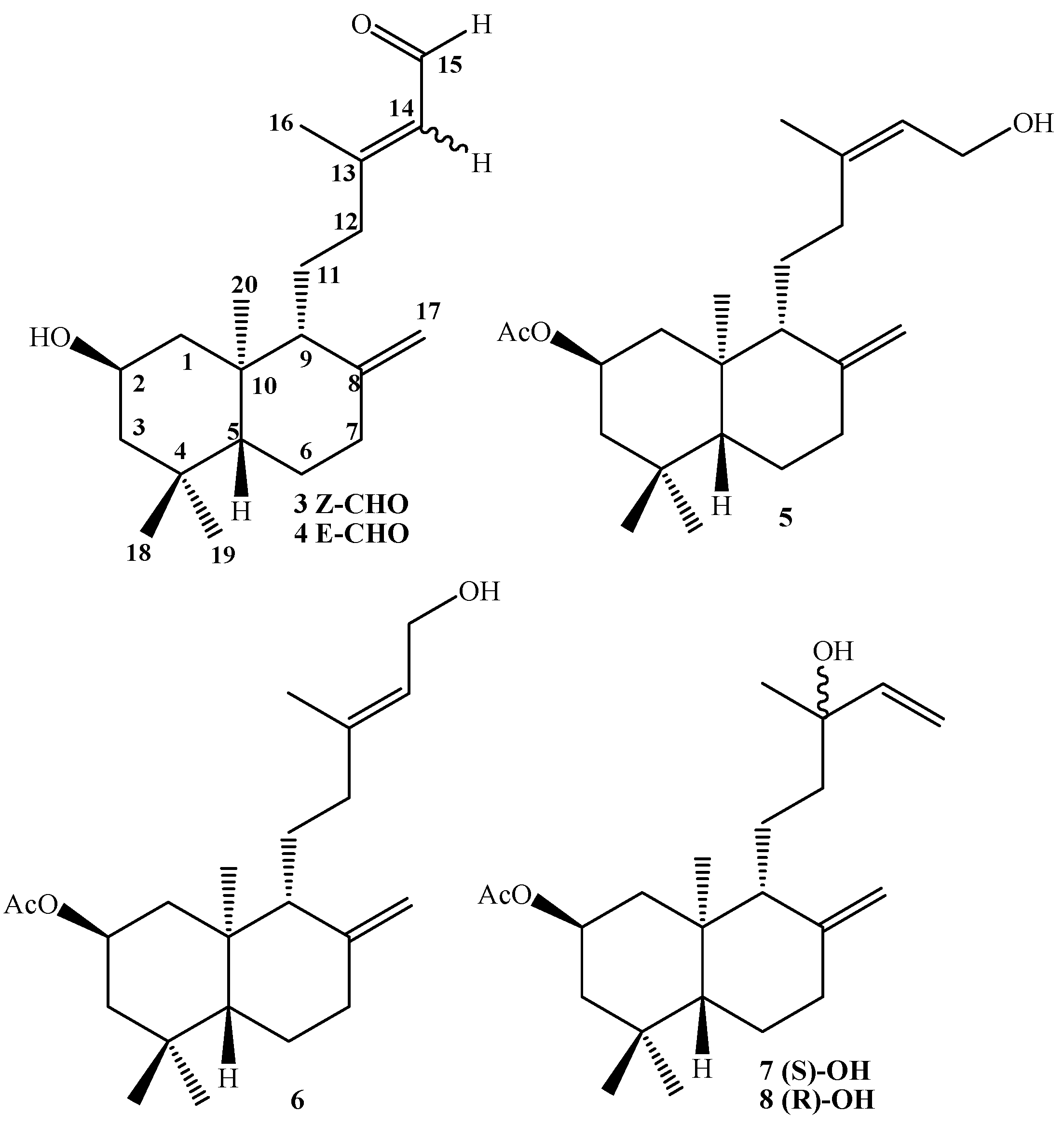
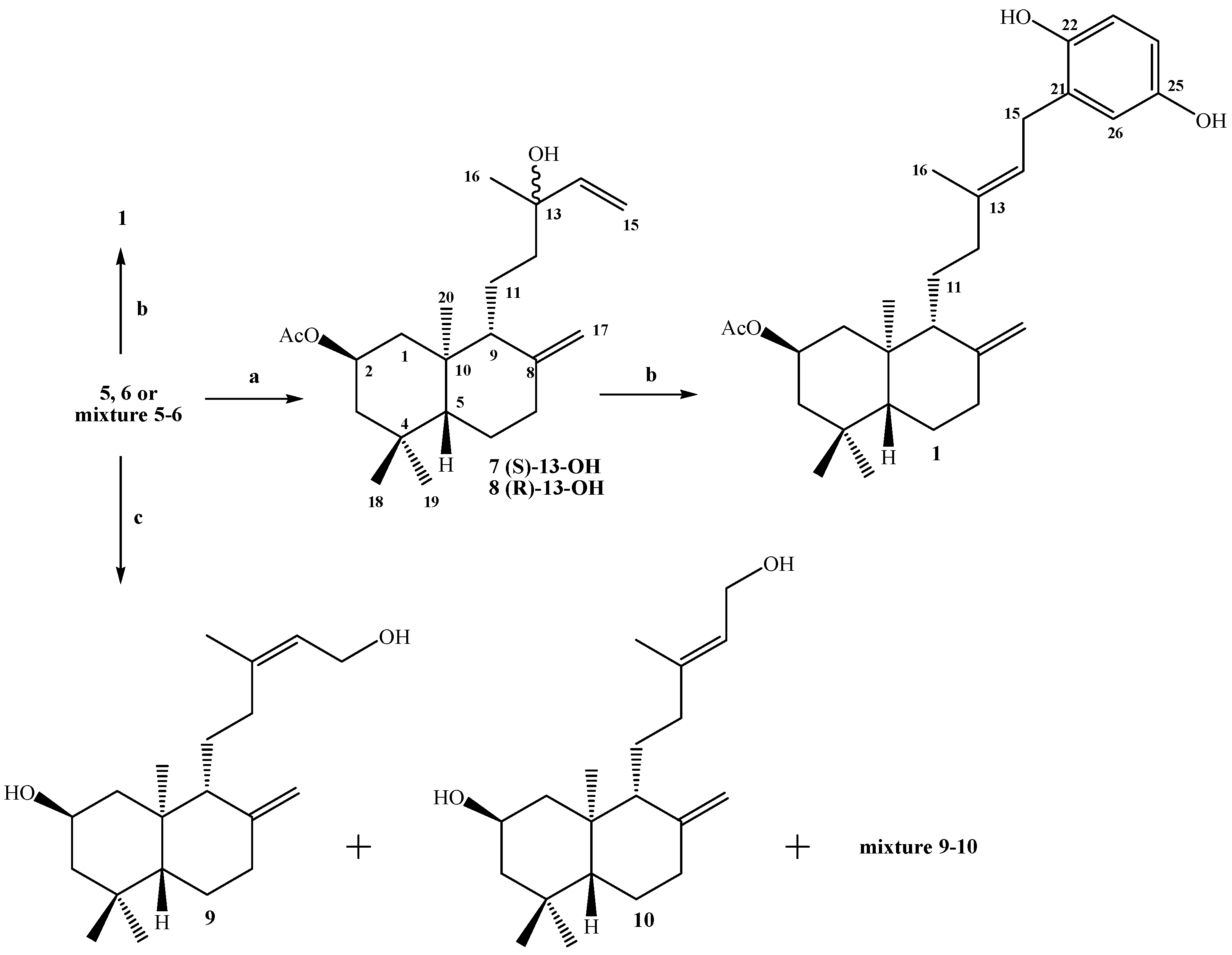
Synthesis of 2β-acetoxy-(S)-13-hydroxy-ent-labda-8(17), 14-diene (7) and 2β-acetoxy-(R)-13-hydroxy-ent-labda-8(17), 14-diene (8) from 5: A solution of 5 (1.54 g, 0.44 mmol) in dry CH2Cl2 (50 mL) and dry pyridine (1 mL), was prepared under a N2 atmosphere and cooled to -10°C (ice/acetone/brine bath). Then SOCl2 (0.6 mL, 8.26 mmol) was slowly added dropwise while maintaining slow agitation. After one hour, the completion of the reaction were verified by TLC. Then saturated aqueous solution of NaHCO3 (50 mL) was added and the mixture was extracted with EtOAc (2 x 25 mL) and the combined organic layers were washed with water (2 x 20 mL), dried over Na2SO4, filtered and evaporated. The crude was redissolved in CH2Cl2 (5 mL) and chromatographed on silica-gel with petroleum ether/EtOAc mixtures of increasing polarity (19.8:0.2→8.8:11.2). Two fractions were obtained: Fraction I: 1.08 g of a non-polar unidentified complex mixture (colorless viscous oil) Fraction II: colorless viscous oil, 0.34 g (22.1%) of a mixture of 7-8.
Synthesis of 7-8 mixture from 6: From 2.23 g (6.40 mmol) of alcohol 6, 0.8 mL (11.0 mmol) of SOCl2 and 1.5 mL of pyridine, 1.32 g of non-polar complex mixture and 0.75 g (33.7%) of 7-8 mixture were obtained.
Synthesis of 7-8 mixture from a mixture of 5-6: From 2.38 g (6.83 mmol) of 5-6 mixture, 0.8 mL (11.0 mmol) of SOCl2 and 1.5 mL of pyridine, 1.57 g of non-polar complex mixture and 0.72 g (30.3%) of 7-8 mixture were obtained.
Compound 7: 1H-NMR: 5.90 (dd, J= 17.3 and 10.8 Hz, 1H, H-14); 5.20 (dd, J= 17.3 and 1.5 Hz, 1H, H-15b); 5.05 (dd, J= 10.8 and 1.5 Hz, 1H, H-15a); 5.02 (ddt, J= 11.7, 11.7 and 3.9 Hz, 1H, H-2); 4.89 (s, 1H, H-17b); 4.55 (s, 1H, H-17a); 2.39 (ddd, J= 12.7, 4.4 and 2.5 Hz, 1H, H-7α); 2.07 (m, 1H, H-1α); 2.03 (s, 3H, CH3CO2); 1.95 (ddd, J= 13.2, 12.7 and 4.9 Hz, 1H, H-7β); 1.70 (m, 3H, H-3α, H-12b and H-6β); 1.61 (bd, J= 10.3 Hz, 1H, H-9); 1.43 (m, 1H, H-11b); 1.32 (m, 1H, H-11a); 1.29 (m, 2H, H-12a and H-6α); 1.27 (s, 3H, H-16); 1.23 (m, 1H, H-3β); 1.09 (dd, J= 17.7 and 2.5 Hz, 1H, H-5); 1.07 (dd, J= 11.7 and 11.7 Hz, 1H, H-1β), 0.93 (s, 3H, H-18); 0.88 (s, 3H, H-19); 0.76 (s, 3H, H-20); 13C-NMR: 44.1 (C-1), 69.3 (C-2), 46.8 (C-3), 34.9 (C-4), 55.0 (C-5), 23.9 (C-6), 38.0 (C-7), 147.4 (C-8), 57.1 (C-9), 41.2 (C-10), 17.9 (C-11), 41.1 (C-12), 73.4 (C-13), 145.2 (C-14), 111.7 (C-15), 27.5 (C-16). 107.6 (C-17), 33.6 (C-18), 22.4 (C-19), 15.2 (C-20), 170.6 (CH3CO), 21.5 (CH3CO); M.S.(m/z, %): M+ 348 (< 1%), 202 (13.2); 201 (13.9); 189 (13.9); 188 (20.8); 187 (33.0); 175 (16.0); 173 (8.9); 161 (10.5); 159 (12.8); 147 (11.4); 136 (14.5); 135 (100); 134 (15.9); 133 (17.2); 131 (12.2); 122 (10.5); 121 (27.1); 120 (15.0); 119 (28.0); 109 (13.8); 107 (32.3); 105 (14.8); 95 (18.3); 94 (9.4); 93 (29.9); 91 (18.1); 81 (21.1); 80 (8.9); 79 (18.0); 71 (22.5).
Compound 8: 1H-NMR: 5.90 (dd, J= 17.4 and 10.8 Hz, 1H, H-14); 5.21 (dd, J= 17.4 and 1.0 Hz, 1H, H-15α); 5.06 (dd, J= 10.8 and 1.0 Hz, 1H, H-15α); 5.02 (ddt, J= 11.7, 11.7 and 3.9 Hz, 1H, H-2); 4.84 (s, 1H, H-17b); 4.50 (s, 1H, H-17α); 2.39 (ddd, J= 12.7, 4.4 and 2.5 Hz, 1H, H-7a); 2.07 (m, 1H, H-1a); 2.03 (s, 3H, CH3CO2); 1.95 (ddd, J= 13.2, 12.7 and 4.9 Hz, 1H, H-7β); 1.70 (m, 3H, H-3α, H-12b and H-6β); 1.61 (bd, J= 10.3 Hz, 1H, H-9); 1.43 (m, 1H, H-11b); 1.32 (m, 1H, H-11a); 1.27 (m, 2H, H-12a and H-6β); 1.27 (s, 3H, H-16); 1.23 (m, 1H, H-3β); 1.09 (dd, J= 17.7 and 2.5 Hz, 1H, H-5); 1.07 (dd, J= 11.7 and 11.7 Hz, 1H, H-1β), 1.04 (s, 3H, H-18); 0.88 (s, 3H, H-19); 0.75 (s, 3H, H-20). 13C-NMR: 44.2 (C-1), 69.3 (C-2), 46.8 (C-3), 34.9 (C-4), 55.0 (C-5), 23.9 (C-6), 38.0 (C-7), 147.5 (C-8), 57.0 (C-9), 41.1 (C-10), 17.8 (C-11), 41.2 (C-12), 73.6 (C-13), 145.0 (C-14), 111.8 (C-15), 28.3 (C-16). 107.4 (C-17), 33.6 (C-18), 22.4 (C-19), 15.2 (C-20), 170.6 (CH3CO), 21.5 (CH3CO). M.S. (m/z, %): M+ 348 (< 1%), 302 (4.9); 270 (10.3); 255 (28.2); 136 (14.8); 135 (100); 134 (16.8); 133 (17); 122 (10.9); 121 (26.9); 120 (14.7); 119 (27.3); 109 (13.2); 107 (31.6); 105 (21.6); 95 (18.1); 93 (29.0); 91 (17.7); 81 (20.2); 79 (17.6); 71 (22.0). Mixture 7-8, IR (cm-1): 3,483, 3,069, 2,960, 1,736, 1,721, 1,644, 1,470, 1,368, 1,255, 1,209.
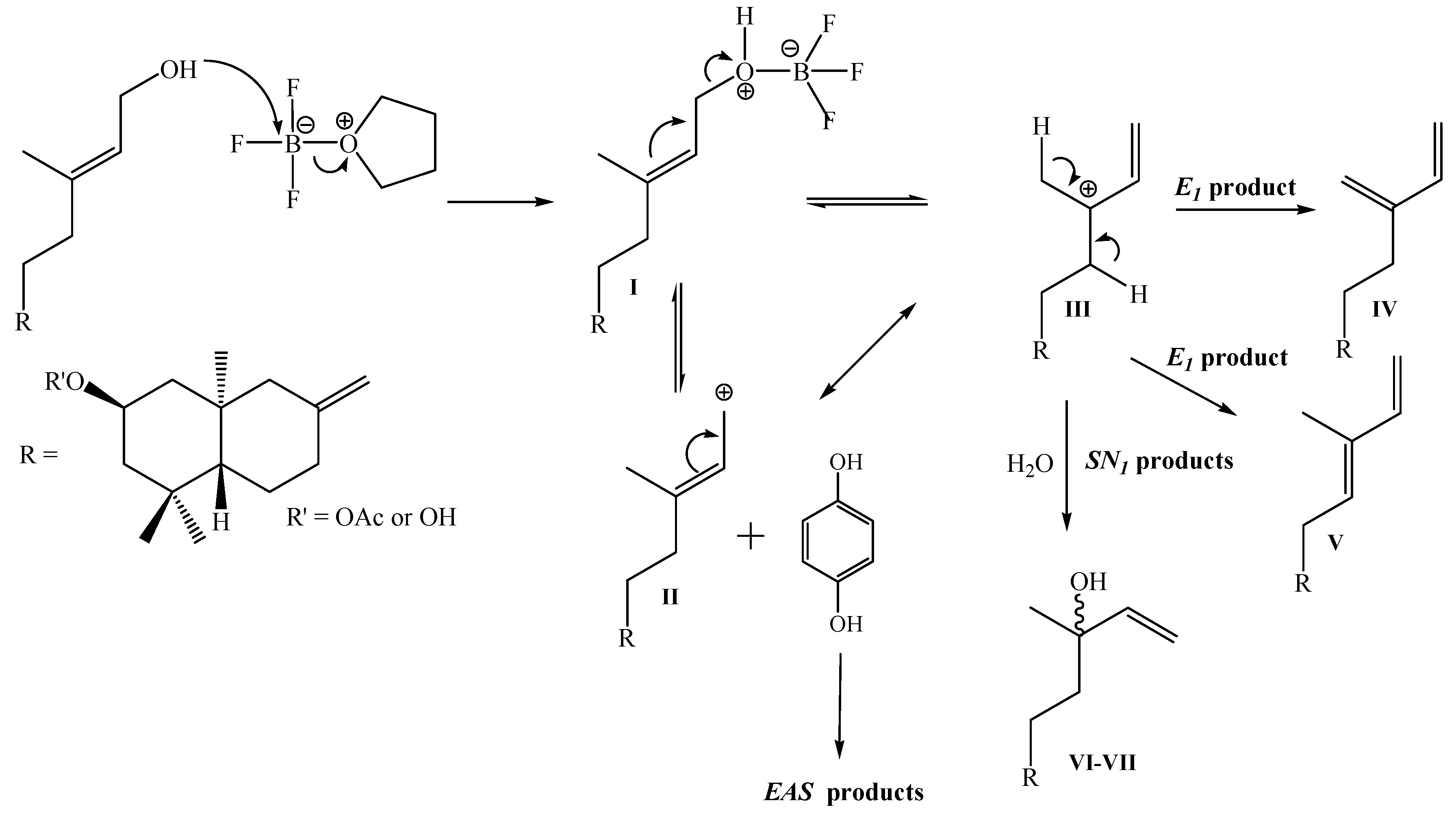
Synthesis of 2β-acetoxy-15-phenyl-(22,25-dihydroxy)-ent-labda-8(17),13(E)-diene (1) from 7-8 mixture: To a solution of 1,4-hydroquinone (0.30 g, 2.27 mmol) and BF3.THF (0.5 mL, 3.98 mmol) in freshly distilled 1,4-dioxane (5 mL) was slowly added dropwise, with stirring at room temperature and under a N2 atmosphere, a solution of 7-8 mixture (0.91 g, 2.61 mmol) in 1,4-dioxane (5 mL). After the addition was complete, stirring at room temperature and under N2 atmosphere was continued overnight. When the completion of the reaction was verified by TLC, the mixture was poured onto crushed ice (app. 30 g) and the organic layer extracted with diethyl ether (3 x 30 mL), the ethereal layer was washed with 5% NaHCO3 (30 mL), then with water (2 x 20 mL) and dried over Na2SO4, filtered and evaporated. The crude was redissolved in CH2Cl2 (5 mL) and chromatographed on silica-gel with petroleum ether/EtOAc mixtures of increasing polarity (19.8:0.2→11.4:8.6). Three fractions were obtained: Fraction I: 0.678 g of non-polar non-identified complex mixture (colorless viscous oil) Fraction II: colorless viscous oil, 0.127 g (11%) of compound 1, and Fraction III: 8.0 mg of unreacted 1,4-hydroquinone.
Synthesis of 1 from 5: 1,4-hydroquinone (0.50 g, 4.54 mmol), BF3.THF (0.5 mL, 3.98 mmol) and 5 (1.39 g, 3.96 mmol) were used. After workup and CC purification: 1.33 g of complex mixture, 0.175 g (10%) of 1 and 0.37 g of unreacted 1,4-hydroquinone were obtained.
Synthesis 1 from 6: 1,4-hydroquinone (0.50 g, 4.54 mmol), BF3.THF (0.7 mL, 5.59 mmol) and 6 (1.54 g, 4.42 mmol) were used. After workup and CC purification: 0.983 g of complex mixture, 0.39 g (20%) of 1 and 0.28 g of unreacted 1,4-hydroqinone, were obtained.
Synthesis 1 from 5-6 mixture: 1,4-hydroquinone (1.30 g, 11.8 mmol), BF3.THF (1.5 mL, 11.9 mmol) and 5-6 mixture (3.92 g, 11.25 mmol) were used. After workup and CC purification: 2.47 g of complex mixture, 1.29 g (26%) of 1 and 0.963 g of unreacted 1,4-hydroqinone, were obtained.
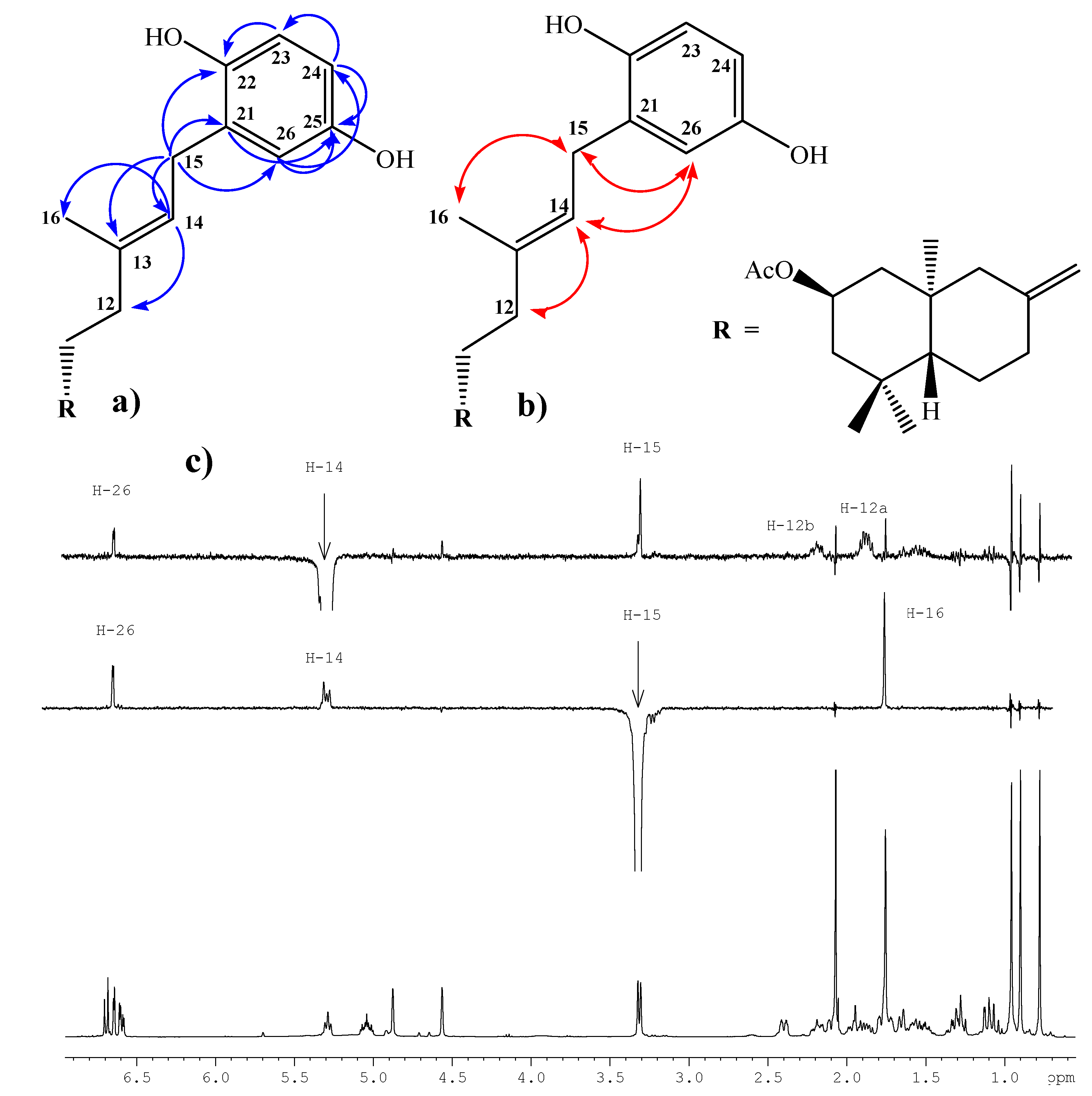
Compound 1, colorless viscous oil, [α]D23 = -2.6º (c 1.85, CHCl3); 1H-NMR: 6.67 (d, J= 8.6 Hz, 1H, H-23); 6.62 (d, J= 3.0 Hz, 1H, H-26); 6.57 (dd, J= 8.6 and 3.0 Hz, 1H, H-24); 5.26 (t, J= 7.1 Hz, 1H, H-14); 5.03 (ddt, J= 12.0, 12.0 and 4.2 Hz, 1H, H-2); 4.85 (s, 1H, H-17b); 4.54 (s, 1H, H-17a); 3.29 (d, J= 7.1 Hz, 2H, H-15); 2.38 (ddd, J= 13.0, 4.4 and 2.5 Hz, 1H, H-7α); 2.17 (ddd, J= 13.9, 8.8 and 4.4 Hz, 1H, H-12b); 2.06 (m, 1H, H-1α); 2.05 (s, 3H, CH3CO2); 1.92 (ddd, J= 13.0, 12.5 and 4.4 Hz, 1H, H-7β); 1.84 (m, 1H, H-12a); 1.75 (m, 1H, H-3α); 1.73 (s, 3H, H-16); 1.71 (m, 1H, H-6β); 1.63 (bd, J= 10.3 Hz, 1H, H-9); 1.51 (m, 2H, H-11a and H-11b); 1.29 (dd, J= 13.2 and 3.9 Hz, 1H, H-6α); 1.26 (dd, J= 12.0 and 12.0 Hz, 1H, H-3β); 1.09 (dd, J=13.2 and 2.5 Hz, 1H, H-5); 1.05 (dd, J= 12.0 and 12.0 Hz, 1H, H-1β); 0.93 (s, 3H, H-18); 0.88 (s, 3H, H-19); 0.75 (s, 3H, H-20). 13C-NMR: 44.0 (C-1), 69.8 (C-2), 46.7 (C-3), 34.9 (C-4), 54.8 (C-5), 23.8 (C-6), 37.9 (C-7), 147.4 (C-8), 55.8 (C-9), 40.9 (C-10), 22.1 (C-11), 38.3 (C-12), 138.5 (C-13), 121.3 (C-14), 29.3 (C-15), 16.3 (C-16), 107.3 (C-17), 33.5 (C-18), 22.4 (C-19), 15.2 (C-20), 128.3 (C-21), 147.8 (C-22), 116.3 (C-23), 113.6 (C-24), 149.5 (C-25), 116.5 (C-26), 171.1 (CH3CO), 21.6 (CH3CO). IR (cm-1): 3,401, 2,940, 1,701, 1,609, 1,501, 1,450, 1,367, 1,265, 1,199, 1,020, 958, 892, 753. M.S. (m/z, %): M+ 440 < 1%, 288 (9.0), 274 (11.8), 273 (55.6), 255 (31.8), 202 (14.2), 188 (11.4), 187 (34.1), 175 (21.3), 161 (14.0), 159 (13.5), 135 (100), 119 (36.9), 107 (45.9), 93 (42.1), 91 (26.7), 81 (29.6), 79 (24.6), 77 (11.4), 69 (21.4), 67 (19.1), 55 (21.1).
Synthesis of 2β-15-dihydroxy-ent-labda-8(17), 13-(Z)-diene (9) and 2β-15-dihydroxy-ent-labda-8(17), 13-(E)-diene (10) from 5-6mixture: To a solution of 5-6 mixture (2.93g, 8.41 mmol) in MeOH (60 mL), finely divided K2CO3 (1.20g, 8.68 mmol) was added and the mixture stirred at room temperature for 0.5 h. After the TLC analysis indicated the completion of the reaction, the solvent was removed until a volume of approximately 5 mL remained and water (30 mL) was added, then 5% HCl (15 mL) was added, the mixture was extracted with EtOAc (3 x 30 mL) and the combined organic layers were washed successively with 10% NaHCO3 and water, dried over Na2SO4, filtered and evaporated. The crude (2.45 g) was redissolved in CH2Cl2 (10 mL) and chromatographed eluting with mixtures of petroleum ether/EtOAc of increasing polarity (19.8:0.2→12.2:7.8) to give three fractions.
Fraction I: compound 9 (0.292 g, 11.3%) colorless viscous oil, [α]D23 = -43.5º (c 0.96, CHCl3); 1H- NMR: 5.44 (bt, J= 7.1 Hz, 1H, H-14); 4.92 (s, 1H, H-17b); 4.61 (s, 1H, H-17a); 4.08 (dd, J= 7.1 and 2.5 Hz, 2H, H-15); 3.90 (ddt, J= 11.5, 11.5 and 4.2 Hz, 1H, H-2); 2.43 (ddd, J= 13.0, 4.2 and 2.5 Hz, 1H, H-7α); 2.09 (m, 3H, H-12a, H-12b and H-1α); 1.98 (dd, J= 13.0 and 5.1 Hz, 1H, H-7β); 1.77 (m, 1H, H-3α); 1.76 (s, 3H, H-16); 1.73 (m, 1H, H-6β); 1.63 (bd, J= 9.8 Hz, 1H, H-9); 1.61 (m, 1H, H-11b), 1.48 (m, 1H, H-11a); 1.31 (dd, J= 13.0 and 4.2 Hz, 1H, H-6α); 1.17 (dd, J= 11.5 and 11.5 Hz, 1H, H-3β); 1.08 (dd, J=12.5 and 2.7 Hz, 1H, H-5); 0.97 (dd, J= 11.5 and 11.5 Hz, 1H, H-1β); 0.91 (s, 3H, H-18); 0.86 (s, 3H, H-19); 0.73 (s, 3H, H-20). 13C-NMR: 48.0 (C-1), 65.6 (C-2), 51.0 (C-3), 35.0 (C-4), 54.8 (C-5), 23.9 (C-6), 38.0 (C-7), 147.7 (C-8), 55.7 (C-9), 40.9 (C-10), 21.9 (C-11), 30.4 (C-12), 140.3 (C-13), 124.6 (C-14), 59.0 (C-15), 23.4 (C-16), 107.3 (C-17), 33.6 (C-18), 22.6 (C-19), 15.4 (C-20). IR (cm-1): 3,380, 2,940, 1,641, 1,470, 1,445, 1,388, 1,035, 888, 754. M.S. (m/z, %): M+ 306 < 1%, 291 (12.8), 288 (12.8), 273 (57.2), 270 (13.4), 257 (12.9), 255 (50.2), 245 (13.3), 207 (19.9), 205 (18.5), 203 (14.1), 202 (15.1), 199 (12.9), 190 (13.4), 189 (20.5), 188 (13.4), 187 (40.9), 175 (36.1), 173 (17.8), 163 (14.3), 150 (21.5), 149 (18.5), 148 (13.5), 147 (43.6), 145 (25.0), 137 (18.7), 136 (19.7), 135 (100.0), 134 (27.1), 133 (44.0), 131 (21.8), 123 (28.5), 122 (24.1), 121 (68.7), 120 (32.0), 119 (57.5), 117 (16.6), 111 (13.9), 109 (47.8), 108 (22.2), 107 (84.9), 106 (19.4), 105 (59.3), 97 (23.8), 96 (14.5), 95 (67.5), 94 (22.1), 93 (96.2), 92 (14.3), 91 (59.0), 85 (20.4), 84 (23.2), 83 (27.7), 81 (71.2), 80 (16.8), 79 (61.5), 77 (32.6), 71 (25.0), 69 (61.9), 68 (16.9), 67 (47.5), 57 (41.6), 55 (59.0), 53 (24.1).
Fraction II: (0.527 g, 20.4%) viscous oil, mixture of 9-10.
Fraction III: compound 10 (1.43 g, 55.4%) white needles, mp = 104.4-106.2°C (Et2O/MeOH), [α]D23 = -20.9º (c 1.05, CHCl3); 1H-NMR: 5.38 (bt, J= 6.9 Hz, 1H, H-14); 4.86 (s, 1H, H-17b); 4.54 (s, 1H, H-17a); 4.15 (d, J= 6.9 Hz, 2H, H-15); 3.88 (ddt, J= 12.0, 12.0 and 4.4 Hz, 1H, H-2); 2.40 (ddd, J= 12.6, 4.4 and 2.5 Hz, 1H, H-7α); 2.16 (ddd, J= 12.0, 9.5 and 4.2 Hz, 1H, H-12b); 2.10 (ddd, J= 11.6, 4.4 and 2.5 Hz, 1H, H-1α); 1.97 (ddd, J= 13.9, 12.6 and 5.1 Hz, 1H, H-7β); 1.84 (dd, J= 9.5 and 6.7 Hz, 1H, H-12a); 1.75 (m, 2H, H-3α and H-6β); 1.67 (s, 3H, H-16); 1.64 (m, 1H, H-11b); 1.63 (bd, J= 10.3 Hz, 1H, H-9); 1.49 (m, 1H, H-11a); 1.29 (dd, J= 12.6 and 4.2 Hz, 1H, H-6α); 1.15 (dd, J= 12.0 and 12.0 Hz, 1H, H-3β); 1.07 (dd, J=12.6 and 2.5 Hz, 1H, H-5); 0.96 (dd, J= 12.0 and 12.0 Hz, 1H, H-1β); 0.93 (s, 3H, H-18); 0.84 (s, 3H, H-19); 0.72 (s, 3H, H-20). 13C-NMR: 48.2 (C-1), 65.7 (C-2), 51.1 (C-3), 35.0 (C-4), 54.9 (C-5), 23.9 (C-6), 38.0 (C-7), 147.7 (C-8), 56.2 (C-9), 41.0 (C-10), 22.0 (C-11), 38.3 (C-12), 140.3 (C-13), 123.2 (C-14), 59.4 (C-15), 16.4 (C-16), 107.2 (C-17), 33.7 (C-18), 22.6 (C-19), 15.4 (C-20). IR (cm-1): 3,339, 2,935, 1646, 1,464, 1,440, 1,388, 1,363, 1,030, 888. M.S. (m/z, %): M+ 306 1.3%, 291 (22.7), 274 (14.1), 273 (68.2), 270 (11.8), 255 (48.3), 245 (15.1), 203 (13.9), 202 (18.4), 199 (11.9), 190 (14.4), 189 (13.2), 187 (41.2), 175 (33.8), 173 (13.2), 163 (12.4), 161 (27.0), 159 (19.3), 149 (13.3), 148 (13.2), 137 (14.5), 136 (18.3), 135 (100.0), 134 (20.6), 133 (38.5), 131 (19.4), 123 (24.4), 122 (19.3), 121 (62.7), 120 (31.9), 119 (44.1), 109 (42.9), 108 (17.9), 107 (73.9), 106 (13.9), 105 (45.0), 97 (15.8), 95 (50.2), 94 (16.1), 93 (77.6), 92 (11.8), 91 (44.7), 85 (13.4), 83 (20.5), 81 (58.3), 79 (48.9), 77 (23.9), 71 (18.0), 69 (51.2), 68 (14.9), 67 (37.9), 57 (32.7), 55 (42.4), 53 (18.2).
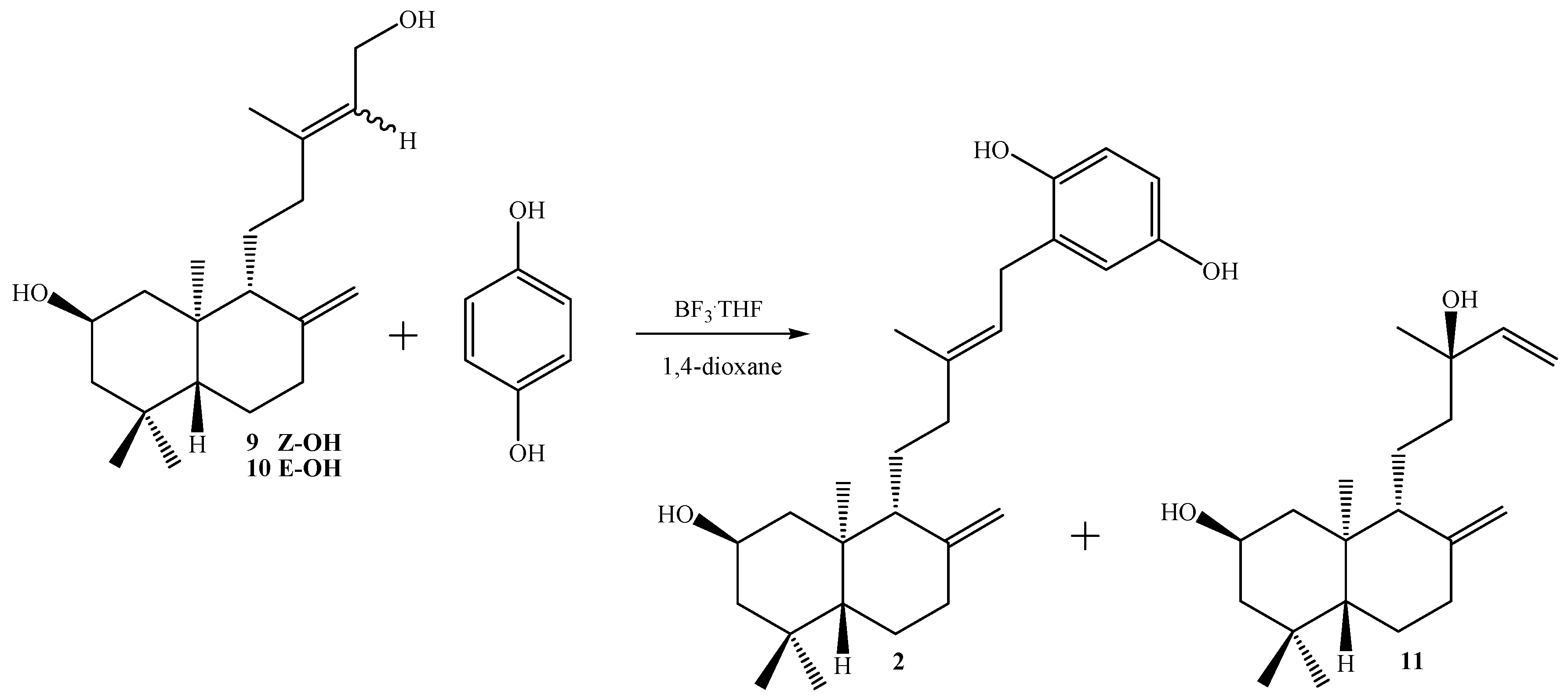
Synthesis of 2β-hydroxy-15-phenyl-(22,25-dihydroxy)-ent-labda-8(17), 13(E)-diene (2) from 9: To a solution of 1,4-hydroquinone (0.086 g, 0.781 mmol) and BF3.THF (0.2 mL, 1.59 mmol) in freshly distilled 1,4-dioxane (5 mL) was slowly added dropwise with stirring at room temperature and under a N2 atmosphere, a solution of 9 (0.238 g, 0.777 mmol) in 1,4-dioxane (5 mL). After the addition was complete, stirring at room temperature and under N2 atmosphere was continued overnight. When the end of the reaction was verified by TLC, the mixture was poured onto crushed ice (app. 30 g) and the organic layer extracted with diethyl ether (2 x 30 mL), the ethereal layer was washed with 5% NaHCO3 (30 mL), then with water (2 x 20 mL) and dried over Na2SO4, filtered and evaporated. The crude was redissolved in CH2Cl2 (5 mL) and chromatographed on silica-gel with petroleum ether/EtOAc mixtures of increasing polarity (19.8:0.2→ 10.2:9.8). Two main fractions were obtained: Fraction I: 0.183 g of non-polar unidentified complex mixture (colorless viscous oil) and Fraction II:colorless viscous oil, 40 mg (13%) of compound 2.
Synthesis of 2 from 10: 1,4-hydroquinone (0.35 g, 3.18 mmol), BF3.THF (0.4 mL, 3.18 mmol) and 10 (0.897 g, 2.93 mmol) were used. After workup and CC purification: 0.420 g of complex mixture, 0.246 g (21%) of 2 and 0.172 g of unreacted 1,4-hydroqinone were obtained.
Synthesis of 2 from 9-10: 1,4-hydroquinone (0.093 g, 0.85 mmol), BF3.THF (0.2 mL, 1.59 mmol) and 9-10 mixture (0.258 g, 0.85 mmol) were used. After workup and CC purification: 0.122 g of complex mixture, 0.057 g (17%) of diol 11 and 0.094 g (28%) of 2 were obtained.
Compound 11, colorless viscous oil, [α]D23 = -48.6º (c 0.14, CHCl3); 1H-NMR: 5.90 (dd, J= 17.4 and 10.8 Hz, 1H, H-14); 5.20 (dd, J= 17.4 and 1.2 Hz, 1H, H-15b); 5.06 (dd, J= 10.8 and 1.2 Hz, 1H, H-15a); 4.86 (s, 1H, H-17b); 4.56 (s, 1H, H-17a); 3.88 (ddt, J= 11.3, 11.3 and 4.4 Hz, 1H, H-2); 2.39 (ddd, J= 12.7, 4.4 and 2.5 Hz, 1H, H-7α); 2.10 (ddd, J= 11.3, 4.4 and 2.5 Hz, 1H, H-1α); 1.96 (ddd, J= 13.5, 12.7 and 5.4 Hz, 1H, H-7β); 1.75 (m, 3H, H-3α, H-12b and H-6β); 1.59 (m, 2H, H-11b and H-9); 1.38 (m, 1H, H-11a); 1.30 (m, 1H, H-12a); 1.27 (s, 3H, H-16); 1.25 (m, 1H, H-6α); 1.43 (dd, J= 11.0 and 11.0 Hz, 1H, H-3β); 1.07 (dd, J= 12.5 and 2.7 Hz, 1H, H-5); 0.97 (dd, J= 11.3 and 11.3 Hz, 1H, H-1β), 0.94 (s, 3H, H-18); 0.84 (s, 3H, H-19); 0.72 (s, 3H, H-20). 13C-NMR: 48.2 (C-1), 65.7 (C-2), 51.1 (C-3), 35.0 (C-4), 54.9 (C-5), 23.9 (C-6), 38.1 (C-7), 147.7 (C-8), 57.1 (C-9), 41.3 (C-10), 18.0 (C-11), 41.3 (C-12), 75.3 (C-13), 145.1 (C-14), 111.7 (C-15), 27.8 (C-16), 107.4 (C-17), 33.7 (C-18), 22.6 (C-19), 15.3 (C-20). IR (cm-1): 3,375, 2,935, 1,445, 1,368, 1,035, 887. M.S. (m/z, %): M+ 306 < 1%, 273 (26.5), 260 (18.1), 255 (50.8), 202 (20.7), 201 (17.9), 189 (18.2), 188 (21.0), 187 (46.8), 175 (30.6), 173 (17.4), 161 (21.1), 159 (18.8), 145 (18.8), 137 (18.0), 136 (18.6), 135 (100.0), 134 (30.9), 133 (30.8), 123 (17.4), 122 (16.8), 121 (49.6), 120 (29.7), 119 (36.9), 109 (32.1), 108 (14.4), 107 (66.1), 105 (36.7), 95 (45.0), 94 (24.5), 93 (73.8), 91 (37.4), 83 (16.5), 81 (47.8), 80 (22.3), 79 (43.1), 77 (18.5), 71 (49.7), 69 (40.9), 67 (33.2), 57 (23.8), 55 (41.0).
Compound 2, colorless viscous oil, [α]D23 = -18.1º (c 0.38, CHCl3); 1H-NMR: 6.67 (d, J= 8.6 Hz, 1H, H-23); 6.64 (d, J= 2.9 Hz, 1H, H-26); 6.57 (dd, J= 8.6 and 2.9 Hz, 1H, H-24); 5.28 (t, J= 7.1 Hz, 1H, H-14); 4.84 (s, 1H, H-17b); 4.54 (s, 1H, H-17a); 3.91 (ddt, J= 12.0, 12.0 and 4.2 Hz, 1H, H-2); 3.29 (d, J= 7.1 Hz, 2H, H-15); 2.37 (ddd, J= 12.0, 4.7 and 2.2 Hz, 1H, H-7α); 2.17 (m, 1H, H-12b); 2.10 (m, 1H, H-1α); 1.89 (ddd, J= 15.2, 12.7 and 4.4 Hz, 1H, H-7β); 1.83 (m, 1H, H-12a); 1.75 (m, 1H, H-3α); 1.72 (s, 3H, H-16); 1.68 (m, 1H, H-6β); 1.60 (bd, J= 11.3 Hz, 1H, H-9); 1.55 (m, 2H, H-11a and H-11b); 1.26 (dd, J= 11.5 and 3.9 Hz, 1H, H-6α); 1.17 (dd, J= 12.0 and 12.0 Hz, 1H, H-3β); 0.97 (dd, J= 12.7 and 2.5 Hz, 1H, H-5); 0.94 (dd, J= 12.0 and 12.0 Hz, 1H, H-1β); 0.91 (s, 3H, H-18); 0.82 (s, 3H, H-19); 0.69 (s, 3H, H-20). 13C-NMR: 47.9 (C-1), 66.1 (C-2), 50.7 (C-3), 35.0 (C-4), 54.6 (C-5), 23.8 (C-6), 38.0 (C-7), 147.6 (C-8), 55.3 (C-9), 40.8 (C-10), 22.0 (C-11), 37.7 (C-12), 138.1 (C-13), 121.7 (C-14), 29.0 (C-15), 16.8 (C-16), 107.2 (C-17), 33.6 (C-18), 22.6 (C-19), 15.3 (C-20), 128.7 (C-21), 147.7 (C-22), 116.3 (C-23), 113.7 (C-24), 149.5 (C-25), 116.6 (C-26). IR (cm-1): 3,375, 2,935, 1,455, 1,199, 1,025. M.S. (m/z, %): M+ 398 < 1%, 280 (2.6), 279 (14.5), 168 (3.3), 167 (36.4), 149 (100.0), 113 (8.4), 112 (5.3), 104 (5.5), 84 (2.8), 83 (5.1), 76 (2.7), 71 (13.8), 70 (12.2), 69 (33.0), 57 (17.8), 55 (7.0).

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





