Radiolabeled Small Molecule Protein Kinase Inhibitors for Imaging with PET or SPECT

Abstract
:1. Introduction

2. Imaging Protein Kinase Expression with Small Molecule Radiolabeled Inhibitors for Cancer


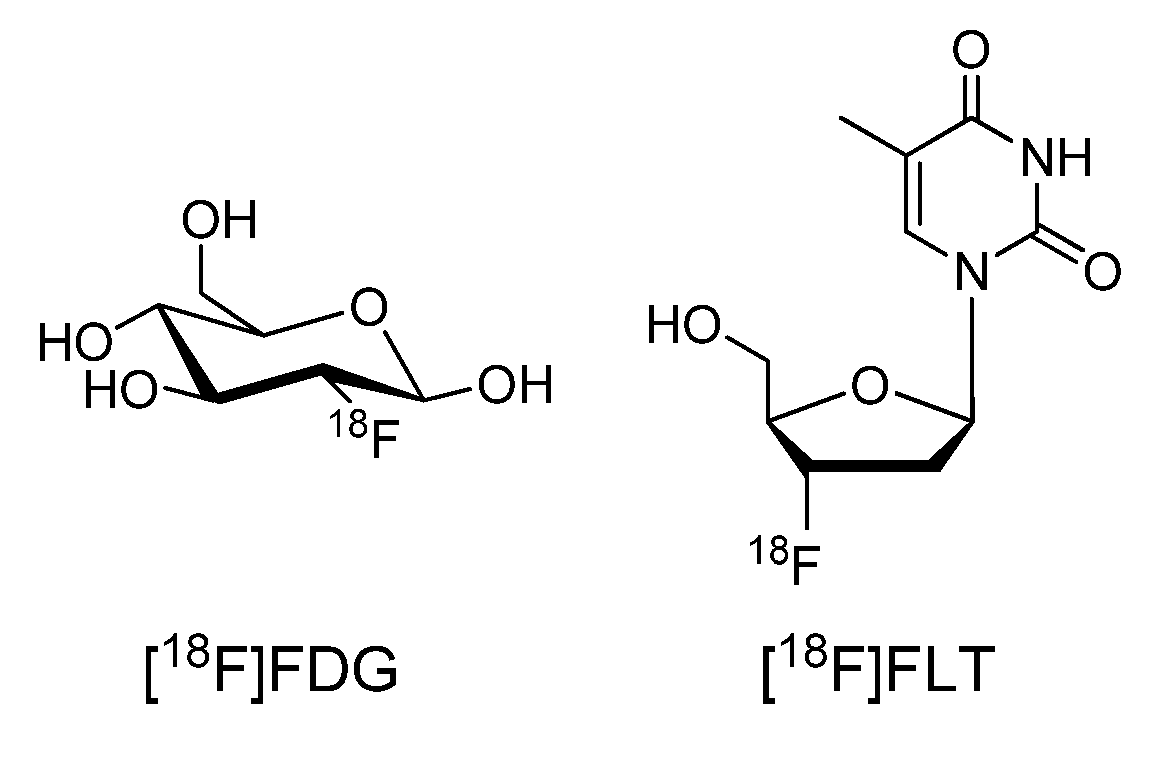{kind=link}
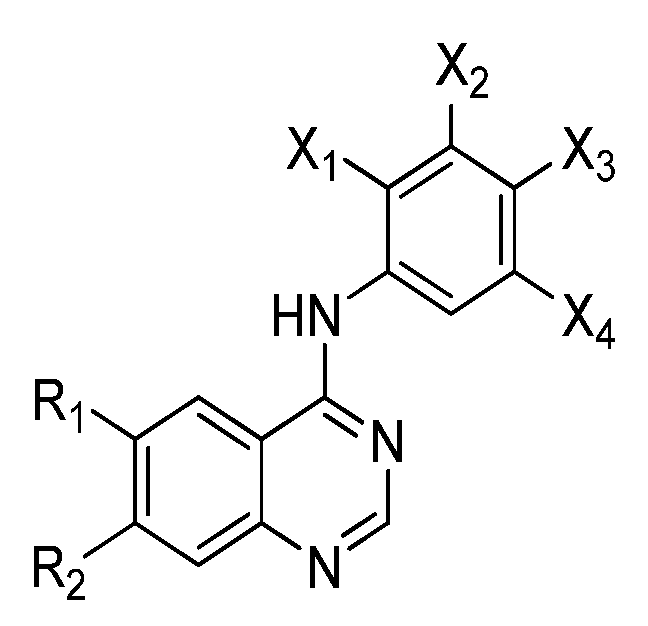{kind=link}
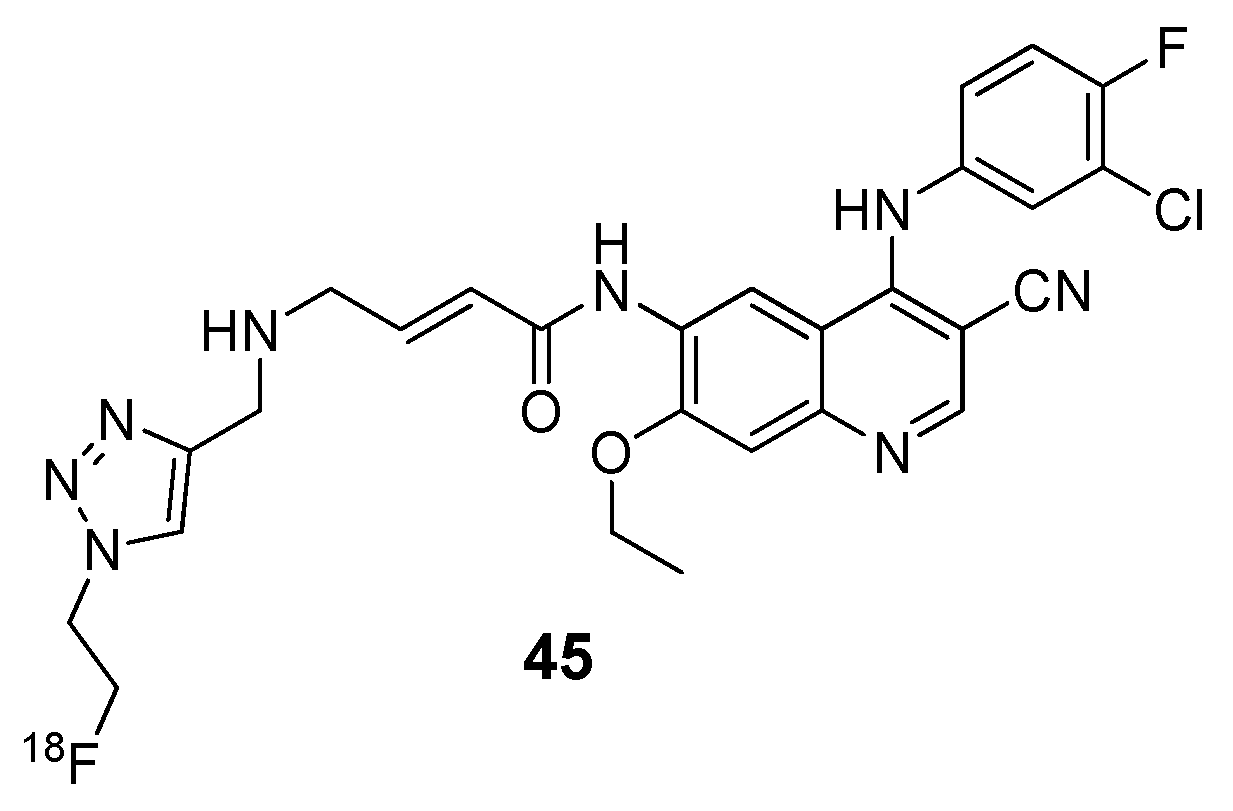{kind=link}
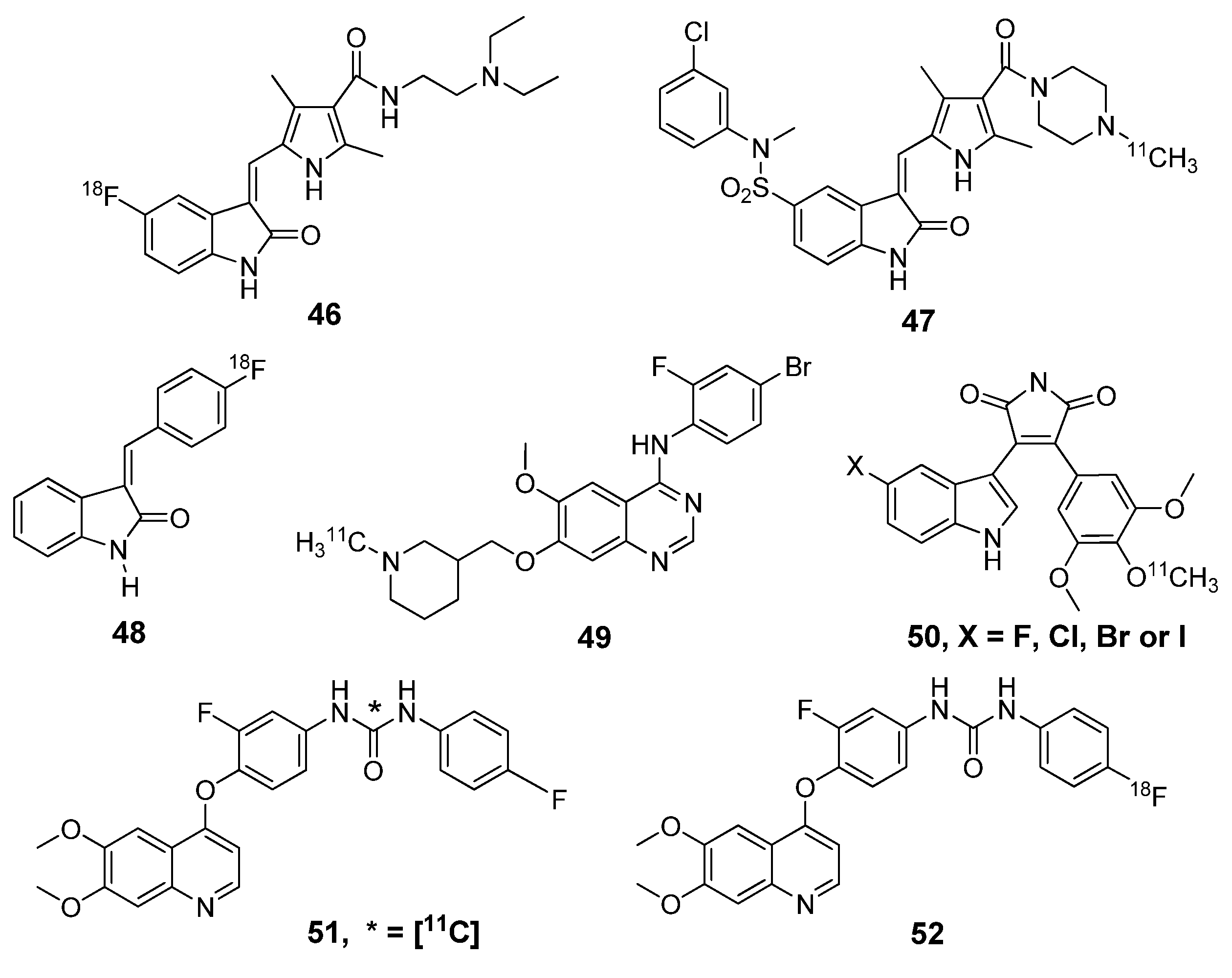{kind=link}
{kind=link}
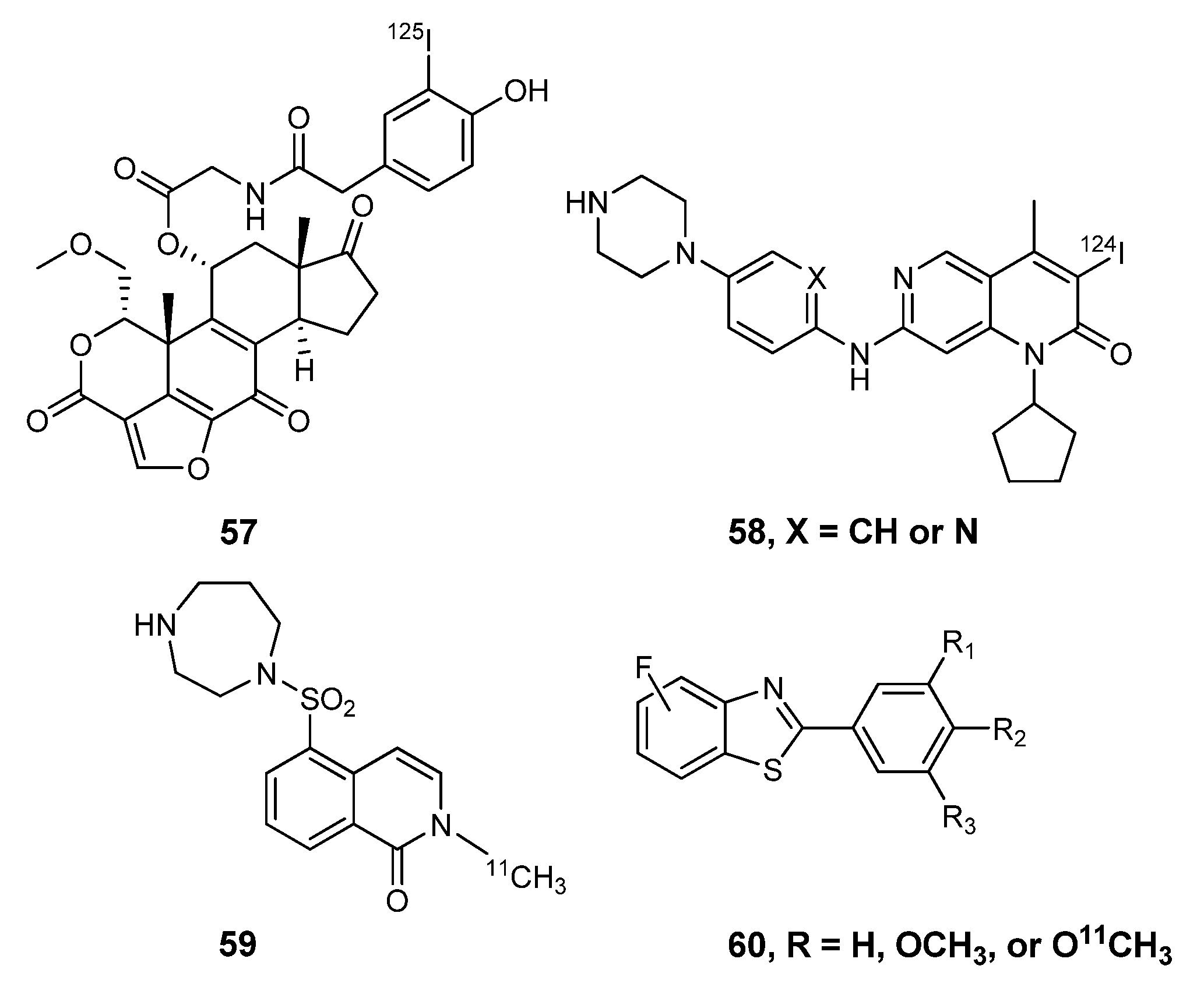{kind=link}
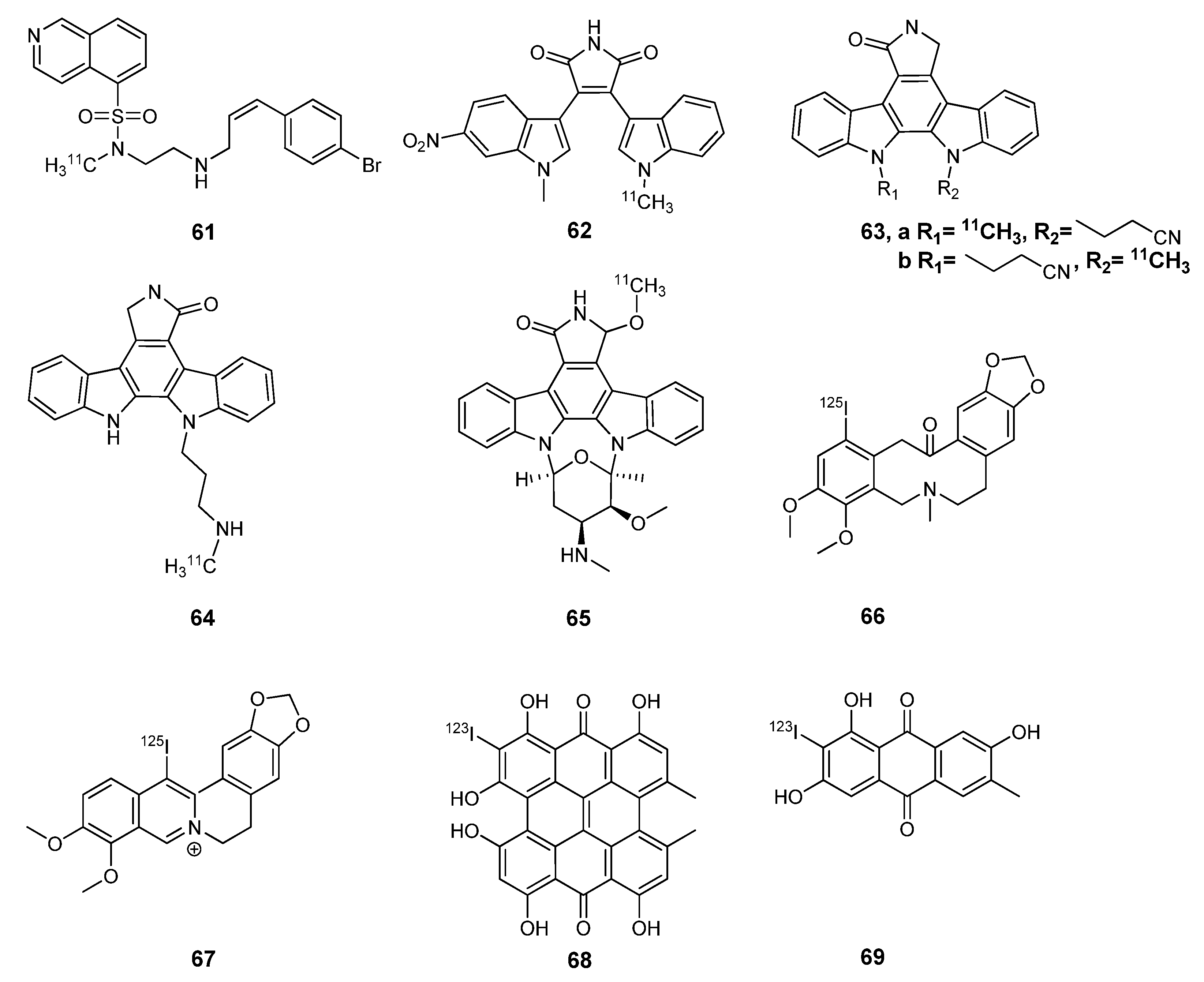{kind=link}
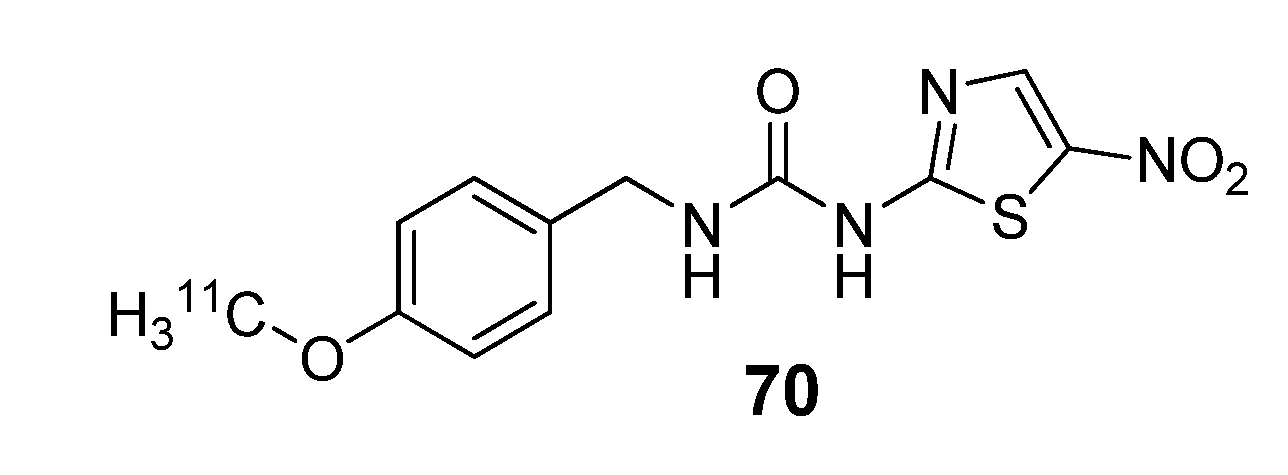{kind=link}
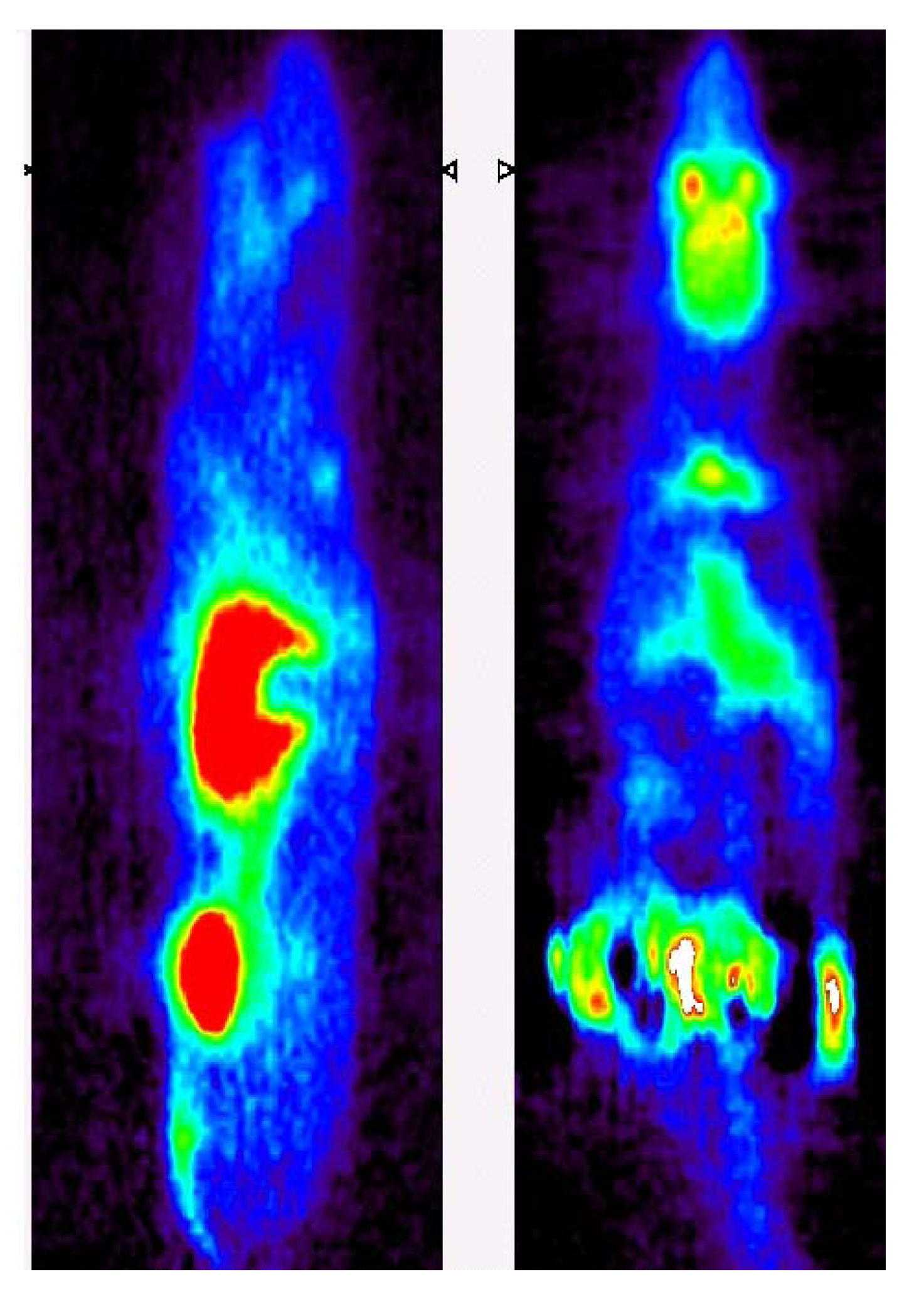{kind=link}
| # | R1 | R2 | X1 | X2 | X3 | X4 | Ref |
| 1 |  |  | H | H | F | Cl | [22,23] |
| 2 | |  | H | H | 18F | Cl | [24,25] |
| 3 |  |  | H | H | H |  | [26] |
| 4 | | | H | H | H | Br | [27,28,29] |
| 5 | | | H | H | H | Br | [30] |
| 6 | | | H | H | H | Cl | [33] |
| 7 |  | | H | H | F | Cl | [34] |
| 8 | |  | H | H | H | Br | [27] |
| 9 |  | | H | H | 18F | Cl | [25] |
| 10 |  | | H | H | 18F | Cl | [25] |
| 11 | | | H | H | 18F | Cl | [35] |
| 12 | | | 18F | H | Cl | Cl | [36] |
| 13 | | | H | CF3 | H | 18F | [36] |
| 14 | | | H | H | 18F | H | [36,37] |
| 15 | | | H | 18F | H | H | [37] |
| 16 | | | 18F | H | H | H | [37] |
| 17 |  | | H | H | 18F | H | [37] |
| 18 | | | H | 18F | H | H | [37] |
| 19 | | | 18F | H | H | H | [37] |
| 20 |  | H | H | H | F | Cl | [38] |
| 21 | | | H | H | H | 125I | [39] |
| 22 | | | H | H | H | 123I | [40] |
| 23 |  | H | H | H | H | 124I | [41] |
| 24 |  | H | H | H | F | Cl | [42] |
| 25 | | H | H | H | H | Br | [42] |
| 26 |  | H | H | H | F | Cl | [42] |
| # | R1 | R2 | X1 | X2 | X3 | X4 | Ref |
|---|---|---|---|---|---|---|---|
| 27 |  | H | F | H | Cl | Cl | [43] |
| 28 | | H | H | H | H | Br | [43] |
| 29 |  | H | 18F | H | H | H | [44] |
| 30 |  | H | 18F | H | H | H | [45] |
| 31 | | H | H | H | H | (CH2)2[18F] | [46] |
| 32 |  | H | 18F | H | Cl | Cl | [47] |
| 33 | | H | H | H | H | 124I | [41] |
| 34 | | H | H | H | H | 124I | [41] |
| 35 |  | H | H | H | H | 124I | [48] |
| 36 |  | H | F | H | Cl | Cl | [49] |
| 37 | | H | H | H | H | I | [49] |
| 38 | | H | H | H | H | Br | [49] |
| 39 |  |  | F | H | Cl | Cl | [50] |
| 40 | |  | F | H | Cl | Cl | [50,51] |
| 41 | | | H | H | H | 124I | [50,51] |
| 42 | | | F | H | Cl | Cl | [51,52] |
| 43 | |  | H | H | H | I | [53] |
| 44 | |  | H | H | F | Cl | [54] |




3. Radiolabeled Small Molecule Protein Kinase Inhibitors for Imaging in the CNS
3.1. Protein kinase A, B and C

3.2. Glycogen synthase kinase-3β


4. Conclusions
Acknowledgements
References
- Liao, J.J. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J. Med. Chem. 2007, 50, 409–424. [Google Scholar] [CrossRef]
- Aaronson, S.A. Growth factors and cancer. Science 1991, 254, 1146–1153. [Google Scholar]
- Cohen, P. Protein kinases - the major drug targets of the twenty first century? Nat. Rev. Drug Discovery 2001, 1, 309–315. [Google Scholar] [CrossRef]
- Tolmachev, V.; Stone-Elander, S.; Orlova, A. Radiolabelled receptor-tyrosine-kinase targeting drugs for patient stratification and monitoring of therapy response: prospects and pitfalls. Lancet Oncol. 2010, 11, 992–1000. [Google Scholar] [CrossRef]
- Mishani, E.; Abourbeh, G. Cancer molecular imaging: radionuclide-based biomarkers of the epidermal growth factor receptor (EGFR). Curr. Top. Med. Chem. 2007, 7, 1755–1772. [Google Scholar]
- McLarty, K.; Reilly, R.M. Molecular imaging as a tool for personalized and targeted anticancer therapy. Clin. Pharm. Ther. 2007, 81, 420–424. [Google Scholar]
- Cai, W.; Niu, G.; Chen, X. Multimodality imaging of the HER-kinase axis in cancer. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 186–208. [Google Scholar]
- Mishani, E.; Abourbeh, G.; Eiblmaier, M.; Anderson, C.J. Imaging of EGFR and EGFR tyrosine kinase overexpression in tumors by nuclear medicine modalities. Curr. Pharm. Des. 2008, 14, 2983–2998. [Google Scholar]
- Gelovani, J.G. Molecular imaging of epidermal growth factor receptor expression-activity at the kinase level in tumors with positron emission tomography. Cancer Metast. Rev. 2008, 27, 645–653. [Google Scholar]
- Mankoff, D.A. Molecular imaging to select cancer therapy and evaluate treatment response. Quart. J. Nucl. Med. 2009, 53, 181–192. [Google Scholar]
- Pantaleo, M.A.; Nannini, M.; Maleddu, A.; Fanti, S.; Nanni, C.; Boschi, S.; Lodi, F.; Nicoletti, G.; Landuzzi, L.; Lollini, P.L.; Biasco, G. Experimental results and related clinical implications of PET detection of epidermal growth factor receptor (EGFr) in cancer. Ann. Oncol. 2009, 20, 213–226. [Google Scholar]
- Mishani, E.; Hagooly, A. Strategies for molecular imaging of epidermal growth factor receptor tyrosine kinase in cancer. J. Nucl. Med. 2009, 50, 1199–1202. [Google Scholar]
- Reilly, R.M. Monoclonal Antibodies and Peptide-Targeted Radiotherapy of Cancer, 1st ed; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Wadas, T.J.; Wong, E.H.; Weisman, G.R.; Anderson, C.J. Coordinating radiometals of copper, gallium, indium, yttrium, and zirconium for PET and SPECT imaging of disease. Chem. Rev. 2010, 110, 2858–2902. [Google Scholar] [CrossRef]
- Fowler, J.S.; Ido, T. Design and synthesis of 2-deoxy-2-[18F]fluoro-D-glucose (18FDG). In Handbook of Radiopharmaceuticals; Welch, M.J., Redvanly, C.S., Eds.; John Wiley and Sons, Ltd.: West Sussex, England, 2003; pp. 307–322. [Google Scholar]
- Kelloff, G.J.; Hoffman, J.M.; Johnson, B.; Scher, H.I.; Siegel, B.A.; Cheng, E.Y.; Cheson, B.D.; O'Shaughnessy, J.; Guyton, K.Z.; Mankoff, D.A.; Shankar, L.; Larson, S.M.; Sigman, C.C.; Schilsky, R.L.; Sullivan, D.C. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development. Clin. Cancer Res. 2005, 11, 2785–2808. [Google Scholar]
- Van den Abbeele, A.D. The lessons of GIST - PET and PET/CT: A new paradigm for imaging. Oncologist 2008, 13, 8–13. [Google Scholar]
- George, S.; Merriam, P.; Maki, R.G.; Van den Abbeele, A.D.; Yap, J.T.; Akhurst, T.; Harmon, D.C.; Bhuchar, G.; O'Mara, M.M.; D'Adamo, D.R.; Morgan, J.; Schwartz, G.K.; Wagner, A.J.; Butrynski, J.E.; Demetri, G.D.; Keohan, M.L. Multicenter phase II trial of sunitinib in the treatment of nongastrointestinal stromal tumor sarcomas. J. Clin. Oncol. 2009, 27, 3154–3160. [Google Scholar]
- Shields, A.F. PET imaging with 18F-FLT and thymidine analogs: promise and pitfalls. J. Nucl. Med. 2003, 44, 1432–1434. [Google Scholar]
- Sohn, H.-J.; Yang, Y.-J.; Ryu, J.-S.; Oh, S.J.; Im, K.C.; Moon, D.H.; Lee, D.H.; Suh, C.; Lee, J.-S.; Kim, S.-W. [18F]Fluorothymidine Positron Emission Tomography before and 7 Days after Gefitinib treatment predicts response in patients with advanced adenocarcinoma of the lung. Clin. Cancer Res. 2008, 14, 7423–7429. [Google Scholar]
- Willman, J.K.; van Bruggen, N.; Dinkelborg, L.M.; Gambhir, S.S. Molecular imaging in drug development. Nat. Rev. Drug Discov. 2008, 7, 591–607. [Google Scholar]
- Holt, D.P.; Ravert, H.T.; Dannals, R.F.; Pomper, M.G. Synthesis of [C-11]gefitinib for imaging epidermal growth factor receptor tyrosine kinase with positron emission tomography. J. Labelled Compd. Radiopharm. 2006, 49, 883–888. [Google Scholar]
- Wang, J.Q.; Gao, M.Z.; Miller, K.D.; Sledge, G.W.; Zheng, Q.H. Synthesis of [C-11]Iressa as a new potential PET cancer imaging agent for epidermal growth factor receptor tyrosine kinase. Bioorg. Med. Chem. 2006, 16, 4102–4106. [Google Scholar]
- DeJesus, O.T.; Murali, D.; Flores, L.G.; Converse, A.K.; Dick, D.W.; Oakes, T.R.; Roberts, A.D.; Nickles, R.J. Synthesis of [18F]-ZD1839 as a PET imaging agent for epidermal growth factor receptors. J. Labelled Compd. Radiopharm. 2003, 46, S1. [Google Scholar]
- Seimbille, Y.; Phelps, M.E.; Czernin, J.; Silverman, D.H.S. Fluoride-18 labeling of 6,7-disubstituted anilinoquinazoline derivatives for positron emission tomography (PET) imaging of tyrosine kinase receptors: synthesis of F-18-Iressa and related molecular probes. J. Labelled Compd. Radiopharm. 2005, 48, 829–843. [Google Scholar]
- Memon, A.A.; Jakobsen, S.; Dagnaes-Hansen, F.; Sorensen, B.S.; Keiding, S.; Nexo, E. Positron emission tomography (PET) imaging with [11C]-labeled erlotinib: a micro-PET study on mice with lung tumor xenografts. Cancer Res. 2009, 69, 873–878. [Google Scholar]
- Mulholland, G.K.; Zheng, Q.H.; Winkle, W.L.; Carlson, K.A. Synthesis and biodistribution of new C-11 and F-18 labeled epidermal growth factor receptor ligands. J. Nucl. Med. 1997, 38, 529–529. [Google Scholar]
- Johnstrom, P.; Fredriksson, A.; Thorell, J.O.; Stone-Elander, S. Synthesis of [methoxy-11C]PD153035, a selective EGF receptor tyrosine kinase inhibitor. J. Labelled Compd. Radiopharm. 1998, 41, 623–629. [Google Scholar]
- Fredriksson, A.; Johnstrom, P.; Thorell, J.O.; von Heijne, G.; Hassan, M.; Eksborg, S.; Kogner, P.; Borgstrom, P.; Ingvar, M.; Stone-Elander, S. In vivo evaluation of the biodistribution of 11C-labeled PD153035 in rats without and with neuroblastoma implants. Life Sci. 1999, 65, 165–174. [Google Scholar] [CrossRef]
- Samén, E.; Thorell, J.-O.; Fredriksson, A.; Stone-Elander, S. The tyrosine kinase inhibitor PD153035: implication of labeling position on radiometabolites formed in vitro. Nucl. Med. Biol. 2006, 33, 1005–1011. [Google Scholar]
- Liu, N.; Li, M.; Li, X.; Meng, X.; Yang, G.; Zhao, S.; Yang, Y.; Ma, L.; Fu, Z.; Yu, J. PET-based biodistribution and radiation dosimetry of epidermal growth factor receptor-selective tracer 11C-PD153035 in humans. J. Nucl. Med. 2009, 50, 303–308. [Google Scholar]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar]
- Ackermann, U.; Tochon-Danguy, H.J.; Scott, A.M. [11C]AG1478 - a potential PET tracer for the molecular imaging of epidermal growth factor receptor (EGFR). J. Nucl. Med. 2004, 45, 112. [Google Scholar]
- Owino, N.O.; Shao, X.; Snyder, S.E. Preparation of a reversible EGFR inhibitor, [11C]CIRC, as a PET radiotracer for tumor imaging. J. Labelled Compd. Radiopharm. 2005, 48, S183. [Google Scholar]
- Snyder, S.E.; Sherman, P.S.; Blair, J.B. 4-(3-chloro-4-[18F]fluorophenylamino)-6,7-dimethoxyquinazoline: A radiolabeled EGF receptor inhibitor for imaging tumor biochemistry with PET. J. Nucl. Med. 2000, 41, 233P. [Google Scholar]
- Bonasera, T.A.; Ortu, G.; Rozen, Y.; Krais, R.; Freedman, N.M.T.; Chisin, R.; Gazit, A.; Levitzki, A.; Mishani, E. Potential 18F-labeled biomarkers for epidermal growth factor receptor tyrosine kinase. Nucl. Med. Biol. 2001, 28, 359–374. [Google Scholar]
- Dorff, P.N.; Vasdev, N.; O'Neil, J.P.; Nandanan, E.; Gibbs, A.R.; VanBrocklin, H.F. Synthesis of 2'-, 3'-, and 4'-[18F]fluoroanilinoquinazoline. Labelled Compd. Radiopharm. 2003, 46, S117. [Google Scholar]
- Fernandes, C.; Oliveira, C.; Gano, L.; Bourkoula, A.; Pirmettis, I.; Santos, I. Radioiodination of new EGFR inhibitors as potential SPECT agents for molecular imaging of breast cancer. Bioorg. Med. Chem. 2007, 15, 3974–3980. [Google Scholar]
- Lim, J.K.; Negash, K.; Hanrahan, S.M.; VanBrocklin, H.F. Synthesis of 4-(3'-[125I]iodoanilino)-6,7-dialkoxyquinazolines: radiolabeled epidermal growth factor receptor tyrosine kinase inhibitors. J. Labelled Compd. Radiopharm. 2000, 43, 1183–1191. [Google Scholar]
- Mattner, F. Radioiodinated epidermal growth factor receptor inhibitors for tumor imaging with SPECT. Quart. J. Nucl. Med. 2001, 45, S6. [Google Scholar]
- Shaul, M.; Abourbeh, G.; Jacobson, O.; Rozen, Y.; Laky, D.; Levitzki, A.; Mishani, E. Novel iodine-124 labeled EGFR inhibitors as potential PET agents for molecular imaging in cancer. Bioorg. Med. Chem. 2004, 12, 3421–3429. [Google Scholar]
- Fernandes, C.; Santos, I.C.; Santos, I.; Pietzsch, H.-J.; Kunstler, J.-U.; Kraus, W.; Rey, A.; Margaritis, N.; Bourkoula, A.; Chiotellis, A.; Paravatou-Petsotas, M.; Pirmettis, I. Rhenium and technetium complexes bearing quinazoline derivatives: progress towards a 99mTc biomarker for EGFR-TK imaging. Dalton Trans. 2008, 3215–3225. [Google Scholar]
- Ben-David, I.; Rozen, Y.; Ortu, G.; Mishani, E. Radiosynthesis of ML03, a novel positron emission tomography biomarker for targeting epidermal growth factor receptor via the labeling synthon: [11C]acryloyl chloride. Appl. Radiat. Isotopes 2003, 58, 209–217. [Google Scholar]
- Vasdev, N.; Dorff, P.N.; Gibbs, A.R.; Nandanan, E.; Reid, L.M.; O'Neill, J.P.; VanBrocklin, H.F. Synthesis of 6-acrylamido-4-(2-[18F]fluoroanilino)quinazoline: a prospective irreversible EGFR binding probe. J. Labelled Compd. Radiopharm. 2005, 48, 109–115. [Google Scholar]
- Vasdev, N.; Dorff, P.N.; Gibbs, A.R.; Nandanan, E.; Reid, L.M.; O'Neil, J.P.; VanBrocklin, H.F. Synthesis of 6-acrylamido-4-(2-[18F]fluoro-anilino)quinazoline. J. Nucl. Med. 2004, 45, 168P. [Google Scholar]
- Waldherr, C.; Satyamurthy, N.; Toyokuni, T.; Wang, S.; Mellinghoff, I.; Tran, C.; Stout, D.; Halpern, B.; Silverman, D.H.; Barrio, J.R.; Phelps, M.E.; Sawyers, C.L.; Czernin, J. Evaluation of N-{4-[(3'-[18F]fluoroethylphenyl)amino]-6-quinazolinyl} acrylamide ([18F]FEQA), a labeled tyrosine kinase inhibitor, for imaging epidermal growth factor receptor density. J. Nucl. Med. 2003, 44, 372. [Google Scholar]
- Abourbeh, G.; Dissoki, S.; Jacobson, O.; Litchi, A.; Ben Daniel, R.; Laki, D.; Levitzki, A.; Mishani, E. Evaluation of radiolabeled ML04, a putative irreversible inhibitor of epidermal growth factor receptor, as a bioprobe for PET imaging of EGFR-overexpressing tumors. Nucl. Med. Biol. 2007, 34, 55–70. [Google Scholar] [CrossRef]
- Pal, A.; Glekas, A.; Doubrovin, M.; Balatoni, J.; Beresten, T.; Maxwell, D.; Soghomonyan, S.; Shavrin, A.; Ageyeva, L.; Finn, R.; Larson, S.M.; Bornmann, W.; Gelovani, J.G. Molecular imaging of EGFR kinase activity in tumors with 124I-labeled small molecular tracer and positron emission tomography. Mol. Imaging Biol. 2006, 8, 262–277. [Google Scholar]
- Mishani, E.; Abourbeh, G.; Rozen, Y. Novel carbon-11 labeled 4-dimethylamino-but-2-enoic acid [4-(phenylamino)-quinazoline-6-yl]-amides: potential PET bioprobes for molecular imaging of EGFR-positive tumors. Nucl. Med. Biol. 2004, 31, 469–476. [Google Scholar]
- Dissoki, S.; Eshet, R.; Billauer, H.; Mishani, E. Modified PEG-anilinoquinazoline derivatives as potential EGFR PET agents. J. Labelled Compd. Radiopharm. 2009, 52, 41–52. [Google Scholar]
- Pantaleo, M.; Mishani, E.; Nanni, C.; Landuzzi, L.; Boschi, S.; Nicoletti, G.; Dissoki, S.; Paterini, P.; Piccaluga, P.; Lodi, F.; Lollini, P.-L.; Fanti, S.; Biasco, G. Evaluation of modified PEG-anilinoquinazoline derivatives as potential agents for EGFR imaging in cancer by small animal PET. Mol. Imaging Biol. 2010. In Press.. [Google Scholar]
- Dissoki, S.; Aviv, Y.; Laky, D.; Abourbeh, G.; Levitzki, A.; Mishani, E. The effect of the [18F]-PEG group on tracer qualification of [4-(phenylamino)-quinazoline-6-yl]-amide moiety--an EGFR putative irreversible inhibitor. Appl. Radiat. Isotopes 2007, 65, 1140–1151. [Google Scholar]
- Pal, A.; Balatoni, J.; Mukhopadhyay, U.; Ogawa, K.; Gonzalez-Lepera, C.; Shavrin, A.; Volgin, A.; Tong, W.; Alauddin, M.; Gelovani, J. Radiosynthesis and initial in vitro evaluation of [18F]F-PEG-IPQA - a novel PET radiotracer for imaging EGFR expression-activity in lung carcinomas. Mol. Imaging Biol. 2010. In Press.. [Google Scholar]
- Kobus, D.; Giesen, Y.; Ullrich, R.; Backes, H.; Neumaier, B. A fully automated two-step synthesis of an 18F-labelled tyrosine kinase inhibitor for EGFR kinase activity imaging in tumors. Appl. Radiat. Isotopes 2009, 67, 1977–1984. [Google Scholar]
- VanBrocklin, H.F.; Vasdev, N.; Dorff, P.N.; O'Neil, J.P.; Taylor, S.E. Metabolism of [18F]fluoroanilinoquinazolines by human hepatocytes. J. Labelled Compd. Radiopharm. 2003, 46, S390. [Google Scholar]
- Pisaneschi, F.; Nguyen, Q.-D.; Shamsaei, E.; Glaser, M.; Robins, E.; Kaliszczak, M.; Smith, G.; Spivey, A.C.; Aboagye, E.O. Development of a new epidermal growth factor receptor positron emission tomography imaging agent based on the 3-cyanoquinoline core: Synthesis and biological evaluation. Bioorg. Med. Chem. 2010, 18, 6634–6645. [Google Scholar]
- Rossi, A.; Maione, P.; Sacco, P.C.; Ambrosio, R.; Falanga, M.; Gridelli, C. Vascular endothelial growth factor receptor as target for advanced non-small cell lung cancer therapy. Curr. Drug Targets 2010, 11, 865–874. [Google Scholar]
- Wang, J.Q.; Miller, K.D.; Sledge, G.W.; Zheng, Q.-H. Synthesis of [18F]SU11248, a new potential PET tracer for imaging cancer tyrosine kinase. Bioorg. Med. Chem. Lett. 2005, 15, 4380–4384. [Google Scholar]
- Kniess, T.; Bergmann, R.; Kuchar, M.; Steinbach, J.; Wuest, F. Synthesis and radiopharmacological investigation of 3-[4'-[18F]fluorobenzylidene]indolin-2-one as possible tyrosine kinase inhibitor. Bioorg. Med. Chem. 2009, 17, 7732–7742. [Google Scholar]
- Wu, C.; Tang, Z.; Fan, W.; Zhu, W.; Wang, C.; Somoza, E.; Owino, N.; Li, R.; Ma, P.C.; Wang, Y. In vivo positron emission tomography (PET) imaging of mesenchymal-epithelial transition (MET) receptor. J. Med. Chem. 2010, 53, 139–146. [Google Scholar] [CrossRef]
- Samen, E.; Thorell, J.-O.; Lu, L.; Tegnebratt, T.; Holmgren, L.; Stone-Elander, S. Synthesis and preclinical evaluation of [11C]PAQ as a PET imaging tracer for VEGFR-2. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1283–1295. [Google Scholar]
- Ilovich, O.; Billauer, H.; Dotan, S.; Mishani, E. Labeled 3-aryl-4-indolylmaleimide derivatives and their potential as angiogenic PET biomarkers. Bioorg. Med. Chem. 2010, 18, 612–620. [Google Scholar]
- Ilovich, O.; Åberg, O.; Långström, B.; Mishani, E. Rhodium-mediated [11C]carbonylation: a library of N-phenyl-N'-{4-(4-quinolyloxy)-phenyl}-[11C]-urea derivatives as potential PET angiogenic probes. J. Labelled Compd. Radiopharm. 2009, 52, 151–157. [Google Scholar] [CrossRef]
- Ilovich, O.; Jacobson, O.; Aviv, Y.; Litchi, A.; Chisin, R.; Mishani, E. Formation of fluorine-18 labeled diaryl ureas--labeled VEGFR-2/PDGFR dual inhibitors as molecular imaging agents for angiogenesis. Bioorg. Med. Chem. 2008, 16, 4242–4251. [Google Scholar]
- Ackermann, U.; Tochon-Danguy, H.J.; Nerrie, M.; Nice, E.C.; Sachinidis, J.I.; Scott, A.M. Synthesis, 11C labeling and biological properties of derivatives of the tyrphostin AG957. Nucl. Med. Biol. 2005, 32, 323–328. [Google Scholar]
- Veach, D.R.; Namavari, M.; Beresten, T.; Balatoni, J.; Minchenko, M.; Djaballah, H.; Finn, R.D.; Clarkson, B.; Gelovani, J.G.; Bornmann, W.G.; Larson, S.M. Synthesis and in vitro examination of [I-124]-, [I-125]- and [I-131]-2-(4-iodophenylamino) pyrido[2,3-d]pyrimidin-7-one radiolabeled Abl kinase inhibitors. Nucl. Med. Biol. 2005, 32, 313–321. [Google Scholar] [CrossRef]
- Doubrovin, M.; Kochetkova, T.; Santos, E.; Veach, D.R.; Smith-Jones, P.; Pillarsetty, N.; Balatoni, J.; Bornmann, W.; Gelovani, J.; Larson, S.M. 124I-Iodopyridopyrimidinone for PET of Abl kinase-expressing tumors in vivo. J. Nucl. Med. 2010, 51, 121–129. [Google Scholar] [CrossRef]
- Kil, K.-E.; Ding, Y.-S.; Lin, K.-S.; Alexoff, D.; Kim, S.W.; Shea, C.; Xu, Y.; Muench, L.; Fowler, J.S. Synthesis and positron emission tomography studies of carbon-11-labeled imatinib (Gleevec). Nucl. Med. Biol. 2007, 34, 153–163. [Google Scholar]
- Veach, D.R.; Namavari, M.; Pillarsetty, N.; Santos, E.B.; Beresten-Kochetkov, T.; Lambek, C.; Punzalan, B.J.; Antczak, C.; Smith-Jones, P.M.; Djaballah, H.; Clarkson, B.; Larson, S.M. Synthesis and biological evaluation of a fluorine-18 derivative of dasatinib. J. Med. Chem. 2007, 50, 5853–5857. [Google Scholar]
- Yuan, H.; Luo, J.; Field, S.; Weissleder, R.; Cantley, L.; Josephson, L. Synthesis and activity of C11-modified wortmannin probes for PI3 kinase. Bioconjugate Chem. 2005, 16, 669–675. [Google Scholar]
- Koehler, L.; Graf, F.; Bergmann, R.; Steinbach, J.; Pietzsch, J.; Wuest, F. Radiosynthesis and radiopharmacological evaluation of cyclin-dependent kinase 4 (Cdk4) inhibitors. Eur. J. Med. Chem. 2010, 45, 727–737. [Google Scholar]
- Valdivia, A.C.; Mason, S.; Collins, J.; Buckley, K.R.; Coletta, P.; Beanlands, R.S.; Dasilva, J.N. Radiosynthesis of N-[11C]-methyl-hydroxyfasudil as a new potential PET radiotracer for rho-kinases (ROCKs). Appl. Radiat. Isotopes 2010, 68, 325–328. [Google Scholar]
- Wang, M.; Gao, M.Z.; Mock, B.H.; Miller, K.D.; Sledge, G.W.; Hutchins, G.D.; Zheng, Q.H. Synthesis of carbon-11 labeled fluorinated 2-arylbenzothiazoles as novel potential PET cancer imaging agents. Bioorg. Med. Chem. 2006, 14, 8599–8607. [Google Scholar]
- Hosoi, R.; Matsumura, A.; Mizokawa, S.; Tanaka, M.; Nakamura, F.; Kobayashi, K.; Watanabe, Y.; Inoue, O. MicroPET detection of enhanced F-18-FDG utilization by PKA inhibitor in awake rat brain. Brain Res. 2005, 1039, 199–202. [Google Scholar]
- Sasaki, T.; Enta, A.; Nozaki, T.; Ishii, S.; Senda, M. Carbon-11-forskolin - a ligand for visualization of the adenylate cyclase-related second messenger system. J. Nucl. Med. 1993, 34, 1944–1948. [Google Scholar]
- Kiesewetter, D.O.; Sassaman, M.B.; Robbins, J.; Jagoda, E.M.; Carson, R.E.; Appel, N.M.; Sutkowski, E.; Herscovitch, P.; Braun, A.; Eckelman, W.C. Synthesis and evaluation of an F-18 analog of forskolin for imaging adenylyl cyclase. J. Fluorine Chem. 2000, 101, 297–304. [Google Scholar]
- Braun, A.P.; Schulman, H. The multifunctional calcium/calmodulin-dependent protein kinase: from form to function. Annu. Rev. Physiol. 1995, 57, 417–445. [Google Scholar]
- Wang, L.H.; Besirli, C.G.; Johnson, E.M. Mixed-lineage kinases: a target for the prevention of neurodegeneration. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 451–474. [Google Scholar]
- Taylor, S.S.; Kim, C.; Vigil, D.; Haste, N.M.; Yang, J.; Wu, J.; Anand, G.S. Dynamics of signaling by PKA. Biochim. Biophys. Acta, Proteins Proteomics 2005, 1754, 25–37. [Google Scholar] [CrossRef]
- Duman, R.S.; Heninger, G.R.; Nestler, E.J. A molecular and cellular theory of depression. Arch. Gen. Psychiat. 1997, 54, 597–606. [Google Scholar]
- Vasdev, N.; LaRonde, F.J.; Woodgett, J.R.; Garcia, A.; Rubie, E.A.; Meyer, J.H.; Houle, S.; Wilson, A.A. Rationally designed PKA inhibitors for positron emission tomography: synthesis and cerebral biodistribution of N-(2-(4-bromocinnamylamino)ethyl)-N-[11C]methyl-isoquinoline-5-sulfonamide. Bioorg. Med. Chem. 2008, 16, 5277–5284. [Google Scholar] [CrossRef]
- Marte, B.M.; Downward, J. PKB/Akt: connecting phosphoinositide 3-kinase to cell survival and beyond. Trends Biochem. Sci. 1997, 22, 355–358. [Google Scholar]
- Wang, M.; Gao, M.; Miller, K.D.; Sledge, G.W.; Hutchins, G.D.; Zheng, Q.-H. The first design and synthesis of [11C]MKC-1 ([11C]Ro 31-7453), a new potential PET cancer imaging agent. Nucl. Med. Biol. 2010, 37, 763–775. [Google Scholar]
- Takahashi, K.; Kudo, K.; Okada, M.; Yanamoto, K.; Hatori, A.; Irie, T.; Suzuki, K.; Miura, S. Synthesis and biodistribution of [11C]methyl-bisindolylmareimide III, an inhibitor of protein kinase C. J. Labelled Compd. Radiopharm. 2007, 50, S398. [Google Scholar]
- Cai, L.; Ozaki, H.; Fujita, M.; Hong, J.S.; Bukhari, M.; Innis, R.B.; Pike, V.W. 11C]GO6976 as a potential radioligand for imaging protein kinase C with PET. J. Labelled Compd. Radiopharm. 2009, 52, S337. [Google Scholar]
- Goekjian, P.G.; Jirousek, M.R. Protein kinase C in the treatment of disease: signal transduction pathways, inhibitors, and agent in development. Curr. Med. Chem. 1999, 6, 887–903. [Google Scholar]
- Sasaki, T.; Ishii, S.I.; Senda, M.; Akinaga, S.; Murakata, C. Synthesis of [7b-methoxy 11C]methoxy staurosporine for imaging protein kinase C localization in the brain. Appl. Radiat. Isotopes 1996, 47, 67–69. [Google Scholar]
- Wei, Y.; Wei, X.H.; Wang, Y.; Liu, X.Q.; Chu, T.W.; Hu, S.W.; Wang, X.Y. Iodination and radiolabeling of alpha-allocryptopine with iodine-125. Appl. Radiat. Isotopes 2005, 62, 55–62. [Google Scholar]
- Li, Z.J.; Wei, Y.; Chu, T.W.; Wang, X.Y.; Liu, X.Q.; Wang, Y.; Hu, S.W. Radioiodination, biodistribution and pharmacokinetics of berberine in mice. J. Radioanal. Nucl. Chem. 2005, 265, 355–359. [Google Scholar]
- Vanbilloen, H.; Bormans, G.; Chen, B.; de Witte, P.; Verbruggen, A.; Verbeke, K. Synthesis and preliminary evaluation of mono-[123I]iodohypericin. J. Labelled Compd. Radiopharm. 2001, 44, S965–S967. [Google Scholar]
- Park, J.H.; Kim, S.W.; Lee, H.J.; Yang, S.D.; Chun, K.S.; Yu, K.H. Synthesis and biological evaluation of [123I]Quinonoids for brain tumor imaging. J. Labelled Compd. Radiopharm. 2005, 48, S304. [Google Scholar]
- Park, J.H.; Kim, S.W.; Hur, S.D.; Yu, K.H. The targeting of HER-2/neu tyrosine kinase expressing breast cancer by 2-[123I]iodoemodin. J. Labelled Compd. Radiopharm. 2007, 50, S416. [Google Scholar]
- Martinez, A.; Castro, A.; Medina, M. Glycogen Synthase Kinase 3 (GSK-3) and Its Inhibitors; John Wiley & Sons, Inc.: Hoboken, Belgium, 2006. [Google Scholar]
- Hernández, F.; Avila, J. The role of glycogen synthase kinase 3 in the early stages of Alzheimers' disease. FEBS Lett. 2008, 582, 3848–3854. [Google Scholar]
- Gould, T.D.; Einat, H.; Bhat, R.; Manji, H.K. AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int. J. Neuropsychopharmacol. 2004, 7, 387–390. [Google Scholar] [CrossRef]
- Vasdev, N.; Garcia, A.; Stableford, W.T.; Young, A.B.; Meyer, J.H.; Houle, S.; Wilson, A.A. Synthesis and ex vivo evaluation of carbon-11 labelled N-(4-methoxybenzyl)-]N '-(5-nitro-1,3-thiazol-2-yl)urea ([C-11]AR-A014418): A radiolabelled glycogen synthase kinase-3 beta specific inhibitor for PET studies. Bioorg. Med. Chem. 2005, 15, 5270–5273. [Google Scholar] [CrossRef]
- Myers, R.; Hume, S.P.; Worth, S.A.; Lammertsma, A.A.; Bloomfield, P.M.; Rajeswaran, S.; Jones, T. Quantification of dopamine receptors and transporter in rat striatum using a small animal PET scanner. In Quantification of Brain Function Using PET; Myers, R., Cunningham, V., Bailey, D., Jones, T., Eds.; Academic Press: San Diego, CA, USA, 1996; pp. 12–15. [Google Scholar]
- All animal experiments were conducted under humane conditions, with approval from the Animal Care Committee at CAMH and in accordance with guidelines set forth by the Canadian Council of Animal Care.
- Vasdev, N.; Wilson, A.A.; Houle, S.; Lough, A.J. N-(4-Methoxybenzyl)-N'-(5-nitro-1,3-thiazol-2-yl) urea (AR-A014418). Acta Cryst. 2007, E63, o1653–o1655. [Google Scholar]
- Lough, A.J.; Hicks, J.W.; Valliant, J.F.; Wilson, A.A.; Vasdev, N. N-(4-Methoxyphenyl)-N'-(5-nitro-1,3-thiazol-2-yl)urea. Acta Cryst. 2010, E66, o2339. [Google Scholar]
- Wilson, A.A.; Garcia, A.; Jin, L.; Houle, S. Radiotracer synthesis from [11C]-iodomethane: a remarkably simple captive solvent method. Nucl. Med. Biol. 2000, 27, 529–532. [Google Scholar]
- Wilson, A.A.; Garcia, A.; Houle, S.; Vasdev, N. Utility of commercial radiosynthetic modules in captive solvent [11C]-methylation reactions. J. Labelled Compd. Radiopharm. 2009, 52, 490–492. [Google Scholar]
- Hooker, J.; Reibel, A.; Hill, S.; Schueller, M.; Fowler, J. One-pot, direct incorporation of [11C]CO2. into carbamates. Angew. Chem., Int. Ed. 2009, 48, 3482–3485. [Google Scholar]
- Wilson, A.A.; Garcia, A.; Houle, S.; Vasdev, N. Direct fixation of [11C]-CO2. by amines: formation of [11C-carbonyl]-methylcarbamates. Org. Biomol. Chem. 2010, 8, 428–432. [Google Scholar] [CrossRef]
- Wilson, A.A.; Garcia, A.; Houle, S.; Vasdev, N. The synthesis and application of isocyanates radiolabeled with carbon-11. Chem. - Eur. J. 2010. In Press.. [Google Scholar]
- Handbook of Radiopharmaceuticals; Welch, M.J.; Redvanly, C.S. (Eds.) John Wiley & Sons Ltd.: West Sussex, England, 2003.
- Yang, D.J.; Chanda, M.; Sims-Mourtada, J.; Azhdarinia, A.; Oh, C.-S.; Bryant, J.; Kim, E.E. Challenges and opportunities in molecular imaging. Curr. Med. Imaging Rev. 2008, 4, 46–50. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hicks, J.W.; VanBrocklin, H.F.; Wilson, A.A.; Houle, S.; Vasdev, N. Radiolabeled Small Molecule Protein Kinase Inhibitors for Imaging with PET or SPECT. Molecules 2010, 15, 8260-8278. https://doi.org/10.3390/molecules15118260
Hicks JW, VanBrocklin HF, Wilson AA, Houle S, Vasdev N. Radiolabeled Small Molecule Protein Kinase Inhibitors for Imaging with PET or SPECT. Molecules. 2010; 15(11):8260-8278. https://doi.org/10.3390/molecules15118260
Chicago/Turabian StyleHicks, Justin W., Henry F. VanBrocklin, Alan A. Wilson, Sylvain Houle, and Neil Vasdev. 2010. "Radiolabeled Small Molecule Protein Kinase Inhibitors for Imaging with PET or SPECT" Molecules 15, no. 11: 8260-8278. https://doi.org/10.3390/molecules15118260
APA StyleHicks, J. W., VanBrocklin, H. F., Wilson, A. A., Houle, S., & Vasdev, N. (2010). Radiolabeled Small Molecule Protein Kinase Inhibitors for Imaging with PET or SPECT. Molecules, 15(11), 8260-8278. https://doi.org/10.3390/molecules15118260