Conazoles

Abstract
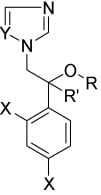
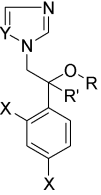
:
Abbreviations
| C.a.: | Candida albicans |
| CMC: | Chronic mucocutaneus candidiasis |
| C.tr.: | Candida tropicalis |
| A.f.: | Aspergillus fumigatus |
| AIDS: | Acquired Immune-Deficiency Syndrome |
| Cr.n.: | Cryptococcus neoformans |
| CYP: | Cytochrome P-450 |
| EMEM: | Eagle’s Minimum Essential Medium |
| HIV: | human immune-deficiency virus |
| MIC90: | Minimum inhibitory concentration that inhibited 90% of the test isolates |
| M.c.: | Microsporum canis |
| MIC: | Minimum inhibitory concentration |
| Muc.: | Mucor species |
| Ph.v.: | Phialophora verrucosum |
| Sap.: | Saprolegnia |
| Sp.s.: | Sporothrix schenckii |
| Spp.: | species |
| T.m.: | Trichophyton mentagrophytes |
| T.r.: | Trichophyton rubrum |
1. Introduction

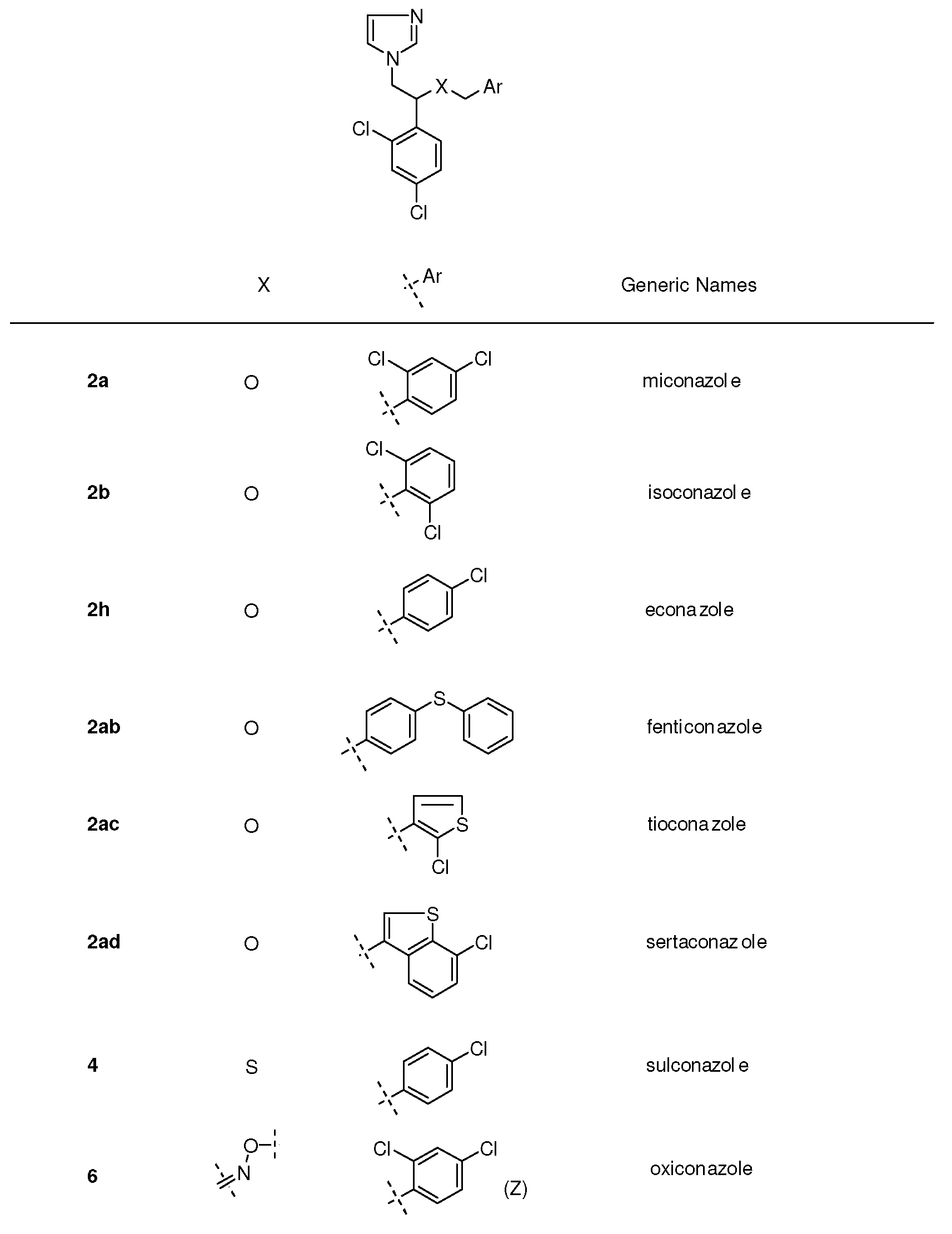
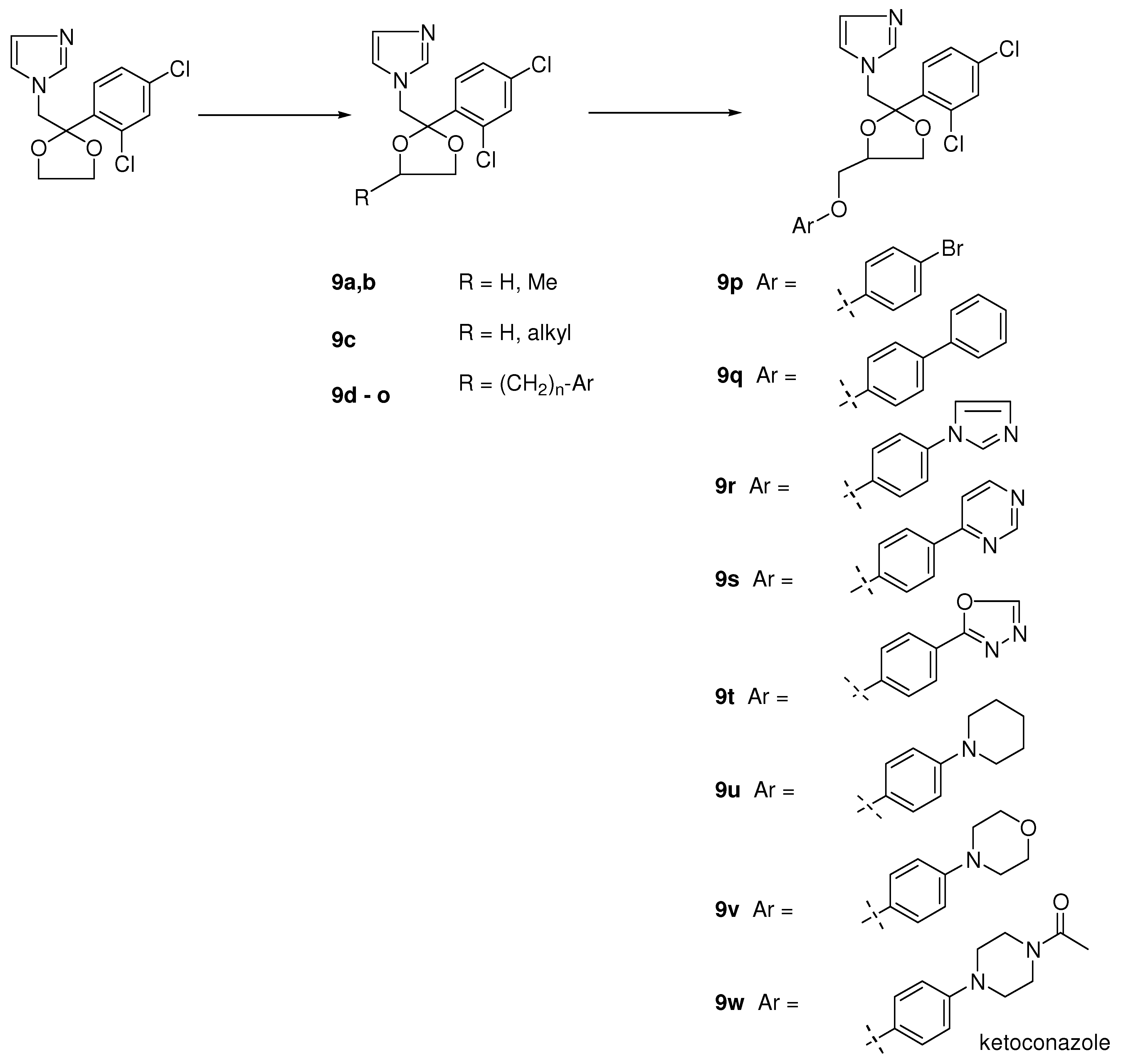
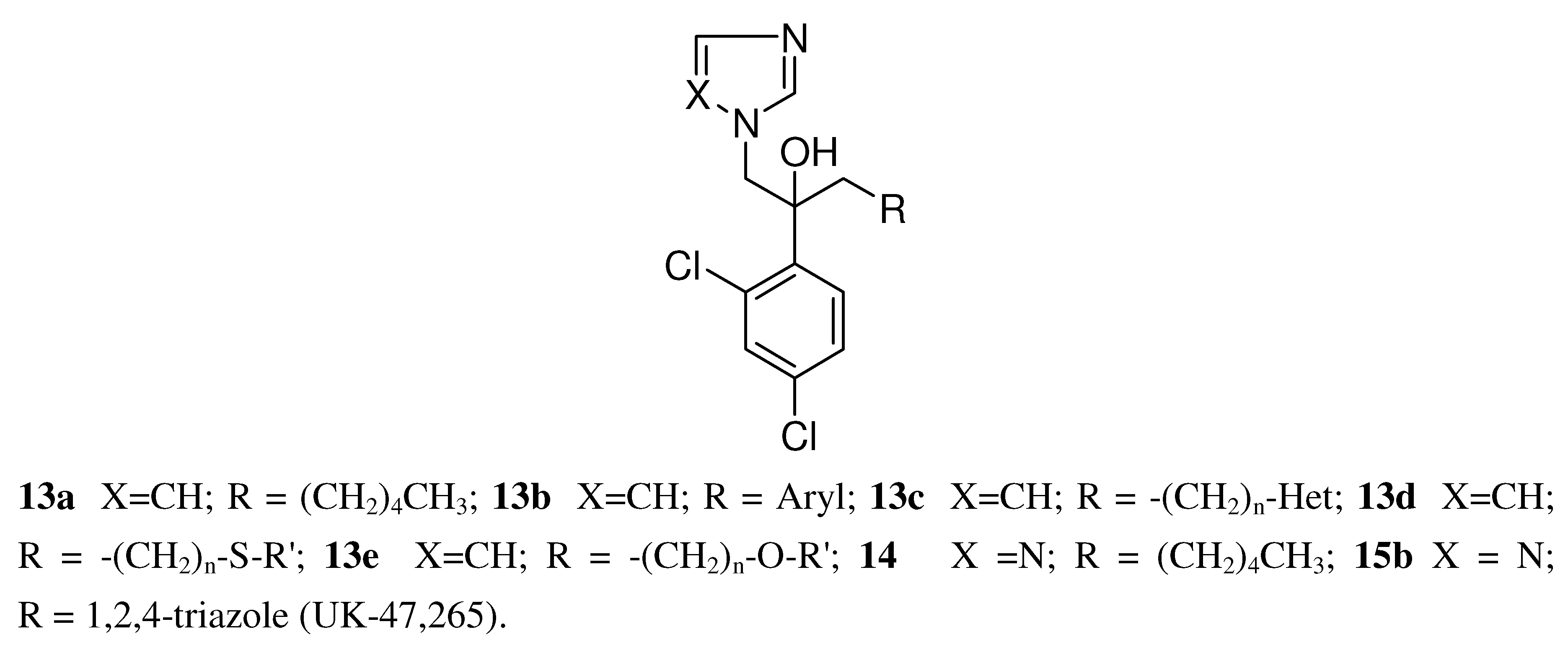
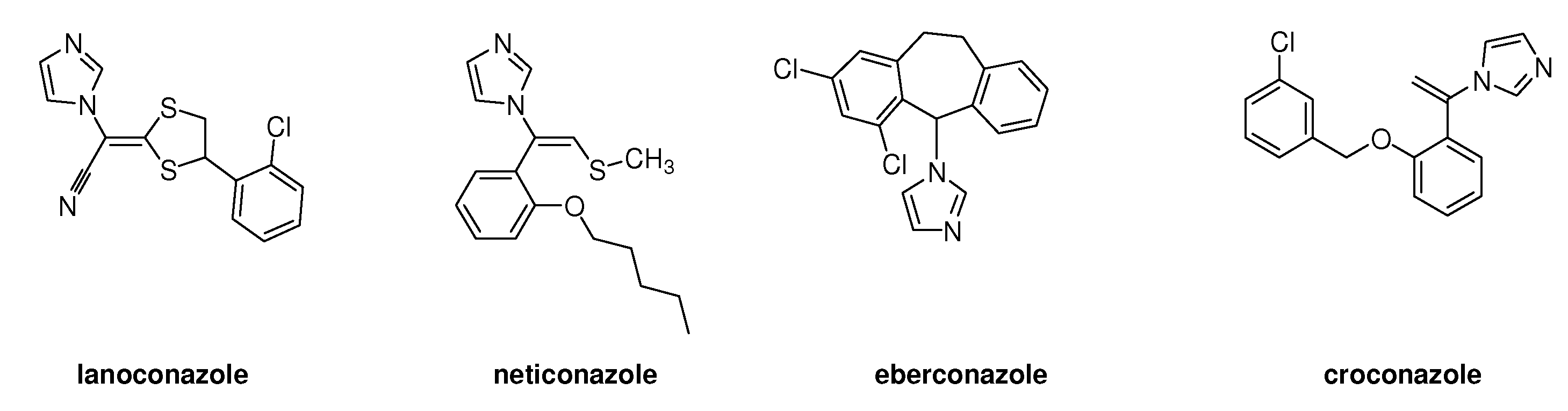
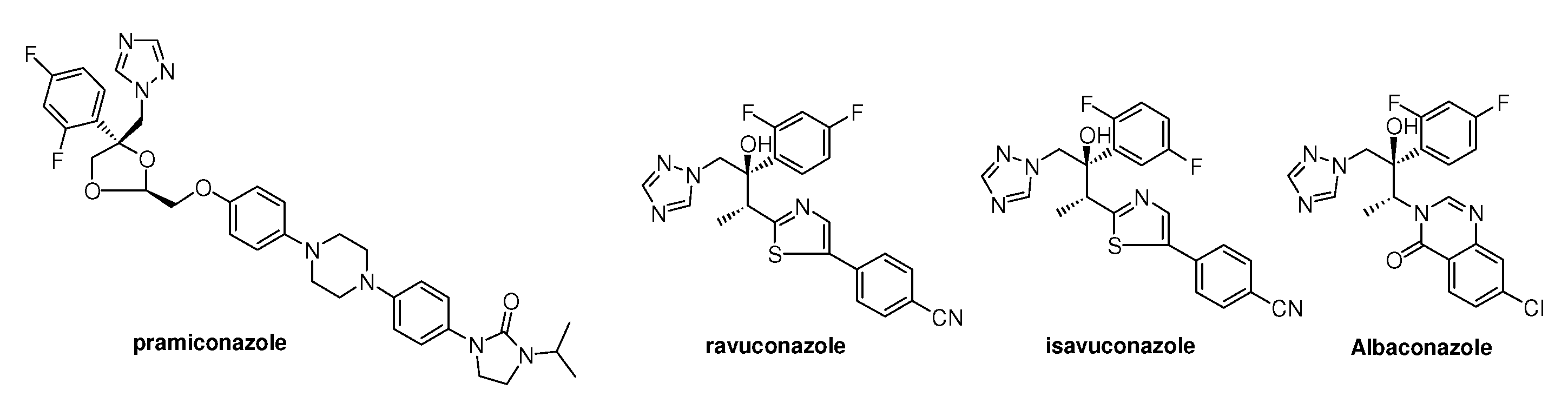
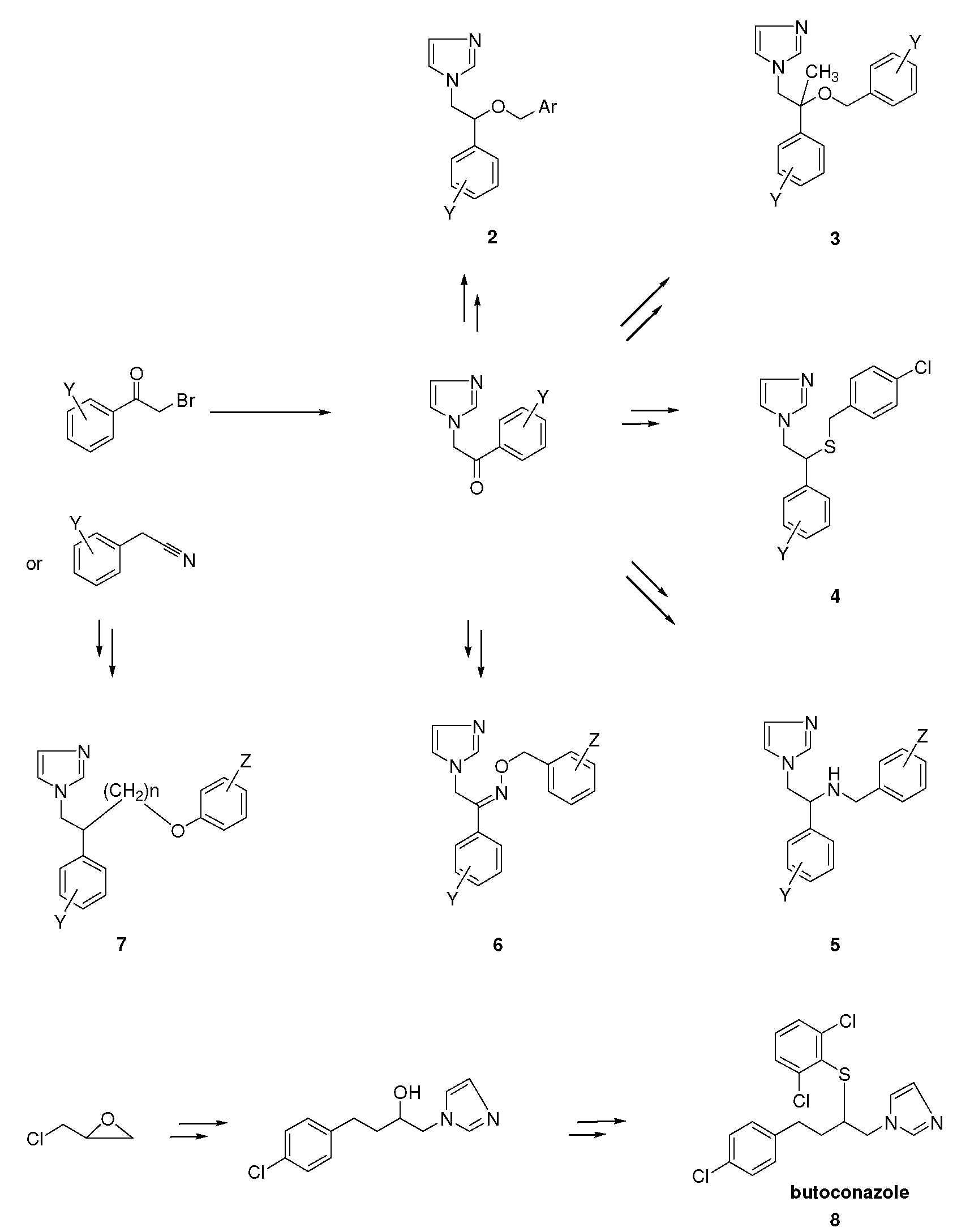
2. Syntheses and Structure-Activity-Relationship



{kind=link}
{kind=link}
{kind=link}
{kind=link}
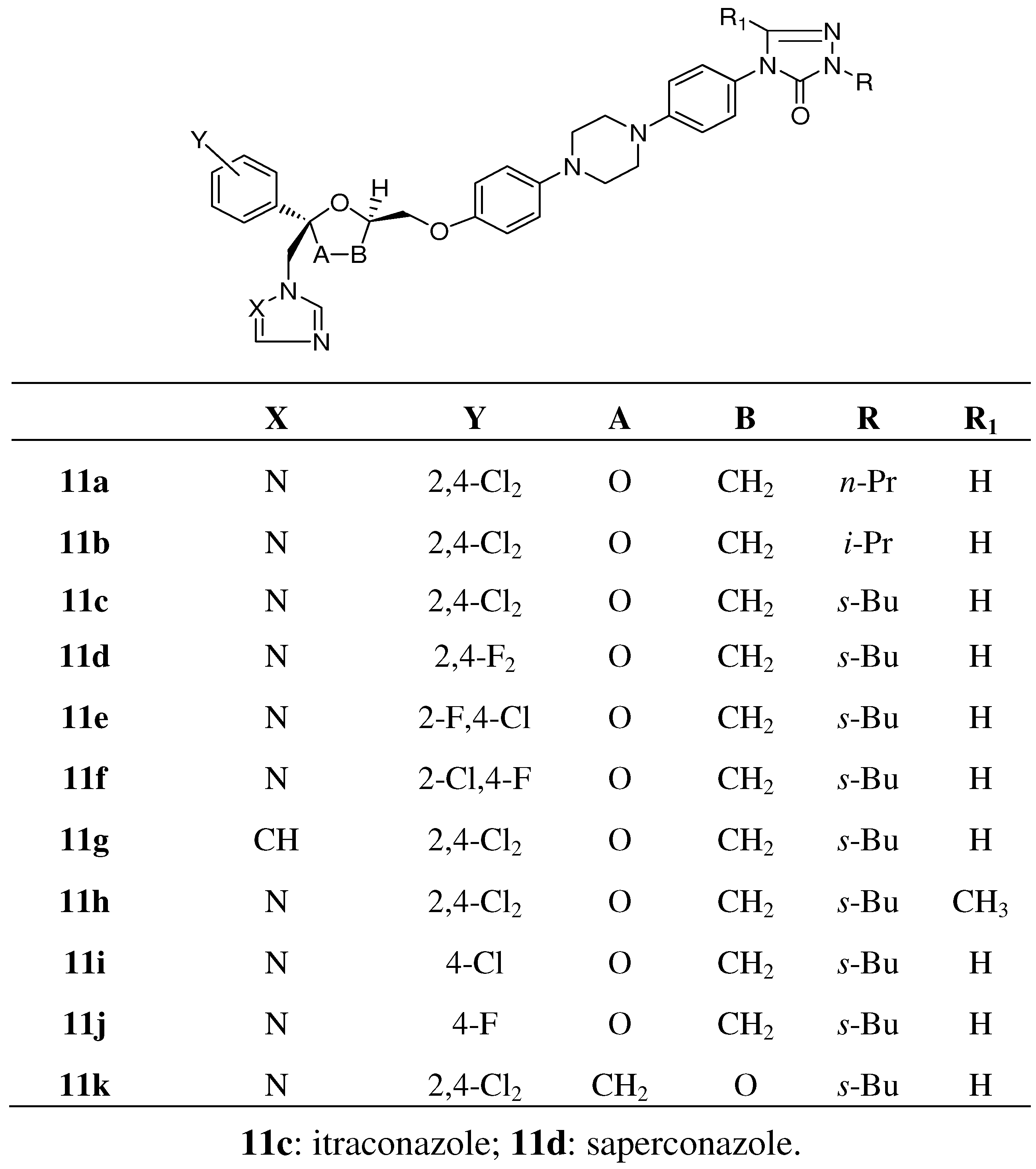{kind=link}
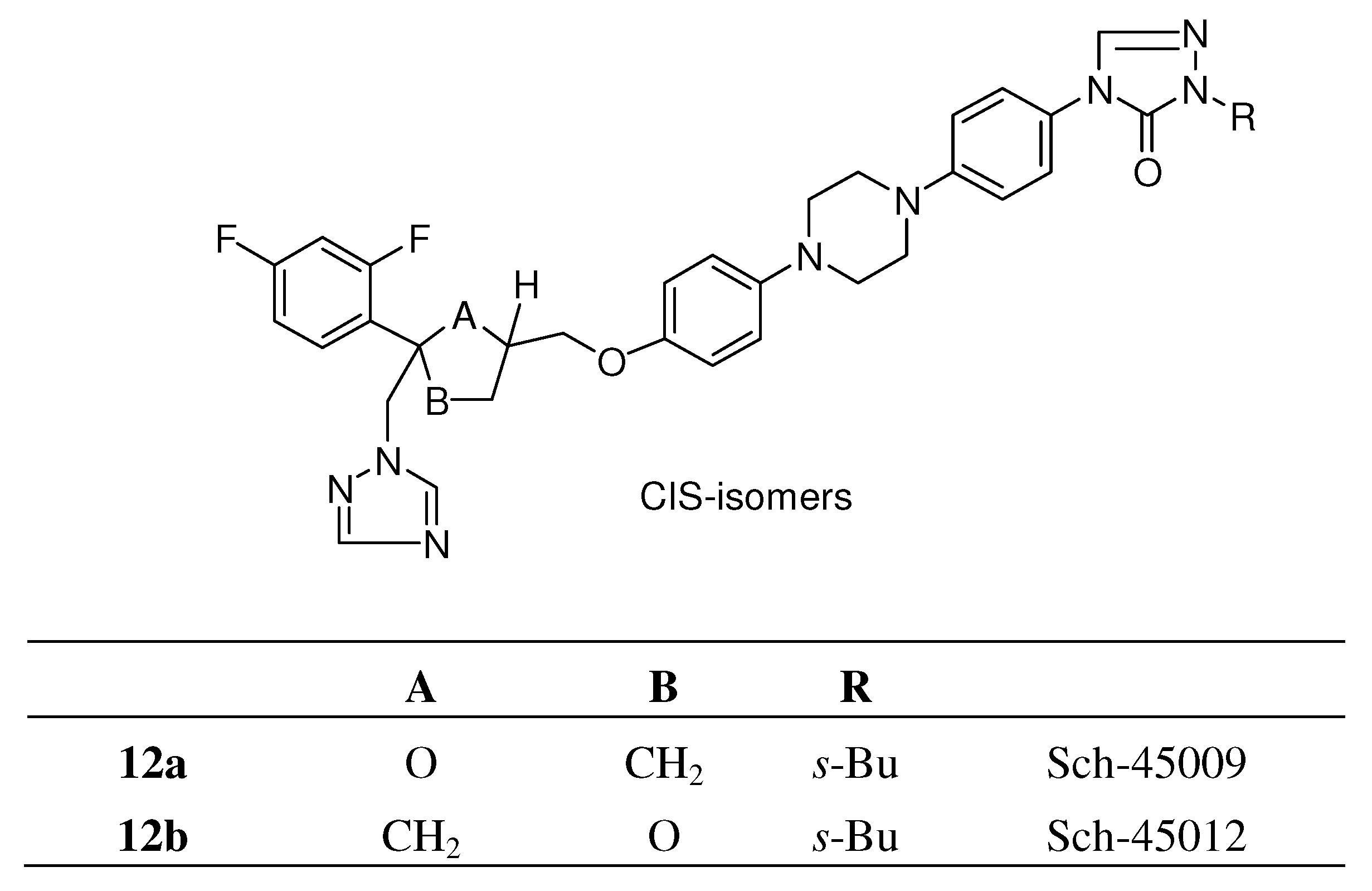{kind=link}
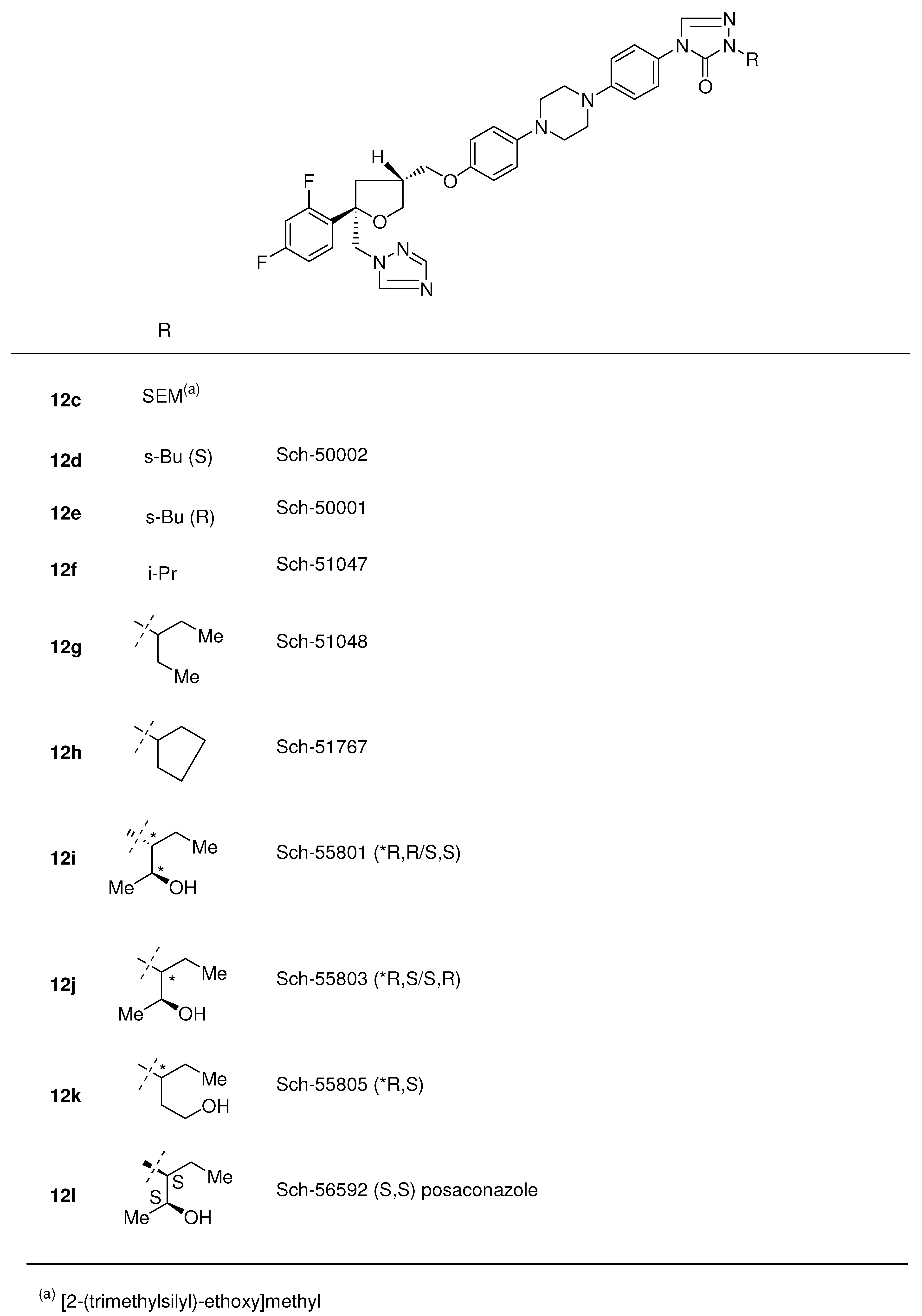{kind=link}
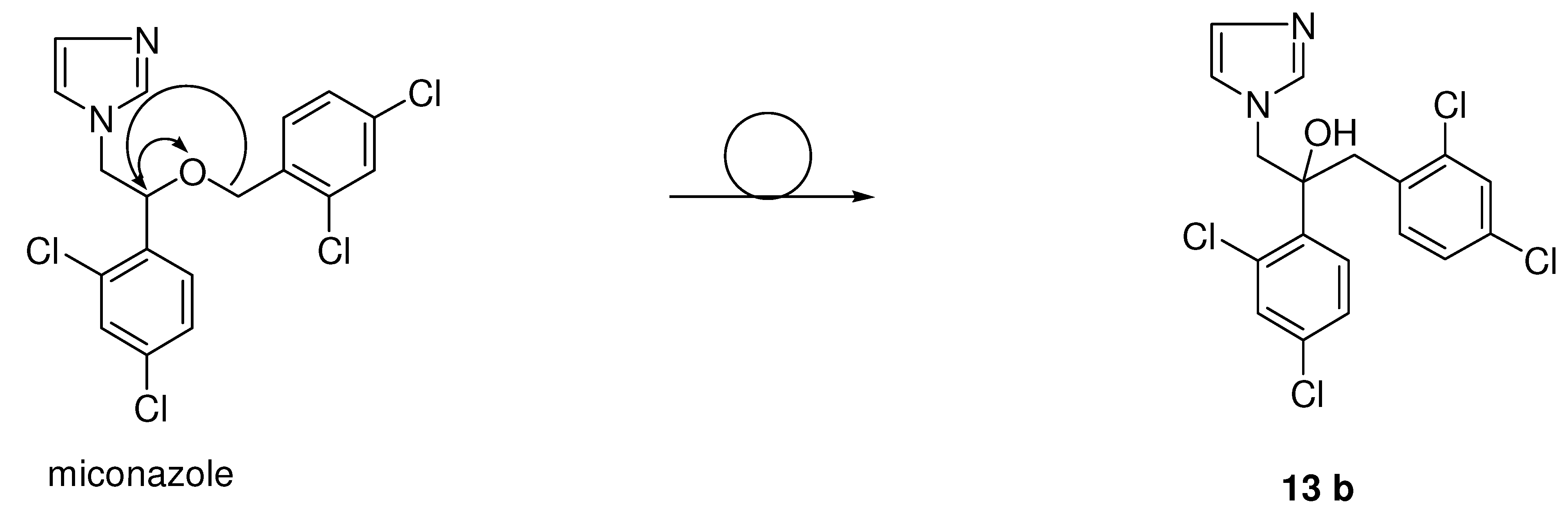{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| R | X | Y | Z | C.a.(a) | M.c. | T.m. | T.r. | A.f.(b) | |
| 2a | H | 0 | 2,4-Cl2 | 2,4-Cl2 | 10 | <1 | 0.1 | <1 | 10 |
| 2b | H | 0 | 2,4-Cl2 | 2,6-Cl2 | 100 | <1 | 0.1 | 0.1 | 10 |
| 2c | H | 0 | 4-F | 2,4-Cl2 | 100 | 10 | <1 | <1 | 10 |
| 2d | H | 0 | 2,4-Cl2 | 4-F | 100 | 10 | 10 | 10 | 10 |
| 2e | H | 0 | 2,4-Cl2 | 2-F | 100 | 10 | 10 | 10 | 100 |
| 2f | H | 0 | 2,4-Cl2 | 2-Cl | 100 | 10 | 0.1 | 0.1 | 10 |
| 2g | H | 0 | 4-Cl | 2,6-Cl2 | 10 | 100 | 10 | 10 | 100 |
| 2h | H | 0 | 2,4-Cl2 | 4-Cl | 100 | 0.1 | 0.01 | 0.1 | <1 |
| 2i | H | 0 | 4-Cl | 2,4-Cl2 | 10 | 10 | <1 | 0.1 | 10 |
| 2j | H | 0 | 2-Cl | 2,4-Cl2 | 100 | <1 | <1 | <1 | 10 |
| 2k | H | 0 | 4-Br | 2-Cl | 100 | 10 | 10 | 10 | 100 |
| 2l | H | 0 | 4-Br | 4-Cl | 10 | <1 | 0.1 | 0.1 | <1 |
| 2m | H | 0 | 4-F | 4-Cl | 100 | <1 | <1 | <1 | 10 |
| 2n | H | 0 | 4-F | 2-Cl | 100 | 100 | 10 | <1 | 100 |
| 2o | H | 0 | 4-Cl | 2-Cl | 100 | 100 | 10 | 10 | 100 |
| 2p | H | 0 | 4-Cl | 4-Cl | 10 | <1 | 0.1 | 0.1 | 10 |
| 2q | H | 0 | H | 4-Cl | 100 | 10 | <1 | <1 | 10 |
| 2r | H | 0 | H | H | >100 | 100 | 10 | 10 | 100 |
| 2s | H | 0 | 4-Me | 2,4-Cl2 | 100 | 100 | 10 | 10 | 100 |
| 2t | H | 0 | 2-Me | 2,4-Cl2 | 100 | 10 | 10 | <1 | 10 |
| 2u | H | 0 | 2,4-Cl2 | 2-Me | 100 | 10 | 0.1 | 0.1 | 10 |
| 2v | H | 0 | 2-Me | 2,4-Cl2 | 100 | <1 | <1 | <1 | <1 |
| 2w | H | 0 | 2,4-Cl2 | 3-MeO | 100 | 10 | 10 | 10 | 100 |
| 2x | H | 0 | 2,4-Cl2 | 4-MeO | 100 | 10 | 10 | 10 | 100 |
| 2y | H | 0 | 4-Cl | 2-Me | 100 | 100 | 10 | 10 | 100 |
| 2z | H | 0 | 4-Me | 2-Cl | 100 | 100 | 10 | 10 | 100 |
| 2aa | H | 0 | 4-Me | 4-Cl | 100 | 10 | <1 | <1 | 10 |
| 3a | Me | 0 | 4-Cl | 2,4-Cl2 | >100 | 10 | 10 | 10 | 100 |
| 3b | Me | 0 | H | 2,4-Cl2 | >100 | 100 | 10 | 10 | >100 |
| 3c | Me | 0 | 4-Cl | 4-Cl | 100 | 100 | 10 | 10 | 100 |
| 5 | H | NH | 4-Cl | 2-Cl | >100 | 100 | 100 | 100 | ND(c) |
| tolnaftate | >100 | >100 | 10 | <1 | >100 | ||||
| nystatin | 333 | ND | ND | ND | ND | ||||
 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | X | Y | Z | M.c.(a,b) | T.m. | T.r. | Cr.n. | C.tr.(c) | C.a. | Muc. | A.f. | In vivo (d) |
| 2a miconazole | O | CH2 | 2,4-Cl2 | 1 | <1 | <1 | 1 | 100 | 10 | >100 | 10 | 4/13 |
| 7a | CH2 | O | 2,4-Cl2 | <1 | <1 | <1 | <1 | >100 | >100 | >100 | >100 | NT(e) |
| 7b | (CH2)2 | O | 2,4-Cl2 | 10 | <1 | <1 | <1 | >100 | 100 | 100 | 100 | 2/2 |
| 7c | (CH2)2 | O | 4-F | 10 | <1 | <1 | <1 | >100 | 10 | 100 | 100 | 2/2 |


 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| X | Y | n | M.c.(a,b) | T.m. | T.r. | Ph.v.(c) | Cr.n. | C.tr. | C.a. | Muc. | A.f. | Rat(d,e) | Guinea Pig (f) | |
| 9d | 4-Cl | 2-Cl2 | 0 | 10 | <1 | <1 | 100 | 10 | >100 | >100 | 100 | 100 | NT | NT |
| 9e | 4-Cl | 2,4-Cl2 | 0 | 10 | <1 | <1 | 100 | 10 | >100 | 100 | 100 | 100 | 1/2 | 2/2 |
| 9f | 2,4-Cl2 | H | 0 | <1 | <1 | <1 | 100 | 100 | >100 | 100 | 100 | <1 | 0/2 | NT |
| 9g | 2,4-Cl2 | 2-Cl | 0 | 10 | <1 | <1 | 100 | <1 | 100 | 100 | 100 | 100 | - | NT |
| 9h | 2,4-Cl2 | 4-Cl | 0 | <1 | <1 | <1 | 100 | <1 | 10 | 100 | 10 | <1 | NT | 1/2 |
| 9i | 2,4-Cl2 | 2,4-Cl2 | 0 | 10 | <1 | <1 | 100 | <1 | >100 | >100 | 10 | <1 | 2/2 | NT |
| 9j | 2,4-Cl2 | 4-Br | 0 | <1 | <1 | <1 | 100 | <1 | 10 | 100 | 10 | <1 | 0/2 | 3/4 |
| 9k | 2,4-Cl2 | H | 1 | <1 | <1 | <1 | 100 | <1 | 10 | 100 | 10 | <1 | 0/2 | 2/2 |
| 9l | 2,4-Cl2 | 4-Cl | 1 | <1 | <1 | <1 | 100 | 1 | 10 | 10 | 10 | 10 | 0/2 | 2/2 |
| 9m | 2,4-Cl2 | 4-Br | 1 | <1 | <1 | <1 | 100 | <1 | >100 | 10 | <1 | 10 | 0/2 | NT |
| 9n | 2,4-Cl2 | 4-Ph | 1 | 10 | <1 | <1 | >100 | 100 | >100 | >100 | 10 | 100 | 0/2 | NT |
| 9o | 2,4-Cl2 | 4-Ph | 2 | <1 | <1 | <1 | >100 | 10 | >100 | >100 | >100 | 10 | 0/2 | NT |
| 2a | miconazole | 1 | <1 | <1 | 100 | 1 | 100 | 10 | >100 | 10 | 0/6 | 4/13 | ||
| Sabourand broth, inhibition (μg/mL) | Sabourand broth + 10% inact. Bovine serum, inhibition (μg/mL) | |||
|---|---|---|---|---|
| Organism | Complete | Marked(a) | Complete | Marked (a) |
| Microsporum canus | 100 | 100 | 100 | 10 |
| Trichophyton mentagrophytes | 1 | 1 | 1 | 0.1 |
| Trichophyton rubrum | 100 | 10 | 10 | 1 |
| Cryptococcus neoformans | 10 | 1 | 1 | 0.1 |
| Candida albicans | >100 | 10 | 100 | 0.1 |
| Candida tropicalis | 10 | 1 | 1 | 0.1 |
| Phialophora verrucosa | 100 | 10 | 10 | 1 |
| Sporothrix schenckii | 100 | 100 | 100 | 1 |
| Aspergillus fumigatus | >100 | >100 | >100 | 100 |
| Mucor spp. | >100 | >100 | 100 | 100 |
| Saprolegnia spp. | 100 | 10 | 10 | 1 |






 |
 |
 |
 |

3. Pharmacology and Clinical Results
3.1. Topical agents
3.1.1. Miconazole (2a, Figure 2)
| Strains testesd | Number of isolates | Antifungal Agent | MIC range | MIC90 | Ref. |
|---|---|---|---|---|---|
| Trichophyton | 7 | Fenticonazole | 0.28 – 20 | [20,21] | |
| mentagrophytes | 2 | Miconazole | 0.312 – 80 | [20,21] | |
| 2 | Econazole | 0.60 – 0.80 | [20] | ||
| Trichophyton | 2 | Fenticonazole | 0.33 – 0.625 | [20,21] | |
| rubrum | 2 | Miconazole | 0.312 – 0.55 | [20,21] | |
| 1 | Econazole | 0.58 | [20] | ||
| Trichophyton | 1 | Fenticonazole | 1.25 | [21] | |
| verrucosum | 1 | Miconazole | 10 | [21] | |
| Trichophyton | 1 | Fenticonazole | 0.25 | [20] | |
| tonsurans | 1 | Miconazole | 8 | [20] | |
| 1 | Econazole | 4 | [20] | ||
| Microsporum | 2 | Fenticonazole | 5 – 20 | [21] | |
| Canis | |||||
| Microsporum | 2 | Fenticonazole | 8 – 9.2 | [20] | |
| audouini | 2 | Miconazole | 9 – 10 | [20] | |
| 2 | Econazole | 10 | [20] | ||
| Microsporum | 2 | Fenticonazole | 5 – 8.8 | [20,21] | |
| gypseum | 1 | Miconazole | 10 – 12 | [20,21] | |
| 1 | Econazole | 10 | [20] | ||
| Epidermophyton | 2 | Fenticonazole | 0.625 – 1.1 | [20,21] | |
| floccosum | 1 | Miconazole | 0.4 – 0.625 | [20,21] | |
| 1 | Econazole | 1.3 | [20] | ||
| Candida | 106 | Fenticonazole | 0.25 – 32 | 8(a) | [33] |
| albicans | 106 | Miconazole | 0.25 – 32 | 16 | [33] |
| 106 | Econazole | 0.25 – 32 | 16 | [33] | |
| 106 | Sulconazole | 0.12 – 8 | 1 | [33] | |
| 106 | Butoconazole | 0.12 – 8 | 1 | [33] | |
| 106 | Isoconazole | 0.12 – 4 | 4 | [33] | |
| 30 | Ketoconazole | 0.03 – 0.25 | 0.5(b) | [25] | |
| 30 | Fluconazole | 0.5 – 64 | 4 | [25] | |
| 30 | Sertaconazole | 0.03 – 1 | 0.06 | [25] | |
| 30 | Itraconazole | 0.03 – 0.125 | 0.03 | [25] | |
| Candida | 24 | Fenticonazole | 0.25 – 1 | 0.5(a) | [33] |
| parapsilosis | 24 | Miconazole | 0.12 – 0.5 | 0.5 | [33] |
| 24 | Econazole | 0.25 – 4 | 4 | [33] | |
| 24 | Sulconazole | 0.12 – 8 | 2 | [33] | |
| 24 | Butoconazole | 0.12 – 1 | 0.25 | [33] | |
| 24 | Isoconazole | 0.12 – 0.25 | 0.25 | [33] | |
| 18 | Ketoconazole | 0.03 – 0.06 | 0.06(b) | [25] | |
| 18 | Fluconazole | 1 – 8 | 8 | [25] | |
| 18 | Sertaconazole | 0.06 – 0.5 | 0.25 | [25] | |
| 18 | Itraconazole | 0.03 – 0.06 | 0.06 | [25] | |
| Candida | 19 | Fenticonazole | 0.5 – 16 | 8(a) | [33] |
| tropicalis | 19 | Miconazole | 0.5 – 16 | 8 | [33] |
| 19 | Econazole | 0.5 – 16 | 8 | [33] | |
| 19 | Sulconazole | 0.25 – 8 | 4 | [33] | |
| 19 | Butoconazole | 0.12 – 1 | 1 | [33] | |
| 19 | Isoconazole | 0.12 – 2 | 2 | [33] | |
| 20 | Ketoconazole | 0.03 – 4 | 4(b) | [25] | |
| 20 | Fluconazole | 4 – 64 | 64 | [25] | |
| 20 | Sertaconazole | 0.125 – 8 | 2 | ||
| 20 | Itraconazole | 0.03 – 64 | 64 | ||
| Candida krusei | 9 | Fenticonazole | 4 – 16 | 16(a) | [33] |
| 9 | Miconazole | 2 – 4 | 4 | [33] | |
| 9 | Econazole | 4 – 8 | 8 | [33] | |
| 9 | Sulconazole | 4 – 8 | 8 | [33] | |
| 9 | Butoconazole | 0.25 – 0.5 | 0.5 | [33] | |
| 9 | Isoconazole | 1 – 2 | 2 | [33] | |
| 15 | Ketoconazole | 0.03 – 2 | 0.5(b) | [25] | |
| 15 | Fluconazole | 4 – 64 | 64 | [25] | |
| 15 | Sertaconazole | 0.06 – 4 | 1 | [25] | |
| 15 | Itraconazole | 0.03 – 64 | 64 | [25] | |
| Candida | 20 | Fenticonazole | 2 – 8 | 4(a) | [33] |
| guilliermondii | 20 | Miconazole | 0.25 – 1 | 1 | [33] |
| 20 | Econazole | 0.5 – 4 | 4 | [33] | |
| 20 | Sulconazole | 0.5 – 2 | 2 | [33] | |
| 20 | Butoconazole | 0.12 – 0.25 | 0.12 | [33] | |
| 20 | Isoconazole | 0.12 – 1 | 0.5 | [33] | |
| 20 | Ketoconazole | 0.5 – 4 | 1 | [33] | |
| Candida | 11 | Ketoconazole | 0.03 – 2 | 2(b) | [25] |
| glabrata | 11 | Fluconazole | 1 – 64 | 64 | [25] |
| 11 | Fenticonazole | 0.5 – 1 | 1 | [25] | |
| 11 | Sertaconazole | 0.125 – 0.5 | 0.25 | [25] | |
| 11 | Itraconazole | 0.03 – 64 | 16 | [25] | |
| 84 | Miconazole | < 0.05 – 3.125 | 1.56(c) | [144 | |
| 84 | Tioconazole | < 0.05 – 12.5 | 0.2 | [144] | |
| 84 | Econazole | < 0.05 – 12.5 | 1.56 | [144] | |
| Cryptococcus | 3 | Fenticonazole | 0.312 – 0.55 | [20,21] | |
| neoformans | 2 | Miconazole | 0.625 – 1.95 | [20,21] | |
| 2 | Econazole | 2.25 – 2.62 | [20] | ||
| Geotrichum | 1 | Fenticonazole | 40 | [20] | |
| candidum | 1 | Miconazole | 40 | [20] | |
| 1 | Econazole | 40 | [20] | ||
| Torulopsis | 1 | Fenticonazole | 40 | [20] | |
| glabrata | 1 | Miconazole | 1.25 | [20] | |
| 1 | Econazole | 0.31 | [20] | ||
| Malassezia | 12 | Ketoconazole | 0.11 – 3.12 | 2.9 | [73] |
| furfur | 12 | Econazole | 0.78 – 6.25 | 4.8 | [73] |
| 12 | Miconazole | 0.88 – 6.25 | 5.2 | [73] | |
| 12 | Itraconazole | 0.05 – 3.12 | 2.7 | [73] | |
| 12 | Fluconazole | 0.07 – 25 | 4.8 | [73] | |
| Aspergillus | 2 | Fenticonazole | 22.3 – 160 | [20,21] | |
| fumigatus | 1 | Miconazole | 33.1 – 160 | [20,21] | |
| Econazole | 28 | [20] | |||
| Aspergillus | 2 | Fenticonazole | 10 –37.5 | [20,21] | |
| niger | 2 | Miconazole | 22 – 40 | [20] | |
| 1 | Econazole | 18 | [20] | ||
| Aspergillus | 1 | Fenticonazole | 10 | [20] | |
| flavus | 1 | Miconazole | 12 | [20] | |
| 1 | Econazole | 2 | [20] |
3.1.2. Econazole (2h, Figure 2)
3.1.3. Isoconazole (2b, Figure 2)
3.1.4. Fenticonazole (2ab, Figure 2)
3.1.5. Oxiconazole (6, Figure 2)
| Fungi | Number tested | MIC90 | Range |
|---|---|---|---|
| Pathogenic yeasts | 47 | ||
| Candida albicans | 17 | 128 | 0.12 – 128 |
| Candida tropicalis | 9 | 32 | 0.06 – 32 |
| Candida parapsilosis | 6 | 4 | 0.06 – 64 |
| Cryptococcus neoformans | 10 | 0.13 | 0.06 – 0.25 |
| Torulopsis glabrata | 10 | 0.50 | 0.25 – 0.50 |
| Filamentous fungi | 81 | ||
| Aspergillus flavus | 10 | 1 | 0.25 – 2 |
| Aspergillus fumigatus | 10 | 2 | 0.50 – 4 |
| Mucorspecies | 5 | 4 | 0.1 – 4 |
| Pseudallescheria boydii | 1 | 32a | |
| Sporothrix schenckii | 9 | 1 | 0.50 – 4 |
| Dermatophytes | 29 | 1 | 0.06 – 4 |
3.1.6. Tioconazole (2ac, Figure 2)
3.1.7. Sulconazole (4, Figure 2)
| Organism | MIC (μg/mL) |
|---|---|
| Trichoiphyton mentagrophytes | 1.56 |
| Trichophyton rubrum | 6.25 |
| Epidermophyton floccosum | 0.78 |
| Microsporum canis | 6.25 |
| Microsporum gypseum | 12.5 |
| Candida albicans | 6.25 |
| Cryptococcus neoformans | 3.13 |
| Aspergillus fumigatus | 6.25 |
| Aspergillus flavus | 25 |
| Aspergillus niger | 12.5 |
3.1.8. Butoconazole (8, Scheme 1).
3.1.9. Sertaconazole (2ad, Figure 2).
3.1.10. Terconazole (10, Scheme 2).
| Lowest active concentration of terconazole, μg/mL | ||||||||
|---|---|---|---|---|---|---|---|---|
| Sab. broth | Sab.broth+serum | EMEM | EMEM + serum | |||||
| C(a) | M (b) | C | M | C | M | C | M | |
| M. canis | 100 | 100 | 10 | 100 | 10 | 100 | 100 | |
| T. mentagrophytes | 1 | 1 | 0.1 | 1 | 0.1 | 1 | 0.1 | |
| T. rubrum | 100 | 10 | 10 | 1 | 10 | 1 | 10 | 0.1 |
| C. albicans str. 1 | 100 | 10 | 100 | 0.1 | 0.01 | 0.01 | ||
| C. albicans str. 2 | 100 | 100 | 0.1 | 0.1 | 0.01 | 10 | 0.1 | |
| C. albicans str. 3 | 100 | 100 | 0.1 | 0.1 | 0.01 | 10 | 0.1 | |
| C. tropicalis | 10 | 1 | 1 | 0.1 | 0.1 | 1 | 0.1 | |
| C. neoformans | 10 | 1 | 1 | 0.1 | ||||
| S. schenckii | 100 | 100 | 1 | 100 | 10 | 10 | ||
| A. fumigatus | 100 | 100 | 100 | 100 | 100 | 100 | 100 | |
| Mucor species | 100 | 100 | 10 | 100 | 10 | |||
3.2. Oral treatment of superficial and deep mycoses
3.2.1. Ketoconazole (9w, Scheme 2) [71]
3.2.2. Itraconazole (11c, Scheme 2)
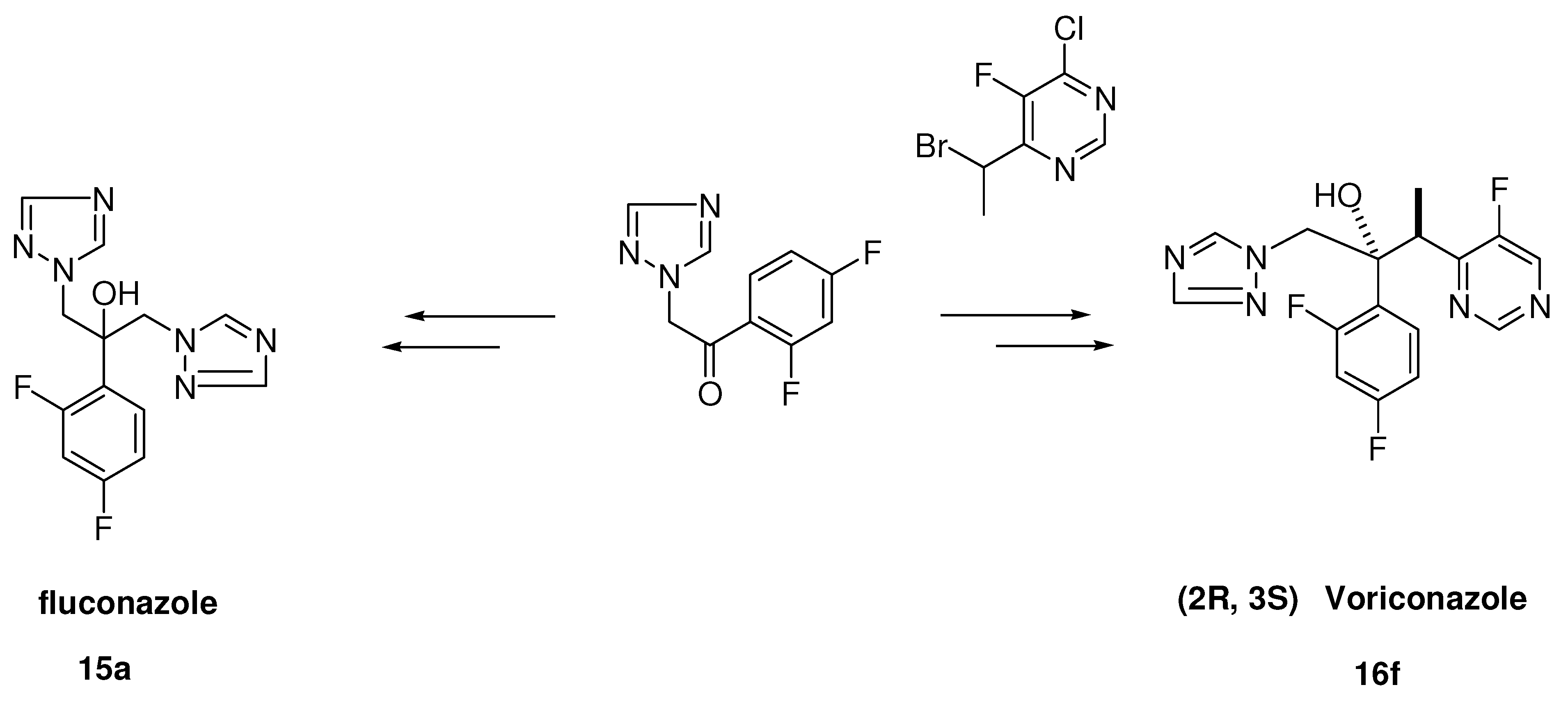
3.2.3. Fluconazole (15a, Scheme 4)
3.2.4. Voriconazole (16f, Scheme 4)
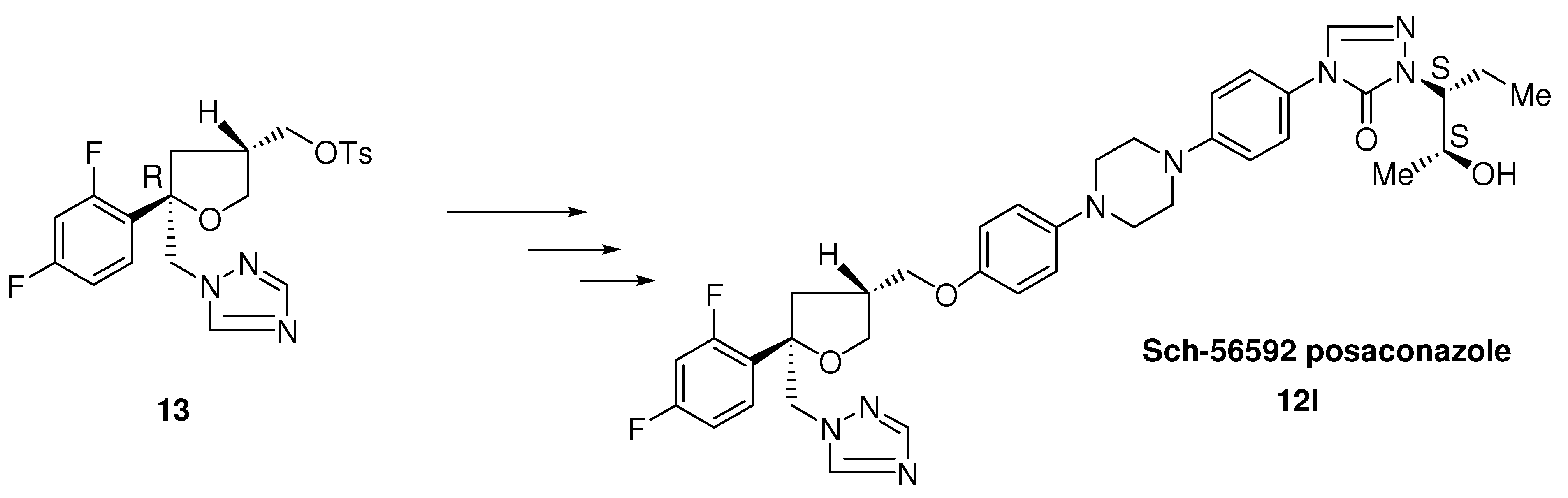
3.2.5. Posaconazole (12l, Scheme 3)
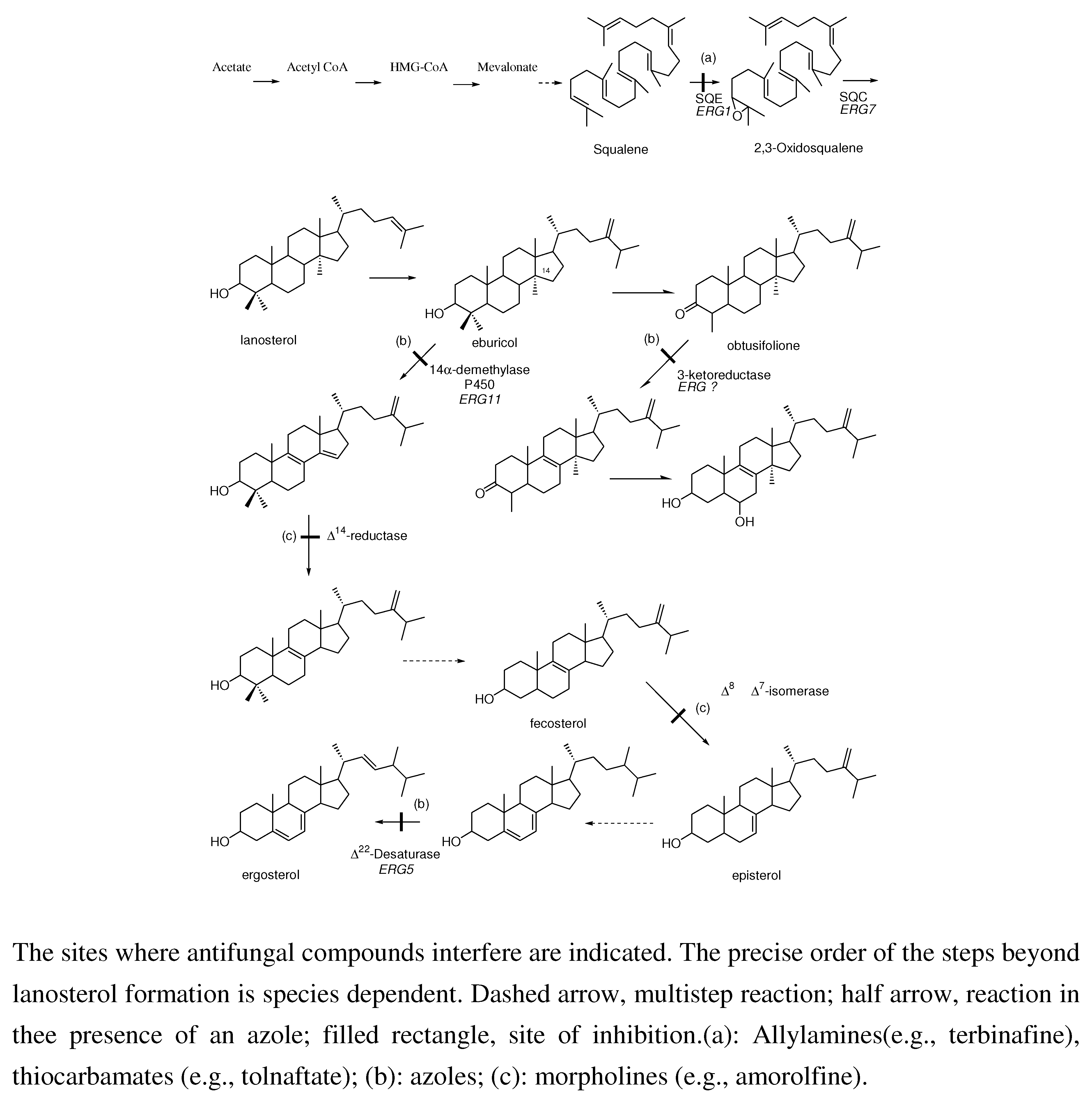
4. Mode of Action
4.1. Ergosterol pathway inhibition

4.2. Antifungal azole drug resistance
4.3. Molecular interaction of azoles antifungal agents with CYP51
 DM with azole ligands is not available, the model originally was based on the interaction of cytochrome P‑450cam with 1-phenylimidazole, 2-phenylimidazole, 4-phenylimidazole and metyrapone, respectively. From these data and other data a model has been developed. In this model it became clear that the halogenated phenyl rings were surrounded and oriented to hydrophobic residues. The azole nitrogen, N3 in the imidazoles and N4 in the triazoles coordinates with the heme iron. The etheral oxygen in miconazole and in ketoconazole can undergo a hydrogen bond interaction. Unfortunately the predictions based on docking analysis in this model could not be confirmed by X-ray crystallography [139]. Furthermore a three-dimensional quantitative structure-activity model has been described [140].
DM with azole ligands is not available, the model originally was based on the interaction of cytochrome P‑450cam with 1-phenylimidazole, 2-phenylimidazole, 4-phenylimidazole and metyrapone, respectively. From these data and other data a model has been developed. In this model it became clear that the halogenated phenyl rings were surrounded and oriented to hydrophobic residues. The azole nitrogen, N3 in the imidazoles and N4 in the triazoles coordinates with the heme iron. The etheral oxygen in miconazole and in ketoconazole can undergo a hydrogen bond interaction. Unfortunately the predictions based on docking analysis in this model could not be confirmed by X-ray crystallography [139]. Furthermore a three-dimensional quantitative structure-activity model has been described [140].5. Drug Interactions in the Clinic
6. Conclusions


Acknowledgements
References
- Schiller, D.S.; Fung, H.B. Posaconazole: An Extended- Spectrum Triazole Antifungal. Clin. Ther. 2007, 29, 1862–1886. [Google Scholar] [CrossRef]
- Godefroi, E.F.; Heeres, J.; Van Cutsem, J.; Janssen, P.A.J. The Preparation and Antimycotic Properties of Derivatives of 1-Phenethylimidazole. J. Med. Chem. 1969, 12, 784–791. [Google Scholar] [CrossRef]
- Godefroi, E.F. Reductive ring cleavage of 1,3-disubstituted imidazolium iodides by sodium borohydride. J. Org. Chem. 1968, 33, 860–862. [Google Scholar] [CrossRef]
- Tajana, A.; Sibilia, C.; Cappelletti, R.; Cova, A.; Nardi, D. Physico-Chemical, Structural and Analytical Studies on Fenticonazole, a New Drug with Antimycotic Properties. Arzneim.-Forsch./Drug Res. 1981, 31, 2127–2133. [Google Scholar]
- Heeres, J.; Backx, L.J.J.; Van Cutsem, J. Antimycotic Imidazoles. 3. Synthesis and Antimycotic Properties of 1-[2-(Aryloxyalkyl)-2-phenylethyl]-1H-imidazoles. J. Med. Chem. 1977, 20, 1516–1521. [Google Scholar] [CrossRef]
- Mixich, G.; Thiele, K. Ein Beitrag zur stereospezifischen Synthese von antimykotisch wirksamen Imidazolyloximäthern. Arzneim.-Forsch./Drug Res. 1979, 29, 1510–1513. [Google Scholar]
- Walker, K.A.M.; Braemer, A.C.; Hitt, S.; Jones, R.E.; Matthews, T.R. 1-[4-(4-Chlorophenyl)-2-(2,6-dichlorophenylthio)-n-butyl]-1H-imidazole Nitrate, a New Potent Antifungal Agent. J. Med. Chem. 1978, 21, 840–843. [Google Scholar] [CrossRef]
- Heeres, J.; Van Cutsem, J. Imidazoles, 5, Synthesis and Antimycotic Properties of 1-[[2-Aryl-4(aralkyl)-1,3-dioxolan-2-yl]methyl]-1H-imidazoles. J. Med. Chem. 1981, 24, 1360–1364. [Google Scholar] [CrossRef]
- Heeres, J.; Backx, L.J.J.; Van Cutsem, J. Antimycotic Imidazoles. Part 4. Synthesis and Antifungal Activity of Ketoconazole, a Potent Orally Active Broad-Spectrum Antifungal Agent. J. Med.Chem. 1979, 22, 1003–1005. [Google Scholar] [CrossRef]
- Heeres, J.; Hendrickx, R.; Van Cutsem, J. Antimycotic Azoles, 6, Synthesis and Antifungal Properties of Terconazole, a Novel Triazole Ketal. J. Med. Chem. 1983, 26, 611–613. [Google Scholar] [CrossRef]
- Heeres, J.; Backx, L.J.J.; Van Cutsem, J. Antimycotic Azoles. 7. Synthesis and Antifungal Properties of a Series of Novel Triazol-3-ones. J. Med. Chem. 1984, 27, 894–900. [Google Scholar] [CrossRef]
- Vanden Bossche, H.; Heeres, J.; Backx, L.J.J.; Marichal, P.; Willemsens, G. Discovery, Chemistry, Mode of Action, and Selectivity of Itraconazole. In Cutaneus Antifungal Agents; Rippon, J.W., Fromtling, R.A., Eds.; Marcel Decker Inc.: New York, NY, USA, 1993; pp. 263–283. [Google Scholar]
- Saksena, A.K.; Girijavallabhan, V.M.; Wang, H.; Liu, Y.-T.; Pike, R.E.; Ganguly, A.K. Consise Asymmetric routes to 2,2,4-trisubstituted tetrahydrofurans via chiral titanium imide enolates: key intermediates towards synthesis of highly active azole antifungals Sch 51048 and Sch 56592. Tetrahedron Lett. 1996, 32, 5657–5660. [Google Scholar]
- Saksena, A.K.; Girijavallabhan, V.M.; Lovery, R.G.; Pike, R.E.; Wang, H.; Liu, Y.T.; Pinto, P.; Bennett, F.; Jao, E.; Patel, N.; Desai, J.A.; Rane, D.F.; Cooper, A.B.; Ganguly, A.H. Advances in the Chemistry of Novel Broad-Spectrum Orally Active Azole Antifungals: Recent Studies Leading to the Discovery of Sch 56592. In Anti-Infectives Recent Advances in Chemistry and Structure-Activity Relationships; Bently, P.H., O’Hanlon, P.J., Eds.; The Royal Society of Chemistry: Cambridge, UK, 1997; pp. 180–199. [Google Scholar]
- Hitchcock, C.A.; Whittle, P.J. Chemistry and Mode of Action of Fluconazole. In Cutaneus Antifungal Agents; Rippon, E.J.W., Fromtling, R.A., Eds.; Marcel Dekker Inc.: New York, NY, USA, 1993. [Google Scholar]
- Butters, M.; Ebbs, J.; Green, S.P.; MacRae, J.; Morland, M.C.; Murtiashaw, C.W.; Pettman, A.J. Process Development of Voriconazole: A Novel Broad-Spectrum Triazole Antifungal agent. Org. Process Res. Dev. 2001, 5, 28–36. [Google Scholar] [CrossRef]
- Jevons, S.; Gymer, G.E.; Brammer, K.W.; Cox, D.A.; Leeming, M.R.G. Antifungal Activity of Tioconazole (UK-20,349), a New Imidazole Derivative. Antimicrob. Agents Chemother. 1979, 15, 597–602. [Google Scholar] [CrossRef]
- Veronese, M.; Salvaterra, M.; Barzaghi, D. Fenticonazole, a New Imidazole Derivative with Antibacterial and Antifungal Activity. Arzneim.-Forsch./Drug Res. 1981, 31, 2133–2137. [Google Scholar]
- Graziani, G.; Cazzulani, P.; Barbadoro, B. Toxicological and Pharmacological Properties of Fenticonazole, a New Topical Antimycotic. Arzneim.-Forsch./Drug Res. 1981, 31, 2145–2151. [Google Scholar]
- Costa, A.L. “In vitro” Antimycotic Activity of Fenticonazole (Rec 15/1476). Mykosen 1982, 25, 47–52. [Google Scholar] [CrossRef]
- Veronese, M. Fenticonazole: A New Antifungal Imidazole Derivative In vitro and In vivo Activity. Mykosen 1984, 27, 194–202. [Google Scholar] [CrossRef]
- Veronese, M.; Barzaghi, D.; Bertonccini, A. Antifungal activity of Fenticonazole in Experimental Dermatomycosis and Candidiasis. Arzneim.-Forsch./Drug Res. 1981, 31, 2137–2139. [Google Scholar]
- Yoshida, H.; Kasuga, O.; Yamaguchi, T. Studies on Antifungal Activities of Sulconazole. Chemotherapy 1984, 32, 477–484. [Google Scholar]
- Ortiz, J.A. Sertaconazole, A New Antifungal agent. Arzneim.-Forsch./Drug Res. 1992, 42, 689. [Google Scholar]
- Palacin, C.; Tarrago, C.; Agut, J.; Guglietta, A. In vitro Activity of Sertaconazole, Ketoconazole, Fenticonazole, Clotrimazole and Itraconazole Against Pathogenic Vaginal Yeast Infections, Methods Find. Exp. Clin. Pharmacol. 2001, 23, 61–64. [Google Scholar]
- Rotstein, D.M.; Walker, K.A.M. The Synthesis and Antifungal Activity of the Enantiomers of Butoconazole Nitrate. Tetrahedron Assymmetry 1993, 4, 1521–1526. [Google Scholar] [CrossRef]
- Heeres, J. New Azole Compounds: From Hypnotics to Itraconazole, a Broad-Spectrum Antifungal Agent. In Actualités Chim. Therapeut; Proceedings of 28emes Rencontre Internationales de Chimie Therapéutique, Toulouse, France, 7-9 July 1993; pp. 149–156.
- Thienpont, D.; Van Cutsem, J.; Van Gerven, F.; Heeres, J.; Janssen, P.A.J. Ketoconazole - a new broad-spectrum orally active antimycotic. Experientia 1979, 35, 606–607. [Google Scholar] [CrossRef]
- Richardson, K. The Discovery of Fluconazole. In Cutaneus Antifungal Agents; Rippon, J.W., Fromtling, R.A., Eds.; Marcel Dekker Inc.: New York, NY, USA, 1993; pp. 171–181. [Google Scholar]
- Dickinson, R.P.; Bell, A.S.; Hitchcock, C.A.; Narayanaswami, S.; Ray, S.J.; Richardson, K.; Troke, P.F. Novel antifungal 2-aryl-1-(1H-1,2,4-triazol-1-yl)butan-2-ol derivatives with high activity against Aspergillus fumigatus. Bioorg. Med. Chem. Lett. 1996, 6, 2031–2036. [Google Scholar]
- Van Cutsem, J.M.; Thienpont, D. Miconazole, a Broad-Spectrum Antimycotic Agent with Antibacterial activity. Chemotherapy 1972, 17, 392–404. [Google Scholar] [CrossRef]
- Heel, R.C.; Brogden, R.N.; Pakes, G.E.; Speight, T.M.; Avery, G.S. Miconazole: A Preliminary Review of its Therapeutic Efficacy In Systemic Fungal Infections. Drugs 1980, 19, 7–30. [Google Scholar] [CrossRef]
- Hernandez Molina, J.M.; Llosa, J.; Martinez Brocal, A.; Ventosa, A. In vitro activity of cloconazole, sulconazole, butoconazole, isoconazole, fenticonazole, and five other antifungal agents against clinical isolates of Candida albicans and Candida spp. Mycopathalogia 1992, 118, 15–21. [Google Scholar] [CrossRef]
- Odds, F.C.; Cheesman, A.B.C.L.; Abbott, A.B. Suppression of ATP in Candida albicans by imidazole and derivative antifungal agents. Sabouraudi: J. Med. Vet. Mycol. 1985, 23, 415–424. [Google Scholar] [CrossRef]
- Brugmans, J.; Van Cutsem, J.; Heykants, J.; Schuurmans, V.; Thienpont, D. Systemic antifungal potential, safety, biotransport and transformation of miconazole nitrate. Eur. J. Clin. Pharmacol. 1972, 5, 93–99. [Google Scholar] [CrossRef]
- Lewi, P.J.; Boelaert, J.; Daneels, R.; De Meyere, R.; Van Landuyt, H.; Heykants, J.; Symoens, J.; Weynants, J. Pharmacokinetic profile of intravenous miconazole in man. Comparison of normal subjects and patients with renal insufficiency. Eur. J. Clin. Pharmacol. 1976, 10, 49–54. [Google Scholar]
- Janssen, P.A.J.; Van Bever, W.F.M. Miconazole. In Pharmacological and Biochemical Properties of Drug Substances; American Pharmaceutical Association, Academy of Pharmaceutical Sciences: Washington, DC, USA, 1979; Volume 2, pp. 333–354. [Google Scholar]
- Thienpont, D.; Van Cutsem, J.; Van Nueten, J.M.; Niemegeers, C.J.E.; Marsboom, R. Biological and toxicological properties of econazole, a broad-spectrum antimycotic. Arzneim.-Forsch. 1975, 25, 224–230. [Google Scholar]
- Levine, H.B. R34000, a dioxolane imidazole in the therapy for experimental coccidioidomycosis. Chest 1976, 70, 755–759. [Google Scholar] [CrossRef]
- Cartwright, R.Y. Absorbtion of econazole from the human gastrointestinal tract. In Curr. Chemothery: Proceedings of International Congress Chemotherapy, Tokyo, Japan; 1978; Volume 1, pp. 231–233. [Google Scholar]
- Heel, R.C.; Brogden, R.N.; Speight, T.M.; Avery, G.S. Econazole: A Review of its Antifungal Activity and Therapeutic Efficacy. Drugs 1978, 16, 177–201. [Google Scholar] [CrossRef]
- Aron-Brunetiere, R.; Dompmartin-Pernot, D.; Drouhet, E. Treatment of pityriasis capitis (dandruff) with econazole nitrate. Acta Dermatovener 1977, 57, 77–80, (Stockholm). [Google Scholar]
- Kessler, H.J. Mikrobiologische Untersuchungen mit Isoconazolnitrat, einem Breitspectrum-Antimykoticum aus der Gruppe der Imidazol-Derivate. Arzneim.-Forsch./Drug Res. 1979, 29, 1344–1351. [Google Scholar]
- Fromtling, R.A. Overview of Medically Important Azole Derivatives. Clin. Microbiol. Rev. 1988, 1, 187–217. [Google Scholar]
- Fark, B.; Földes, M.; Kajtar, I. Results of Isoconazol Nitrate Treatment of Vaginal Mycoses. Mykosen 1982, 25, 629–632. [Google Scholar]
- Herms, E.; Kallischnigg, G. Once-Daily-Application of Isoconazole as Cream, Solution and Spray: Comparative Studies on Patients with Dermatomycoses. Z. Hautkr. 1988, 63, 377–384. [Google Scholar]
- Weitgasser, H.; Herms, E. Comparative Clinical Investigations with the New Antimycotic Agent Isoconazole Nitrate and its Combination with Diflucortolone-21-valerate in the Case of Inflammatory and Eczematised Dermatomycoses. Mykosen 1979, 22, 177–183. [Google Scholar] [CrossRef]
- Oyeka, C.A.; Gugnani, H.C. Isoconazole Nitrate versus clotrimazole in foot and nail infections due to Hendersonula toruloidea, Scytalidium hyalinum and dermatophytes. Mycoses 1992, 35, 357–361. [Google Scholar]
- Eschborn, H.M. Fenticinazole. A local antimycotic. Mykosen 1993, 44, 34–36. [Google Scholar]
- Polak, A. Oxiconazole, a New Imidazole Derivative. Arzneim.-Forsch./Drug Res. 1982, 32, 17–24. [Google Scholar]
- Gebhart, R.J.; Espinel-Ingroff, A.; Shadomy, S. In vitro Susceptibility Studies with Oxiconazole. Chemotherapy 1984, 30, 244–247. [Google Scholar] [CrossRef]
- Jegasothy, B.V.; Pakes, G.E. Oxiconazole Nitrate: Pharmacology, Efficacy, and Safety of a New Imidazole Antifungal Agent. Clin. Ther. 1991, 13, 126–141. [Google Scholar]
- Wagner, W. Vergleich der klinischen Wirksamkeit und Verträglichkeit von Oxiconazol nach 1x täglicher Applikation mit der nach 2x täglicher Application. Mykosen 1986, 29, 280–284. [Google Scholar] [CrossRef]
- Odds, F.C. Laboratory evaluation of antifungal agents: a comparative study of five imidazole derivatives of clinical importance. J. Antimicrob. Chemother. 1980, 6, 749–761. [Google Scholar] [CrossRef]
- Clissold, S.P.; Heel, R.C. Tioconazole, A Review of its Antimicrobial Activity and Therapeutic Use in Superficial Mycoses. Drugs 1986, 31, 29–51. [Google Scholar] [CrossRef]
- Krohn, K.; Vinnerberg, A. Open Comparison of the Efficacy, Toleration and Safety of Tioconazole and Econazole in the 3-Day Treatment of Vaginal Candidiasis. Gynäk. Rdsch. 1983, 23, 29–32. [Google Scholar]
- Artner, J.; Fuchs, G. Open Studies of the Efficacy, Tolerance, Systemic Absorbtion and Vaginal Persistence following a Single Application of Tioconazole Ointment in the Treatment of Patients with vaginal Candidosis. Gynäk. Rdsch. 1983, 23, 12–19. [Google Scholar]
- Clayton, Y.M.; Hay, R.J.; McGibbon, D.H.; Pye, R.J. Double Blind comparison of the efficacy of tioconazole and miconazole for the treatment of fungal infection of the skin orerythrasma. Clin. Exp. Dermatology 1982, 7, 543–551. [Google Scholar]
- Seidman, L.S.; Skokos, C.K. An evaluation of butoconazole nitrate 2% Site Release vaginal cream (Gynazole-1) compared to fluconazole 150 mg tablets (Diflucan) in the time to releif of symptoms in patients with vulvovaginal candidiasis. Infect. Dis. Obstet. Gynecol. 2005, 13, 197–206. [Google Scholar] [CrossRef]
- Palacin, C.; Sacristan, A.; Ortiz, J.A. In vitro Activity of Sertaconazole. Arzneim.-Forsch./Drug Res. 1992, 42, 699–705, Nr. 5a. [Google Scholar]
- Carillo-Munoz, A.J.; Giusiano, G.; Ezkurra, P.A.; Quindos, G. Sertaconazole: updated review of a topical antifungal agent. Expert Rev. Anti Infect. Ther. 2005, 3, 333–342. [Google Scholar] [CrossRef]
- Palacin, C.; Sacristan, A.; Ortiz, J.A. In vivo Activity of Sertaconazole in Experimental Dermatophytosis in Guinea Pigs. Arzneim.-Forsch./Drug Res. 1992, 42, 714–718, Nr. 5a. [Google Scholar]
- Pedragosa, R.; Gonzalez, B.; Martin, M.; Herrero, E.; Roset, P.; Marquez, M.; Torres, J.; Ortiz, J.A. Therapeutic Efficacy and Safety of the New Antimycotic Sertaconazole in the Treatment of Pityriasis versicolor. Arzneim.-Forsch./Drug Res. 1992, 42, 760–763, Nr. 5a. [Google Scholar]
- Van Cutsem, J.; Van Gerven, F.; Zaman, R.; Janssen, P.A.J. Terconazole, a new broad-spectrum antifungal. Chemotherapy 1983, 29, 322–331. [Google Scholar] [CrossRef]
- Tolman, E.I.; Isaacson, D.M.; Rosenthale, M.E.; McGuire, J.L.; Van Cutsem, J.; Borgers, M.; Vanden Bossche, H. Anticandidal Activities of Terconazole, a Broad-Spectrum Antimycotic. J. Antimicrob. Chemother. 1986, 29, 986–991. [Google Scholar] [CrossRef]
- Tolman, E.L.; Isaacson, D.M.; Rosenthale, M.E. In vitro Studies with Terconazole. Gynäk. Rdsch. 1985, 25, 12–25. [Google Scholar]
- Hirsch, H.A. Clinical Evaluation of Terconazole. European Experience. J. Reprod. Med. 1989, 34, 593–596. [Google Scholar]
- Cauwenbergh, G. Terconazole. Pharmacology of a new antimycotic agent. J. Reprod. Med. 1989, 34, 588–592. [Google Scholar]
- Sood, G.; Nyirjesy, P.; Velma Weitz, M.; Chatwani, A. Terconazole cream for Non-Candida Albicans Fungal vaginitis: Results of a Retrospective Analysis. Infect. Dis. Obstet. Gynecol. 2000, 8, 240–243. [Google Scholar]
- Raab, W. Terconazole, ein neues Triazolderivaat zur Behandlung vaginaler Mykosen. Gynäk. Rdsch. 1985, 25, 197–220. [Google Scholar]
- Heel, R.C.; Brogden, R.N.; Carmine, A.; Morley, P.A.; Speight, T.M.; Avery, G.S. Ketoconazole: A Review of its Therapeutic Efficacy in Superficial Fungal Infections. Drugs 1982, 23, 1–36. [Google Scholar] [CrossRef]
- De Brabander, M.; Aerts, F.; Van Cutsem, J.; Vanden Bossche, H.; Borgers, M. The activity of ketoconazole in mixed cultures of leukocytes and Candida albicans. Sabouraudia 1980, 18, 197–210. [Google Scholar] [CrossRef]
- Strippoli, V.; Piacentini, A.; D’Auria, F.D.; Simonetti, N. Antifungal Activity of Ketoconazole and Other Azoles against Mallassezia furfur In vitro and In vivo. Infection 1997, 25, 303–306. [Google Scholar] [CrossRef]
- Borelli, P.; Bran, J.L.; do Fuentes, J.; Legendre, R.; Leiderman, E.; Levine, H.B.; Restrepo, A.; Stevens, D.A. Ketoconazole, an oral antifungal: laboratory and clinical assessment of imidazole drugs. Postgrad. Med. J. 1979, 55, 657–661. [Google Scholar] [CrossRef]
- Levine, H B.; Cobb, J.M. Oral therapy for experimental coccidiodomycosis with R41400 (ketoconazole), a new imidazole. Am. Rev. Respir. Dis. 1978, 118, 715–721. [Google Scholar]
- Williams, D.M.; Graybill, J.R.; Drutz, D.J.; Levine, H.J. Suppression of Cryptococcosis and Histoplasmosis by Ketoconazole in Athymic Nude Mice. J. Infect. Dis. 1980, 141, 76–80. [Google Scholar] [CrossRef]
- Huang, Y.-C.; Colaizzi, J.; Bierman, R.H.; Woestenborghs, R.; Heykants, J. Pharmacokinetics and Dose Proportionality of Ketoconazole in Normal Volunteers. Antimicrob. Agents Chemother. 1986, 30, 206–210. [Google Scholar] [CrossRef]
- Gascoigne, E.W.; Barton, G.J.; Michaels, M.; Meuldermans, W.; Heykants, J. The kinetics of ketoconazole in animals and man. Clin. Res. Rev. 1981, 1, 177–187. [Google Scholar]
- Van Tyle, J.H. Drugs in Perspective, Ketoconazole, Mechanism of Action, Spectrum of Activity, Pharmacokinetics, Drug Interactions, Adverse Reactions and Therapeutic Use. Pharmacotherapy 1984, 4, 343–373. [Google Scholar]
- Rosenblatt, H.M.; Byrne, W.; Ament, M.E.; Graybill, J.; Stiehm, E.R. Successful treatment of chronic mucocuteneous candidiasis with ketoconazole. J. Pediat. 1980, 97, 657–660. [Google Scholar] [CrossRef]
- Smith, E.B.; Henry, J.C. Ketoconazole: An Orally Efective Antifungal Agent, Mechanism of Action, Pharmacology, Clinical Efficacy and Adverse Effects. Pharmacotherapy 1984, 4, 199–204. [Google Scholar]
- Calhoun, D.l.; Waskin, H.; White, M.P.; Bonner, J.R.; Mulholland, J.H.; Rumans, L.W.; Stevens, D.A.; Galgiani, J.N. Treatment of Systemic Sporotrichosis with Ketoconazole. Rev. Infect. Dis. 1991, 13, 47–51. [Google Scholar] [CrossRef]
- Van Cutsem, J.; Van Gerven, F.; Zaman, R.; Heeres, J.; Janssen, P.A.J. Pharmacological and preclinical results with a new oral and topical broad-spectrum antifungal, R51211. In Processdings of 13th International Congress of Chemotherapy, Vienna, Austria, 28 August–2 September; 1983. [Google Scholar]
- Hoffman, H.L.; Ernst, E.J.; Klepser, M.E. Novel triazole antifungal agents. Exp. Opin. Invest. Drugs 2000, 9, 593–605. [Google Scholar] [CrossRef]
- Espinel-Ingroff, A.; Shadomy, S.; Gebhart, R.J. In vitro studies with R 51,211. Antimicrob. Agents Chemother. 1984, 26, 5–9. [Google Scholar] [CrossRef]
- Borgers, M.; Van de Ven, M.-A. Degenerative changes in fungi after itraconazole treatment. Rev. Infect. Dis. 1987, 9, 33–44. [Google Scholar] [CrossRef]
- Van Cutsem, J.; Van Gerven, F. The In vivo Antifungal Activity of Broad-Spectrum Azoles. Drug Dev. Res. 1986, 8, 309–316. [Google Scholar] [CrossRef]
- Lipp, H.-P. Antifungal agents-clinical pharmacokinetics and drug interactions. Mycoses 2008, 51, 7–18. [Google Scholar] [CrossRef]
- Gubbins, P.O. Mould-active azoles: pharmacokinetics, drug interactions in neutropenic patients. Curr. Opin. Infect. Dis. 2007, 20, 579–586. [Google Scholar]
- Kunze, K.L.; Nelson, W.L.; Karasch, E.D.; Thummel, K.E.; Isoherranen, N. Stereochemical aspects of itraconazole metabolism in vitro and in vivo. Drug Metab. Dispos. 2006, 34, 583–590. [Google Scholar] [CrossRef]
- Degreef, H.J.; De Doncker, P.R.G. Current therapy of dermatophytosis. J. Am. Acad. Dermatol. 1994, 31, S25–S30. [Google Scholar] [CrossRef]
- De Doncker, P.; Van Lint, J.; Dockx, P.; Roseeuw, D. Pulse therapy with one-week itraconazole monthly for three or four months in the treatment of onychomycosis. Cutis 1995, 56, 180–183. [Google Scholar]
- Cauwenbergh, G. New and prospective developments in antifungal drugs. Acta Derm. Venereol. Suppl. (Stockh.) 1986, 121, 147–153. [Google Scholar]
- Battegay, M.; Flückiger, U. Therapy schwerer Pilzinfectionen. Internist 2003, 44, 1549–1556. [Google Scholar] [CrossRef]
- Kauffman, C. A. Newer developments in therapy for endemic mycoses. Clin. Infect. Dis. 1994, 19, S28–32. [Google Scholar] [CrossRef]
- Graybill, J.R. Changing Strategies for Treatment of Systemic Mycoses. Braz. J. Infect. Dis. 2000, 4, 47–54. [Google Scholar]
- Hughes, C.E.; Beggs, W.H. Action of fluconazole (UK-49,858) in relation to other systemic antifungal agents. J. Antimicrob. Chemother. 1987, 19, 171–174. [Google Scholar] [CrossRef]
- Odds, F.C.; Cheesman, S.L.; Abbott, A.B. Antifungal effects of fluconazole (UK 49,858), a new triazole antifungal, in vitro. J. Antimicrob. Chemother. 1986, 18, 473–478. [Google Scholar] [CrossRef]
- Odds, F.C.; Webster, C.E. Effects of azole antifungals in vitro on host/parasite interactions relevant to candida infections. J. Antimicrob. Chemother. 1988, 22, 473–481. [Google Scholar] [CrossRef]
- Nguyen, M.H.; Yu, C. Voriconazole against fluconazole-susceptible and –resistant Candida isolates: in vitro efficacy compared with that of itraconazole and ketoconazole. J. Antimicrob. Chemother. 1998, 42, 253–256. [Google Scholar] [CrossRef]
- Nguyen, M.H.; Yu, C. In vitro comparative efficacy of voriconazole and itraconazole against fluconazole-susceptible and -resistant Cryptococcus neoformans isolates. Antimicrob. Agents Chemother. 1998, 42, 253–256. [Google Scholar]
- Classen, D.C.; Burke, J.P.; Smith, C.B. Treatment of Coccidioidal Meningitis with Fluconazole. J. Infect. Dis. 1988, 158, 903–904. [Google Scholar] [CrossRef]
- Nagappan, V.; Deresinsky, S. Posaconazole: A Broad-Spectrum Triazole Antifungal agent. Clin. Infect. Dis. 2007, 45, 1610–1617. [Google Scholar] [CrossRef]
- Manavathu, E.K.; Abraham, O.C.; Chandrasekar, P.H. Isolation of in vitro susceptibility to amphotericin B, itraconazole and posaconazole of voriconazole-resistant laboratory isolates of Aspergillus fumigatus. Clin. Microb. Infect. 2001, 7, 130–137. [Google Scholar] [CrossRef]
- Koltin, Y.; Hitchcock, C.A. The search for new triazole antifungal agents. Curr. Opin. Chem. Biol. 1997, 1, 176–182. [Google Scholar] [CrossRef]
- Gigolashvili, T. Update on Antifungal Therapy. Cancer Pract. 1999, 7, 157–159. [Google Scholar] [CrossRef]
- Aperis, G.; Mylonakis, E. Newer triazole antifungal agents: pharmacology, spectrum, clinical efficacy and limitations. Expert Opin. Invest. Drugs 2006, 15, 579–602. [Google Scholar] [CrossRef]
- Dupont, B. Nouveaux antifongiques: voriconazole et caspofungine. Arch. de pédiatrie 2003, 10, 592s–598s. [Google Scholar] [CrossRef]
- Barchiesi, F.; Arzeni, D.; Camiletti, V.; Simonetti, O.; Cellini, A.; Offidani, A.M.; Scalise, G. In vitro Activity of Posaconazole against Clinical Isolates. J. Clin. Microbiol. 2001, 139, 4208–4209. [Google Scholar]
- Marco, F.; Pfaller, M.A.; Messer, S.A.; Jones, R.N. In vitro activity of a new triazole antifungal agent, Sch 56592, against clinical isolates of filamentous fungi. Mycopathologia 1998, 141, 73–77. [Google Scholar] [CrossRef]
- Sugar, A.M.; Liu, X.-P. In vitro and In vivo Activities of SCH 56592 against Blastomyces dermatitidis. Antimicrob. Agents Chemother. 1996, 40, 1314–1316. [Google Scholar]
- Lutz, J.E.; Clemons, K.V.; Aristizabal, B.H.; Stevens, D.A. Activity of the Triazole SCH 56592 against Disseminated Murine Coccidiodomycosis. Antimicrob. Agents Chemother. 1997, 41, 1558–1561. [Google Scholar]
- Connolly, P.; Wheat, L.J.; Schizlein-Bick, C.; Durkin, M.; Kohler, S.; Smedema, M.; Goldberg, J.; Brizendine, E.; Loebenberg, D. Comparison of a New triazole, Posaconazole with Itraconazole and Amphotericin B for Treatment of Histoplasmosis following Pulmonary Challenge in Immunocompromised Mice. Antimicrob. Agents Chemother. 2000, 44, 2604–2608. [Google Scholar]
- Connolly, P.; Wheat, J.; Schizlein-Bick, C.; Durkin, M.; Kohler, S.; Smedema, M.; Goldberg, J.; Brizendine, E.; Loebenberg, D. Comparison of a New triazole Antifungal agent, Schering 56592 with Itraconazole and Amphotericin B for Treatment of Histoplasmosis in Immunocompetent Mice. Antimicrob. Agents Chemother. 1999, 43, 322–328. [Google Scholar]
- Cacciapuoti, A.; Loebenberg, D.; Corcoran, E.; Menzel, F., Jr.; Moss, E.L., Jr.; Norris, C.; Michalski, M.; Raynor, K.; Halpern, J.; Mendrick, C.; Arnold, B.; Antonacci, B.; Parmegiani, R.; Yarosh-Tomaine, T.; Miller, G.H.; Hare, R.S. In vitro and In vivo Activities of SCH 56592 (Posaconazole), a New Triazole Antifungal Agent, against Aspergillus and Candida. Antimicrob. Agents Chemother. 2000, 44, 2017–2022. [Google Scholar]
- Oakley, K.L.; Morrissey, G.; Denning, D.W. Efficacy of SCH-56592 in a Temporariy Neutropenic Murine Model of Invasive Aspergillosis with an Itraconazole-susceptible and Itraconazole-Resistant Isolate of Aspergillus fumigatus. Antimicrob. Agents Chemother. 1997, 41, 1504–1507. [Google Scholar]
- Graybill, J.R.; Bocanegra, R.; Najvar, L.K.; Luther, M.F.; Loebenberg, D. SCH56592 treatment of murine invasive aspergillosis. J. Antimicrob. Chemother. 1998, 42, 539–542. [Google Scholar] [CrossRef]
- Lozano-Chiu, M.; Arikan, S.; Paetznick, V.L.; Anaissie, E.J.; Loebenberg, D.; Rex, J.H. Treatment of Murine Fusariosis with SCH 56592. Antimicrob. Agents Chemother. 1999, 43, 589–591. [Google Scholar] [CrossRef]
- Nomeir, A.A.; Kumari, P.; Hilbert, M.J.; Gupta, S.; Loebenberg, D.; Cacciapuoti, A.; Hare, R.; Miller, G.H.; Lin, C.C.; Cayen, M.N. Pharmacokinetics of SCH 56592, a New Azole Broad-Spectrum Antifungal Agent, in Mice, Rats, Rabbits, Dogs, and Cynomolgus Monkeys. Antimicrob. Agents Chemother. 2000, 44, 727–731. [Google Scholar] [CrossRef]
- Sansone-Parsons, A.; Krishna, G.; Calzetta, A.; Wexler, D.; Kantesaria, B.; Rosenberg, M.A.; Saltzman, M.A. Effect of a Nutritional Supplement on Posaconazolez Pharmacokinetics following Oral Administration to Healthy Volunteers. Antimicrob. Agents Chemother. 2006, 50, 1881–1883. [Google Scholar] [CrossRef]
- Chhun, S.; Rey, E.; Tran, A.; Lortholary, O.; Pons, G.; Jullien, V. Simultaneous quantification of voriconazole and posaconazole in human plasma by high-performance liquid chromatography with ultra-violet detection. J. Chromatogr. B 2007, 852, 223–228. [Google Scholar] [CrossRef]
- Stevens, D.A.; Rendon, A.; Gaona-Flores, V.; Catanzaro, A.; Anstead, G.M.; Pedicone, L.; Graybill, J.R. Posaconazole Therapy for Chronic Refractory Coccidiodomycosis. Chest 2007, 132, 952–958. [Google Scholar] [CrossRef]
- Walsh, T.J.; Raad, I.; Patterson, T.F.; Chandrasekar, P.; Donowitz, G.R.; Graybill, J.R.; Greene, R.E.; Hachem, R.; Hadley, S.; Herbrecht, R.; Langston, A.; Louie, A.; Ribaud, P.; Segal, B.H.; Stevens, D.A.; van Burik, J.-A.H.; White, C.S.; Corcoran, G.; Gogate, J.; Krishna, G.; Pedicone, L.; Hardalo, C.; Perfect, J.R. Treatment of Invasive Aspergillosis with Posaconazole in Patients Who Are Refractory to or Intolerant of Conventional Therapy: An Externally Controlled Trial. Clin. Infect. Dis. 2007, 44, 2–12. [Google Scholar]
- Raad, I.I.; Graybill, J.R.; Bustamante, A.B.; Cornely, O.A.; Gaona-Flores, V.; Afit, C.; Graham, D.R.; Greenberg, R.N.; Hadley, S.; Langston, A.; Negroni, R.; Perfect, J.R.; Pitisuttithum, P.; Restrepo, A.; Schiller, G.; Pediconi, L.; Ullmann, A.J. Safety of long-term oral posaconazole use in the treatment of refractory invasive fungal infections. Clin. Infect. Dis. 2006, 42, 1726–1734. [Google Scholar] [CrossRef]
- Hof, H. A new, broad-spectrum azole antifungal: posaconazole- mechanism of action and resistance, spectrum of activity. Mycoses 2006, 49, 2–6. [Google Scholar]
- Vanden Bossche, H.; Marichal, P. Mode of action of anti-Candida drugs: Focus on terconazole and other ergosterol biosynthesis inhibitors. Am. J. Obstet. Gynecol. 1991, 165, 1193–1199. [Google Scholar]
- Boiron, P. Azole Antifungals. Infect. Dis. Ther. 1995, 17, 327–349. [Google Scholar]
- Vanden Bossche, H. Biochemical targets for antifungal azole derivatives: hypothesis on the mode of action. In Current Topics in Medical Mycology; McGinnis, M.R., Ed.; Springer-Verlag: NewYork, NY, USA, 1985; Volume 1, pp. 131–351. [Google Scholar]
- Georgopapadakou, N.H.; Dix, B.A.; Smith, S.A.; Freudenberger, J.; Funke, P.T. Effect of Antifungal Agents on Lipid Biosynthesis and Membrane Integraty in Candida albicans. Antimicrob. Agents Chemother. 1987, 31, 46–51. [Google Scholar] [CrossRef]
- Vanden Bossche, H.; Lauwers, W.; Willemsens, G.; Marichal, P.; Cornelissen, F; Cools, W. Molecular Basis for the Antimycotic and Antibacterial Activity of N-Substituted Imidazoles and Triazoles: the Inhibition of Isoprenoid Biosynthesis. Pestic. Sci. 1984, 15, 188–198. [Google Scholar] [CrossRef]
- Sud, J.J.; Feingold, D.S. Mechanism of action of the antimycotic imidazoles. J. Invest. Dermatol. 1981, 76, 438–441. [Google Scholar]
- Beggs, W.H. Mechanism of the rapid methal action of miconazole and sulconazole against Candida albicans. Biochem. Arch. 1994, 10, 117–121. [Google Scholar]
- Beggs, W.H. Rapid fungicidal action of tioconazole and miconazole. Mycopathologica 1987, 97, 187–188. [Google Scholar] [CrossRef]
- De Nollin, S.; Van Belle, H.; Goossens, F.; Thone, F.; Borgers, M. Cytochemical and biochemical studies of yeasts after in vitro exposure to miconazole. Antimicrob. Agents Chemother. 1977, 11, 500–513. [Google Scholar] [CrossRef]
- Shigematsu, M.L.; Arai, T. Effect of ketoconazole on isolated mitochondria from Candida albicans. Antimicrob. Agents Chemother. 1982, 21, 919–924. [Google Scholar] [CrossRef]
- Niemegeers, C.J.E.; Levron, J.C.; Awouters, F.; Janssen, P.A.J. Inhibition and Induction of Microsomal Enzymes in the Rat. A Comparative Study of Four Antimycotics: Miconazole, Econazole, Clotrimazole and Ketoconazole. Arch. Int. Pharmacodyn. 1981, 251, 26–38. [Google Scholar]
- Pfaller, M.A.; Riley, J.; Koerner, T. Effects of Terconazole and Other Azole Antifungal Agents on the Sterol and Carbohydrate Composition of Candida Albicans. Diagn. Microbiol. Infect. Dis. 1990, 13, 31–35. [Google Scholar] [CrossRef]
- Verweij, P.E.; Snelders, E.; Melchers, W.J.G. Azole resistance in Aspergillus fumigatus. In Aspergillus in the Genomic Era; Varga, J., Samson, R.A., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2008; pp. 277–291. [Google Scholar]
- Talele, T.T.; Hariprasad, V.; Kulkarni, V.M. Docking Analysis of a Series of Cytochrome P-45014αDM Inhibiting Azole Antifungals. Drug Des. Discov. 1998, 15, 181–190. [Google Scholar]
- Tatele, T.T.; Kulkarni, V.M. Three-Dimensional Quantitative Structure-Activity Relationship (QSAR) and Receptor Mapping of Cytochrome P-45014αDM Inhibiting Azole Antifungal Agents. J. Chem. Inf. Comput. Sci. 1999, 39, 204–210. [Google Scholar] [CrossRef]
- Lewis, D.F.V.; Wiseman, A.; Tarbit, M.H. Molecular modeling of lanosterol 14α-demethylase (CYP51) from Sacharomyces cerevisiae via homology with CYP102, a unique bacterial cytochrome P450 isoform: Quantitative Structure-Activity Relationships (QSARs) within two related series of antifungal azole derivatives. J. Enzyme Inhib. 1999, 14, 175–192. [Google Scholar] [CrossRef]
- Johnston, A. The pharmacokinetics of voriconazole. Br. J. Clin. Pharmacol. 2003, 56, 1. [Google Scholar] [CrossRef]
- Chiou, C.C.; Groll, A.H.; Walsh, T.J. New Drugs and Novel Targets for Treatment of Invasive Fungal Infections in Patients with Cancer. The Oncologist 2000, 5, 120–135. [Google Scholar] [CrossRef]
- Arias, A.; Arevalo, M.P.; Adreu, A.; Rodriguez, C.; Sierra, A. Candida glabrata: In vitro susceptibility of 84 isolates to Eight Antifungal Agents. Chemotherapy 1996, 42, 107–111. [Google Scholar] [CrossRef]
- Niwano, Y.; Tabuchi, T.; Kanai, K.; Hamaguchi, H.; Uchida, K.; Yamaguchi, H. Short-Term Therapy of Experimental Tinea Pedis in Guinea Pigs with Lanoconazole, a New Imidazole Antimycotic Agent. Antimicrob. Agents Chemother. 1995, 39, 2353–2355. [Google Scholar]
- Niwano, Y.; Seo, A.; Kanai, K.; Hamaguchi, H.; Uchida, K.; Yamaguchi, H. Therapeutic efficacy of Lanoconazole, a New Imidazole Antimycotic Agent, for Experimental Cuteneous Candidiasis in Guinea Pigs. Antimicrob. Agents Chemother. 1994, 38, 2204–2206. [Google Scholar] [CrossRef]
- Ogata, M.; Matsumoto, H.; Hamada, Y.; Takehara, M.; Tawara, K. 1-[1-[2-(3-Chlorobenzyl)oxy]phenyl]vinyl]-1H-imidazole Hydrochloride, a New Potent Antifungal Agent. J. Med. Chem. 1983, 26, 768–770. [Google Scholar]
- Ogata, M.; Matsumoto, H.; Shimizu, S.; Kida, S.; Shiro, M.; Tawara, K. Synthesis and Antifungal Activity of New 1-Vinylimidazoles. J. Med. Chem. 1987, 30, 1348–1354. [Google Scholar] [CrossRef]
- Torres-Rodriguez, J.M.; Mendez, P.; Lopez-Jodra, O.; Morea, Y.; Espasa, M.; Jeminaz, T.; Lagunas, C. In Vitro Susceptibilities of Clinical Yeast Isolates to the New Antifungal Eberconazole Compared with Their Susceptibilities to Clotrimazole and Ketoconazole Antimicrob. Agents Chemother. 1999, 43, 1258–1259. [Google Scholar]
- Enoch, D.A.; Ludlam, H.A.; Brown, N.M. Invasive fungal infections: a review of epidemiology and management options. J. Med. Microbiol. 2006, 55, 809–818. [Google Scholar]
- Sahin, G.O.; Akova, M. Treatment of invasive infections due to rare or emerging yeasts and moulds. Exp. Opin. Pharmacother. 2006, 7, 1181–1190. [Google Scholar] [CrossRef]
- Nucci, M. Emerging moulds: Fusarium, Scedosporium and Zygomycetes in transplant recipients. Curr. Opin. infect. Dis. 2003, 16, 607–612. [Google Scholar] [CrossRef]
- Bartoli, J.; Turmo, E.; Alguero, M.; Boncompte, E.; Vericat, M.L.; Conte, L.; Ramis, J.; Merlos, M.; Garcia-Rafanell, J.; Forn, J. New azole antifungals. 2. Synthesis and antifungal activity of heterocyclecarbocamide derivatives of 3-amino-2aryl-1-azolyl-2-butanol. J. Med. Chem. 1998, 41, 1855–1868. [Google Scholar]
- Bartoli, J.; Turmo, E.; Alguero, M.; Boncompte, E.; Vericat, M.L.; Conte, L.; Ramis, J.; Merlos, M.; Garcia-Rafanell, J.; Forn, J. New azole antifungals. 3. Synthesis and antifungal activity of 3-substituted-4(3H)-quinazolinones. J. Med. Chem. 1998, 41, 1869–1882. [Google Scholar] [CrossRef]
- Pasqualotto, A.C.; Thiele, K.O.; Goldani, L.Z. Novel triazole antifungal drugs: focus on isavuconazole , ravuconazole and albaconazole. Curr. Opin. invest. D 2010, 11, 165–174. [Google Scholar]
- Fung-Tomc, J.C.; Huczko, E.; Minassian, B.; Bonner, D.P. In vitro activity of a new oral triazole, BMS-207147 (ER-30346). Antimicrob. Agents Chemother. 1998, 42, 313–318. [Google Scholar]
- Minassian, B.; Huczko, E.; Washo, T.; Bonner, D.; Fung-Tomc, J. In vitro activity of ravuconazole against zygomycetes, Scedosporium and Fusarium isolates. Clin. Microbiol. Infect. 2003, 9, 1250–1252. [Google Scholar] [CrossRef]
- Warn, P.A.; Sharp, A.; Denning, D.W. In vitro activity of a new triazole BAL4815, the active component of BAL8557 (the water-soluble prodrug), against Aspergillus spp. Antimicrob. Agents Chemother. 2006, 57, 135–138. [Google Scholar]
- Odds, F.C. Drug evaluation: BAL-8557--a novel broad-spectrum triazole antifungal. Curr. Opin. Invest. Drugs 2006, 7, 766–772. [Google Scholar]
- Guinea, J.; Bouza, E. Isavuconazole: a new and promising antifungal triazole for the treatment of invasive fungal infections. Future Microbiol. 2008, 3, 603–615. [Google Scholar] [CrossRef]
- Odds, F.; Ausma, J.; Van Gerven, F.; Woestenborghs, F.; Meerpoel, L.; Heeres, J.; Vanden Bossche, H.; Borgers, M. In vitro and in vivo activities of the novel azole antifungal agent R126638. Antimicrob. Agents Chemother. 2004, 48, 388–391. [Google Scholar]
- Vanden Bossche, H.; Ausma, J.; Bohets, H.; Vermuyten, K.; Willemsens, G.; Marichal, P.; Meerpoel, L.; Odds, F.; Borgers, M. The novel azole R126638 is a selective inhibitor of ergosterol synthesis in Candida albicans, Trichophyton spp. and Microsporum canis. Antimicrob. Agents Chemother. 2004, 48, 3272–3278. [Google Scholar]
- Meerpoel, L.; Backx, L.J.J.; Van der Veken, L.J.E.; Heeres, J.; Corens, D.; De Groot, A.; Odds, F.C.; Van Gerven, F.; Woestenborghs, R.; Van Breda, A.; Oris, M.; Van Dorsselaer, P.; Willemsens, G.H.M.; Vermuyten, K.J.P.; Marichal, P.J.M.G.; Vanden Bossche, H.F.; Ausma, J.; Borgers, M. Synthesis and in vitro and in vivo Structure-Activity Relationships of Novel Antifungal Triazoles for Dermatology. J. Med. Chem. 2005, 48, 2184–2193. [Google Scholar]
- Geria, A.N.; Scheinfeld, N.S. Pramiconazole, a triazole compound for the treatment of fungal infections. IDrugs 2008, 11, 661–670. [Google Scholar]
- Ryder, N.S.; Mieth, H. Allylamine antifungal drugs. Curr. Top. Med. Mycol. 1992, 4, 158–88. [Google Scholar] [CrossRef]
- Krishnan-Natesan, S. Terbinafine: a pharmacological and clinical review. Expert Opin. Pharmacother. 2009, 10, 2723–2733. [Google Scholar] [CrossRef]
- Denning, D.W. Echinocandin antifungal drugs. Lancet 2003, 362, 1142–1151. [Google Scholar] [CrossRef]
- Turner, M.S.; Drew, R.H.; Perfect, J.R. Emerging echinocandins for treatment of invasive fungal infections. Expert Opin. Emerging Drugs 2006, 11, 231–250. [Google Scholar] [CrossRef]
- Odds, F.C. Sordarin antifungal agents. Expert Opin. Ther. Patents 2001, 11, 283–294. [Google Scholar] [CrossRef]
- Gargallo-Viola, D. Sordarins as antifungal compounds. Curr. Opin. Anti-Infective Invest. Drugs 1999, 1, 297–305. [Google Scholar]
- Richardson, M.; Lass-Flörl, C. Changing epidemiology of systemic fungal infections. Clin. Microbiol Infect. 2008, 14, 5–24. [Google Scholar] [CrossRef]
- Pfaller, M.A.; Diekema, D.J. Epidemiology of Invasive Mycoses in North America. Critical Rev. Microbiol. 2010, 36, 1–53. [Google Scholar] [CrossRef]
- Bruggemann, R.J.M.; Alffenaar, J.W.C.; Blijlevens, N.M.A.; Billaud, E.M.; Kosterink, J.G.; Verwey, W.P.E.; Burger, D.M. Clinical Relevance of the Pharmacokinetic Interaction of Azole Antifungal drugs with Other Coadministered Agents. Clin. Infect. Dis. 2009, 48, 1441–1458. [Google Scholar] [CrossRef]
- Hardin, T.C.; Graybill, J.R.; Fetchick, R.; Woestenborghs, R.; Rinaldi, M.G.; Kuhn, J.G. Pharmacokinetics of itraconazole following oral administration to normal volunteers. Antimicrob. Agents Chemother. 1988, 32, 1310–1313. [Google Scholar]
- Zhang, A.Y.; Camp, W.L.; Elewski, B.E. Advances in Topical and Systemic Antifungals. Dermatol. Clin. 2007, 25, 165–183. [Google Scholar]
- Marichal, P.; Koymans, L.; Willemsens, S.; Bellens, D.; Verhasselt, P.; Luyten, W.; Borgers, M.; Ramaekers, F.C.S.; Odds, F.C.; Vanden Bossche, H. Contribution of mutations in the cytochrome-P450 14α -demethylase (Erg11p, cyp51p) to azole resistance in Candida albicans. Microbiology 1999, 145, 2701–2713. [Google Scholar]
- Marichal, P. Mechanisms of resistance to azole antifungal compounds. Curr. Op. Anti-Infect. Invest. Drugs 1999, 1, 318–333. [Google Scholar]
- Cannon, R.D.; Lamping, E.; Holmes, A.R.; Niimi, K.; Baret, P.V.; Keniya, M.V.; Tanabe, K.; Niimi, M.G.A.; Monk, B.C. Efflux-mediated antifungal drug resistance. Clin. Microbiol. Rev. 2009, 22, 291–321. [Google Scholar] [CrossRef]
- Peman, J.; Canton, E.; Espinel-Ingroff, A. Antifungal drug resistance mechanisms. Exp. Rev. Anti-Infect. 2009, 7, 453–460. [Google Scholar] [CrossRef]
- Hof, H. Is there a serious risk of resistance development to azoles among fungi due to the widespread use and long-term application of azole antifungals in medicine? Drug Resist. Updates 2008, 11, 25–31. [Google Scholar] [CrossRef]
- Ferreira da Silva, M.E.; Colombo, A. L.; Paulsen, I.; Ren, Q.; Wortman, J.; Huang, J.; Goldman, M.H.S.; Goldman, G.H. The ergosterol biosynthesis pathway, transporter genes, and azole resistance in Aspergillus fumigates. Med. Mycol. 2005, 43, S313–S319. [Google Scholar]
- Sanglard, D. Resistance of human fungal pathogens to antifungal drugs. Curr. Opin. Microbiol. 2002, 5, 379–385. [Google Scholar]
- Rex, J.H.; Pfaller, M.A.; Galgiani, J.N.; Bartlett, M.S.; Espinel-Ingroff, A.; Ghannoum, M.A.; Lancaster, M.; Odds, F.C.; Rinaldi, M.G.; Walsh, T.J.; Barry, A.L. Development of interpretive breakpoints for anti-fungal susceptibility testing: conceptual framework and analysis of in vitro/in vivo correlation data for fluconazole, itraconazole, and Candida infections. Clin. Infect. Dis. 1997, 24, 235–247. [Google Scholar] [CrossRef]
- Odds, F.C. Should resistance to azole antifungals in vitro be interpreted as predicting clinical non-response? Drug Resist. Updates 1998, 1, 11–15. [Google Scholar] [CrossRef]
- Bates, D.W.; Yu, D.T. Clinical impact of drug-drug interactions with systemic azole antifungals. Drugs Today 2003, 39, 801–813. [Google Scholar] [CrossRef]
- Girmenia, C. New generation azole antifungals in clinical investigation. Expert Opin. Invest. Drugs 2009, 18, 1279–1295. [Google Scholar] [CrossRef]
- Wang, E.-J.; Lew, K.; Casciano, C.N.; Clement, R.P.; Johnson, W.W. Interaction of common azole antifungals with P glycoprotein. Antimicrob. Agents Chemother. 2002, 46, 160–165. [Google Scholar]
- Baciewicz, A.M.; Baciewicz, F.A. Ketoconazole and fluconazole drug interactions. Arch. Int. Med. 1993, 153, 1970–1976. [Google Scholar] [CrossRef]
© 2010 by the authors;
Share and Cite
Heeres, J.; Meerpoel, L.; Lewi, P. Conazoles. Molecules 2010, 15, 4129-4188. https://doi.org/10.3390/molecules15064129
Heeres J, Meerpoel L, Lewi P. Conazoles. Molecules. 2010; 15(6):4129-4188. https://doi.org/10.3390/molecules15064129
Chicago/Turabian StyleHeeres, Jan, Lieven Meerpoel, and Paul Lewi. 2010. "Conazoles" Molecules 15, no. 6: 4129-4188. https://doi.org/10.3390/molecules15064129
APA StyleHeeres, J., Meerpoel, L., & Lewi, P. (2010). Conazoles. Molecules, 15(6), 4129-4188. https://doi.org/10.3390/molecules15064129