Characterization of Amide Bond Conformers for a Novel Heterocyclic Template of N-acylhydrazone Derivatives


Abstract
:1. Introduction


2. Results and Discussion
2.1. Chemical Synthesis
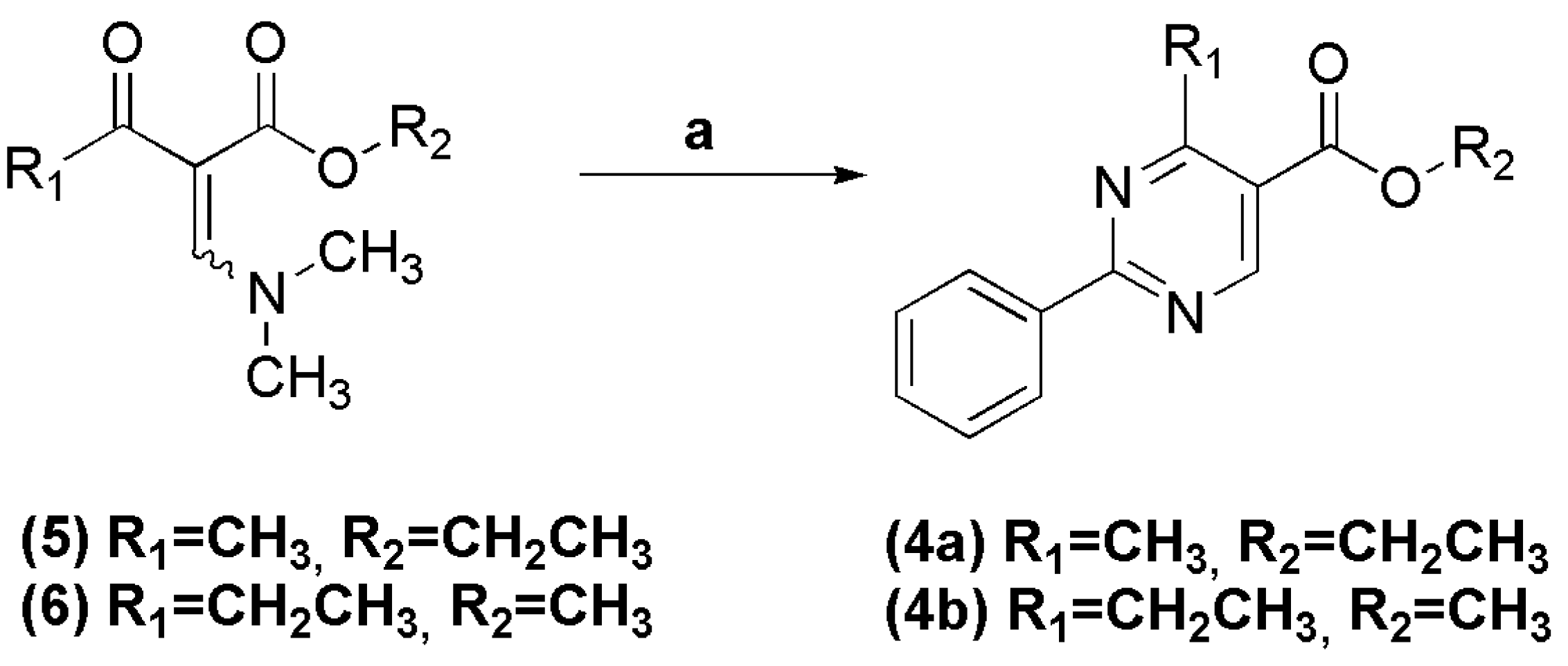
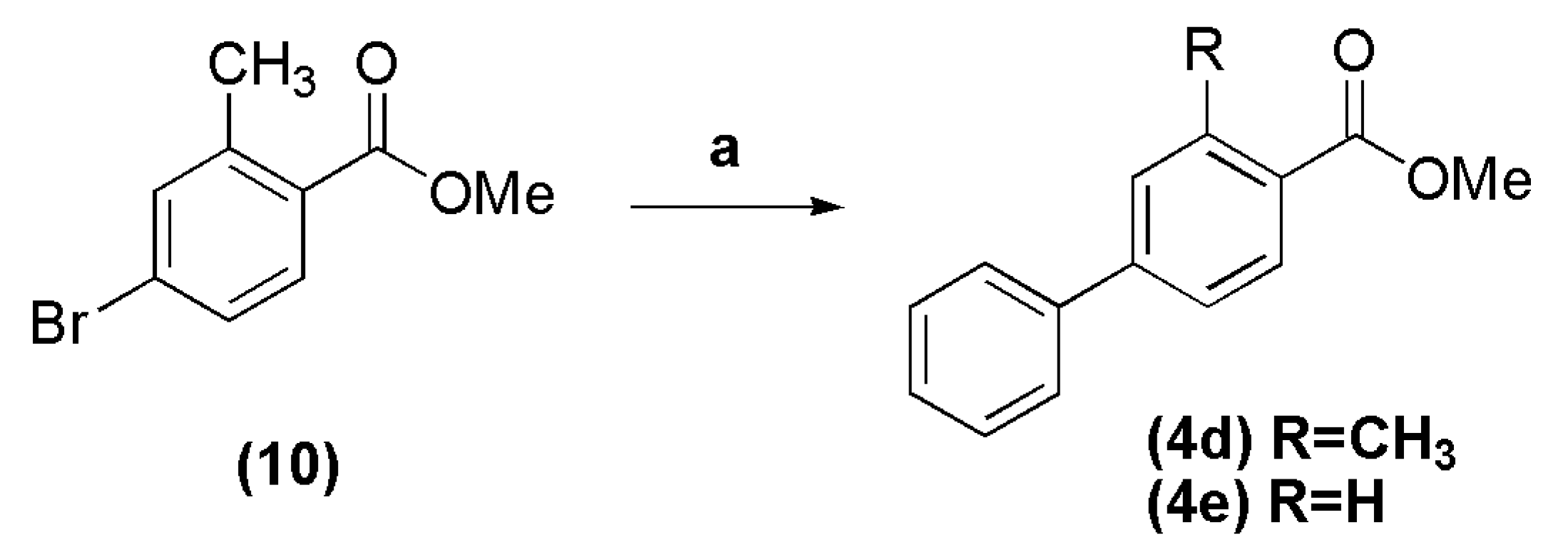
2.1.1. Synthesis of Esters 4a–e



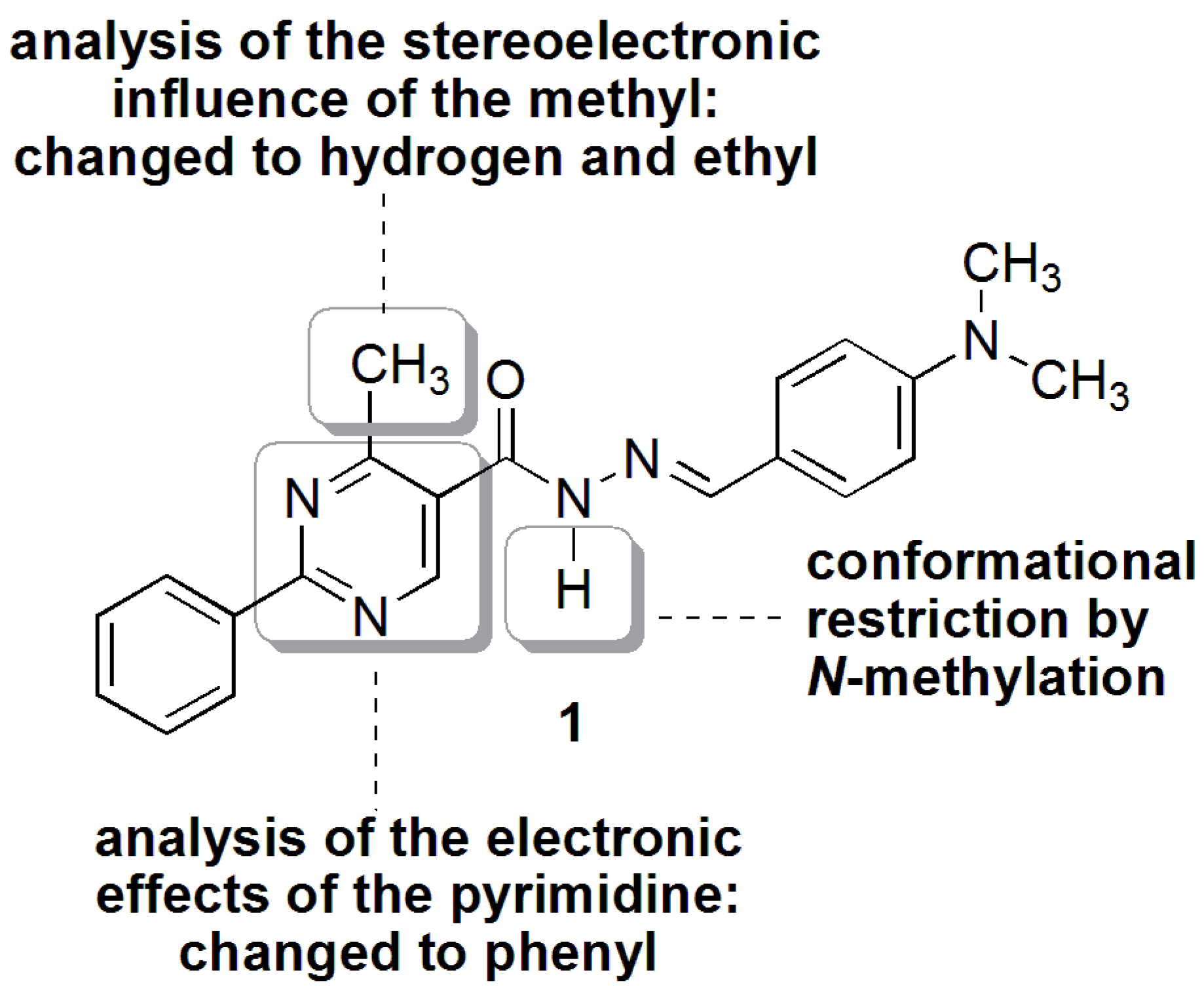
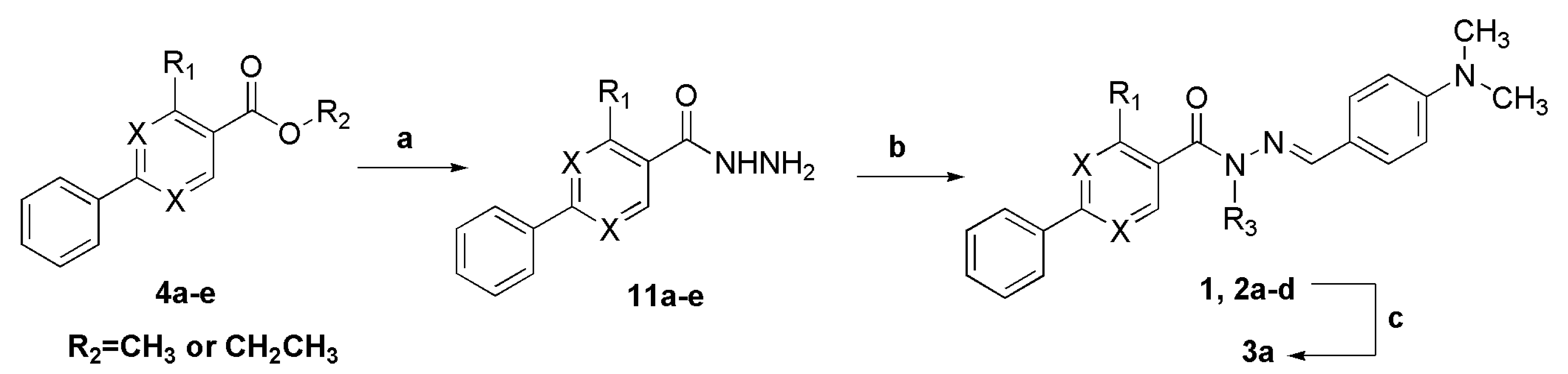
2.1.2. Synthesis of N-acylhydrazone Derivatives 1–3


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | X | R1 | R3 | Yield (%) a,b | Ratio a:b |
|---|---|---|---|---|---|
| 1 | N | CH3 | H | 66 | 2:1 |
| 2a | N | H | H | 21 | 4:1 |
| 2b | N | CH2CH3 | H | 49 | 1.7:1 |
| 2c | C | CH3 | H | 81 | 5:1 |
| 2d | C | H | H | 84 | c |
| 3a | N | CH3 | CH3 | 51 | c |
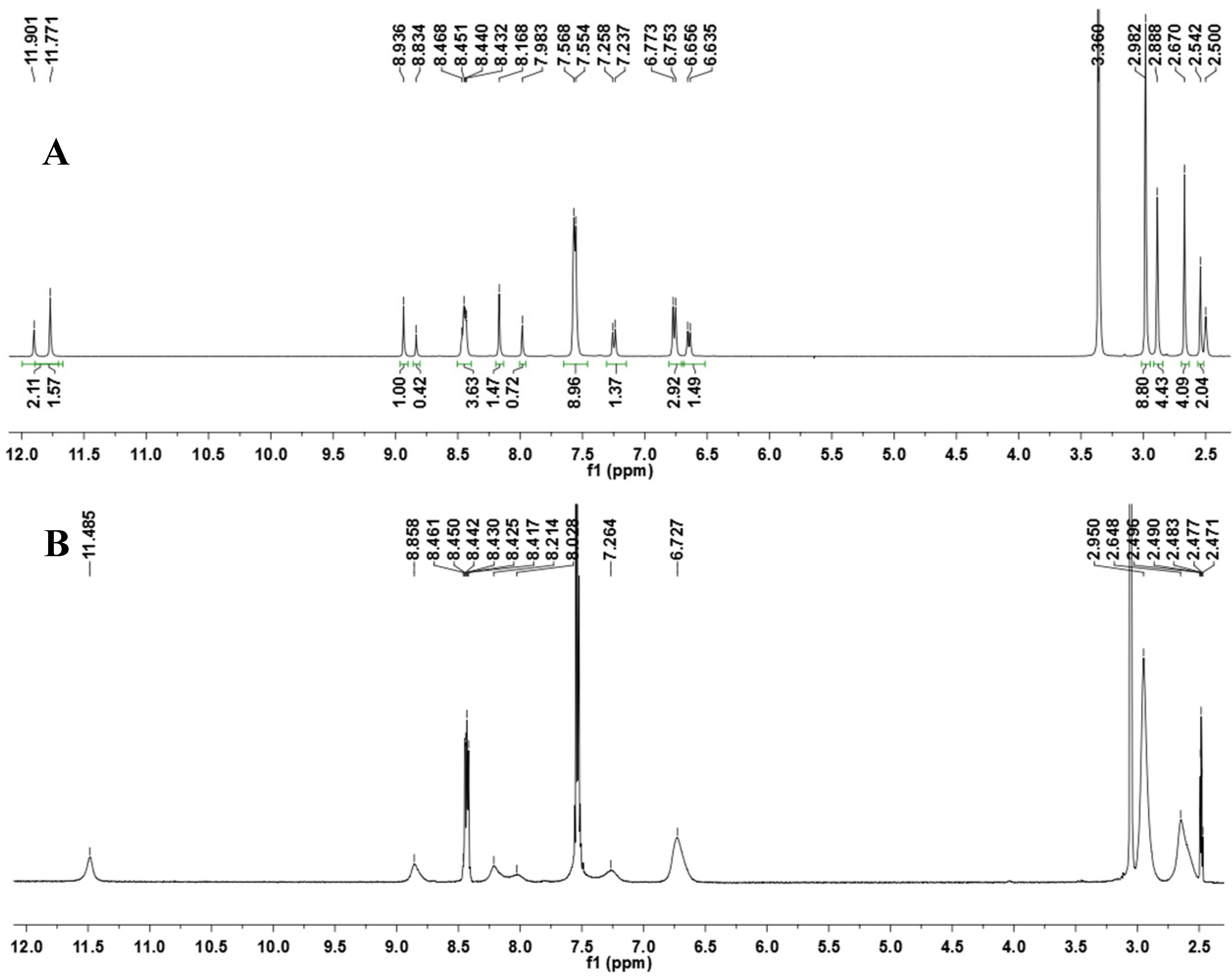
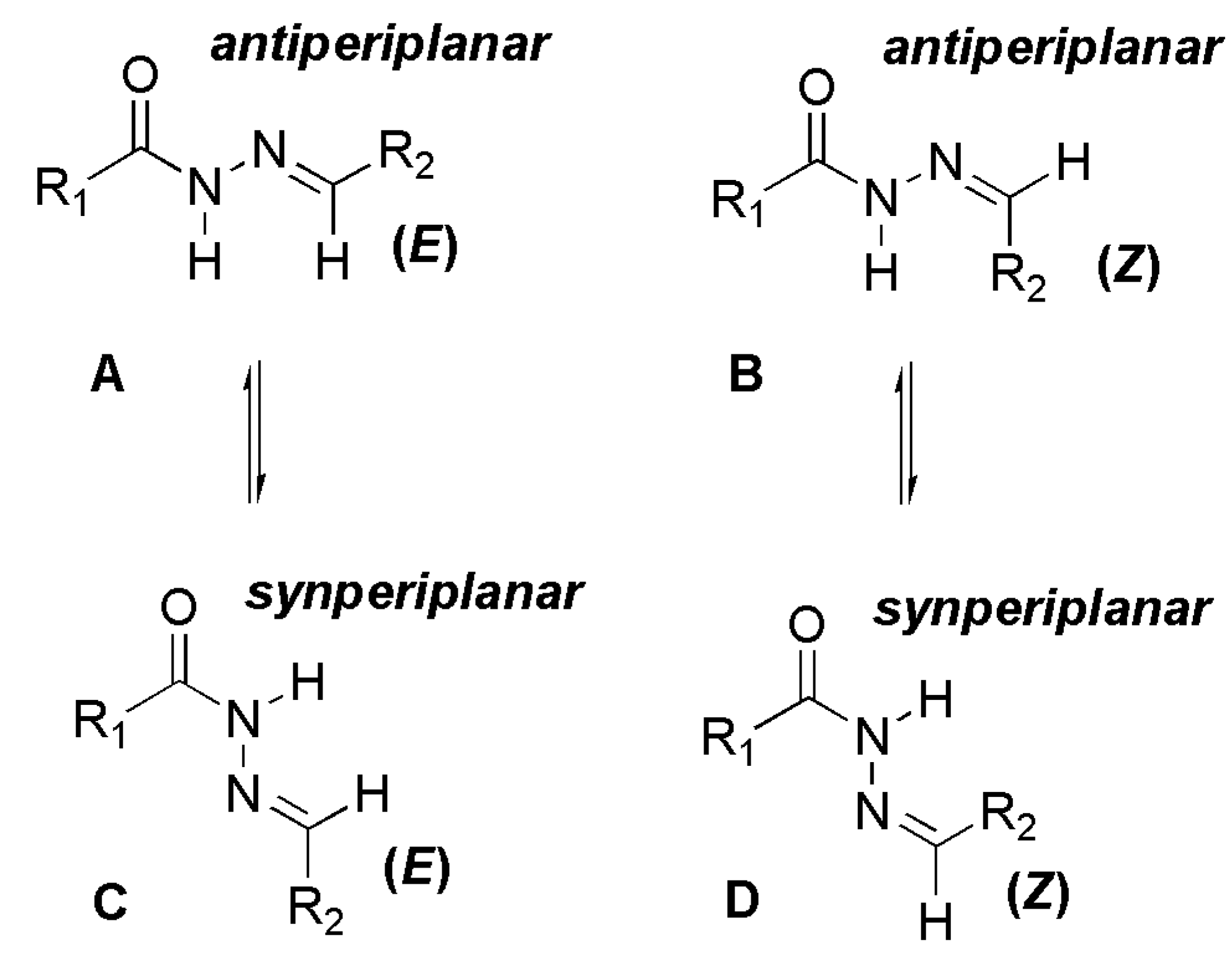
2.2. NAH Stereochemistry Elucidation
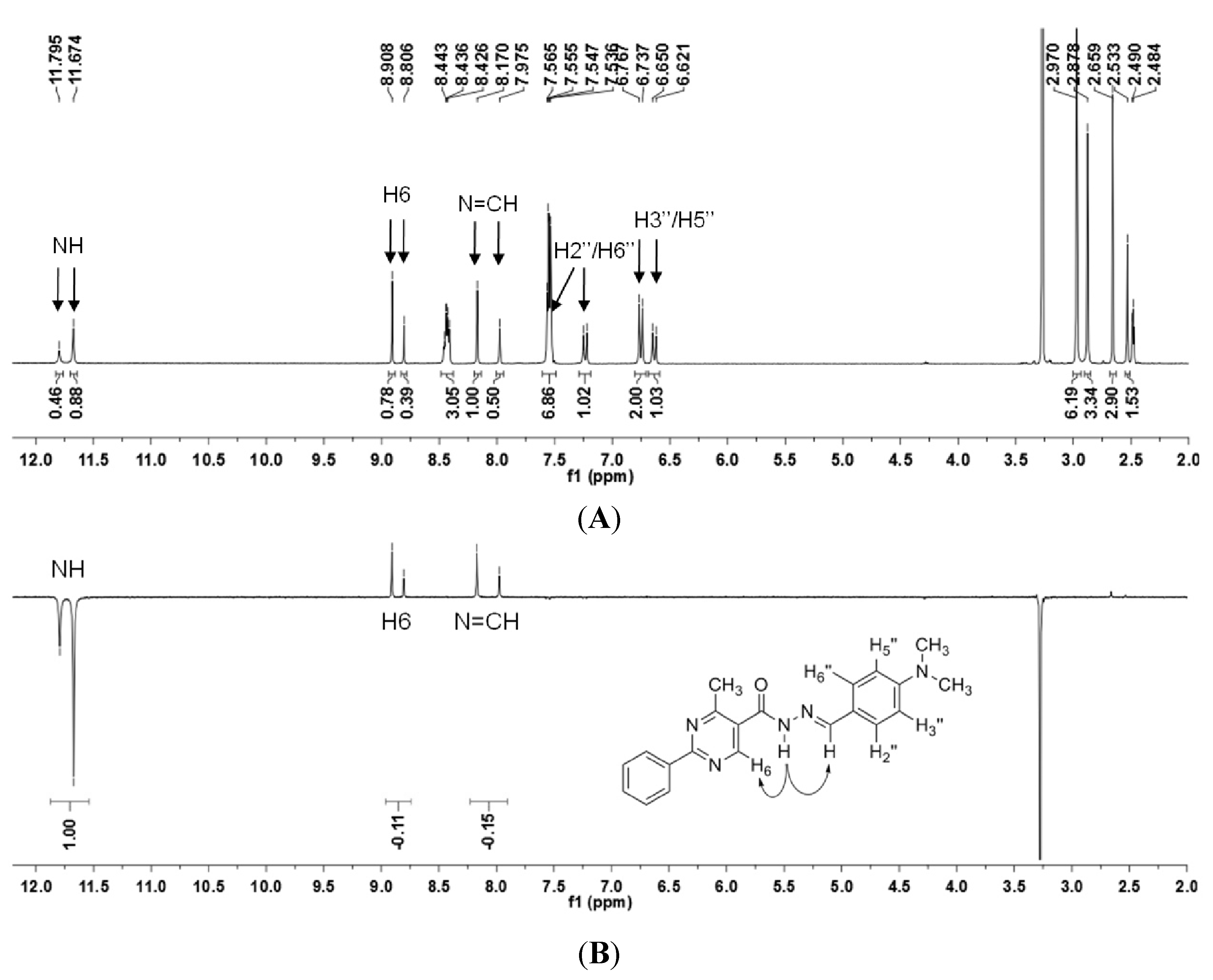
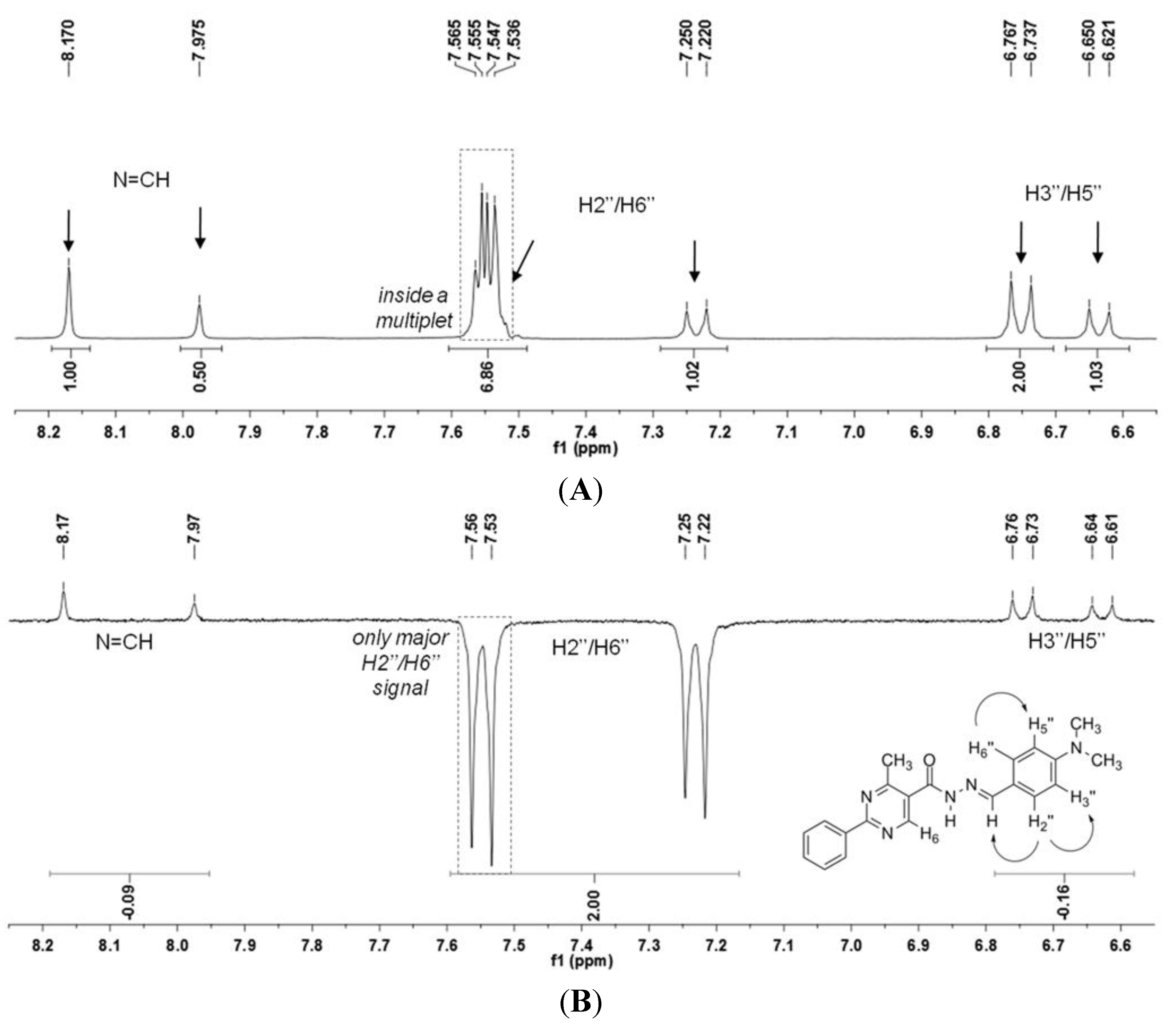
2.2.1. Determination of the Relative Configuration of the Imine Double Bond


| Form | N H–N=CH | N H–H6 | N H–H2′′ a | N H–H3′′ | N H–H6′′ a | N H–CH3 |
|---|---|---|---|---|---|---|
| A | 0.063 | 0.650 | 0.033 | - | - | 0.015 |
| B | 0.746 | 0.497 | 0.006 | - | 0.014 | 0.018 |
| C | 0.064 | 0.586 | 0.811 | 0.023 | 0.022 | 0.017 |

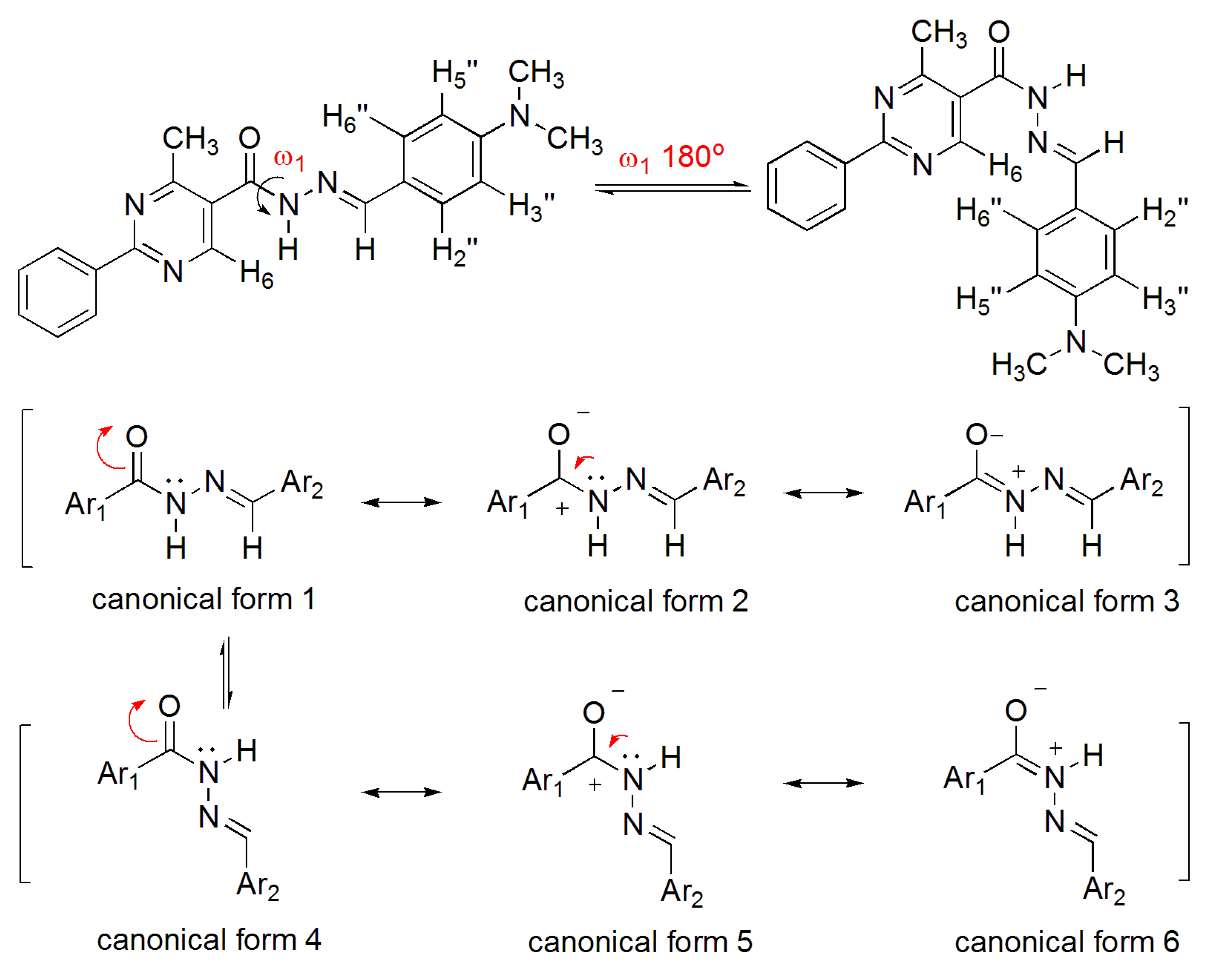
2.2.2. NAH Derivative 1 Conformers Determination



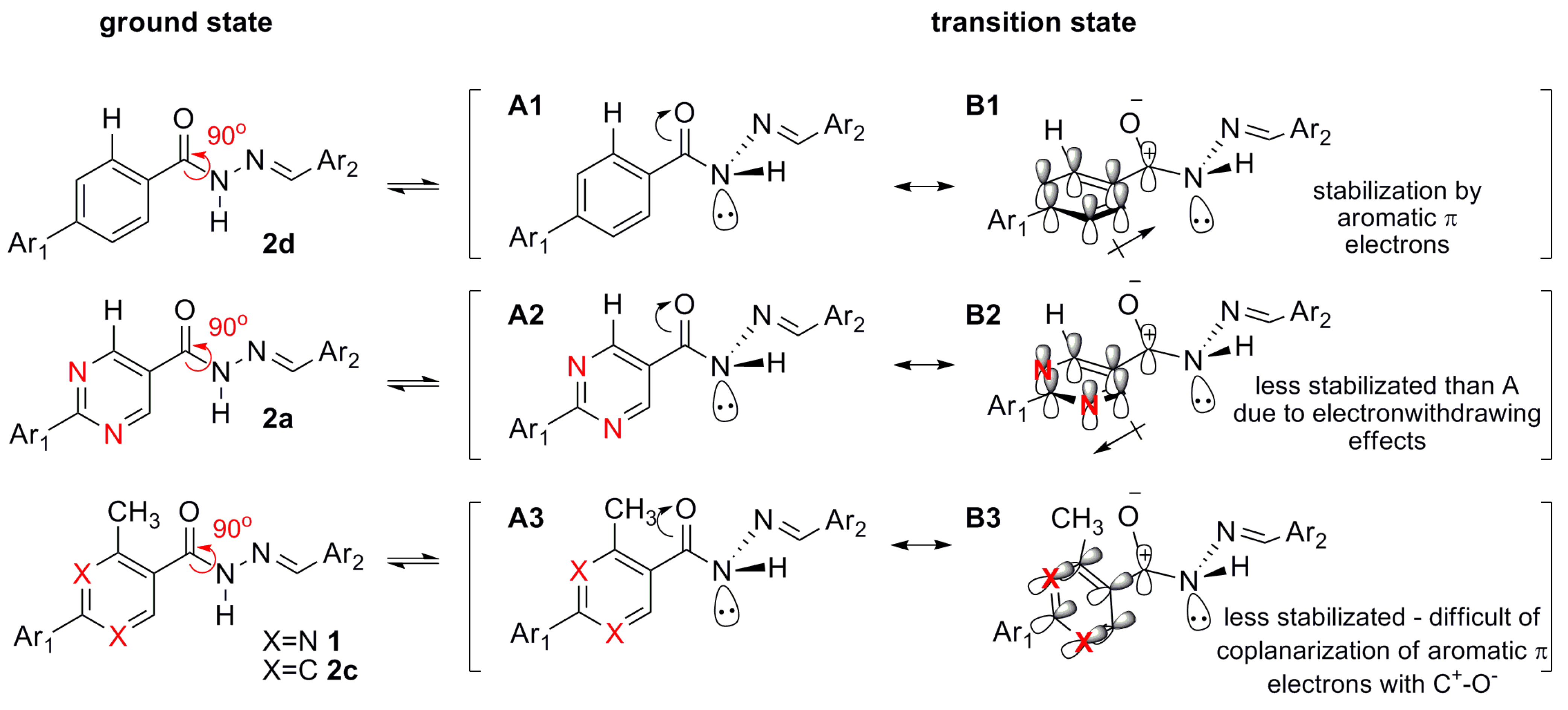
2.2.3. Stereoelectronic Effects on NAH Derivative (1) ap-sp Amide Bond Rotamers

| cpd a | CO-N H | H6 | N=C H | H2′′/H6′′ | H3′′/H5′′ | N(CH3)2 | R1 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 b | 11.67 (66) | 8.81 (34) | 7.98 (34) | 7.55 (66) | 7.24 (34) | 6.64 (34) | 2.88 (34) | 2.53(34) | ||||||
| 2a c | 11.87 (80) | 9.29 e | 8.02 (20) | 7.60–7.58 d (80) g | 7.4 (20) | 6.73 e | 2.95 (20) | - | ||||||
| 2b b | 11.74 (63) | 8.79 (37) | 7.9 (37) | 7.56–7.53 d (63) g | 7.21 (37) | 6.62 (37) | 2.87 (37) | - | ||||||
| 2c c | 11.51 (83) | 7.75–7.70 multiplet | 7.95 (17) | 7.61–7.39 d (83) g | 7.23 (17) | 6.64 (17) | 2.90 (17) | 2.34 (17) | ||||||
| 2d b | 11.60 | 8.00 f | 8.34 | 7.54–7.38 d | 6.77 | 2.98 | 8.00 f | |||||||

| cpd | Ring | Ortho group | δ C=O (ppm) |
|---|---|---|---|
| 1 | pyrimidine | Me | 167.42 |
| 2a | pyrimidine | H | 165.47 |
| 2b | pyrimidine | Et | 169.75 |
| 2c | phenyl | Me | 164.83 |
| 2d | phenyl | H | 162.91 |
| cpd | Long-range correlations for the amide N-H | |||
|---|---|---|---|---|
| 2JCH (maj) | 2JCH (min) | 3JCH (maj) | 3JCH (min) | |
| 1 | C=O | C=O | C=N | C=N, C-5 |
| 2a | C=O | C=O | C=N | C=N, C-5 |
| 2c | C=O | C=O | C=N | C=N, C-4 |
| 2d | C=O | a | C=N | a |
| cpd | Signal | Tc (K) | vc (Hz) | G≠ (kcal mol−1) |
|---|---|---|---|---|
| 1 | N H | 353.15 | 52.0 | 17.4 |
| 2a | N H | 313.15 | 16.52 | 16.1 |
| 2b | N H | 356.15 | 64.3 | 17.4 |
| 2c | N H | 313.15 | 12.12 | 16.3 |
3. Experimental
3.1. General Procedures
3.2. Ethyl 4-methyl-2-phenylpyrimidine-5-carboxylate (4a)
3.3. Methyl 4-Ethyl-2-Phenylpyrimidine-5-Carboxylate (4b)
3.4. Ethyl 2-phenylpyrimidine-5-carboxylate (4c)
3.5. Methyl 3-methyl-biphenyl-4-carboxylate (4d)
3.6. 4-Methyl-2-phenylpyrimidine-5-carbohydrazide (11a)
3.7. 4-Ethyl-2-phenylpyrimidine-5-carbohydrazide (11b)
3.8. 2-Phenylpyrimidine-5-carbohydrazide (11c)
3.9. 3-Methyl-biphenyl-4-carbohydrazide (11d)
3.10. Biphenyl-4-carbohydrazide (11e)
3.11. General Procedure to Synthesize the N-Acylhydrazones 1, 2a–d
3.12. (E)-N′-(4-(Dimethylamino)Benzylidene)-N,4-Dimethyl-2-Phenylpyrimidine-5-Carbohydrazide (3a)
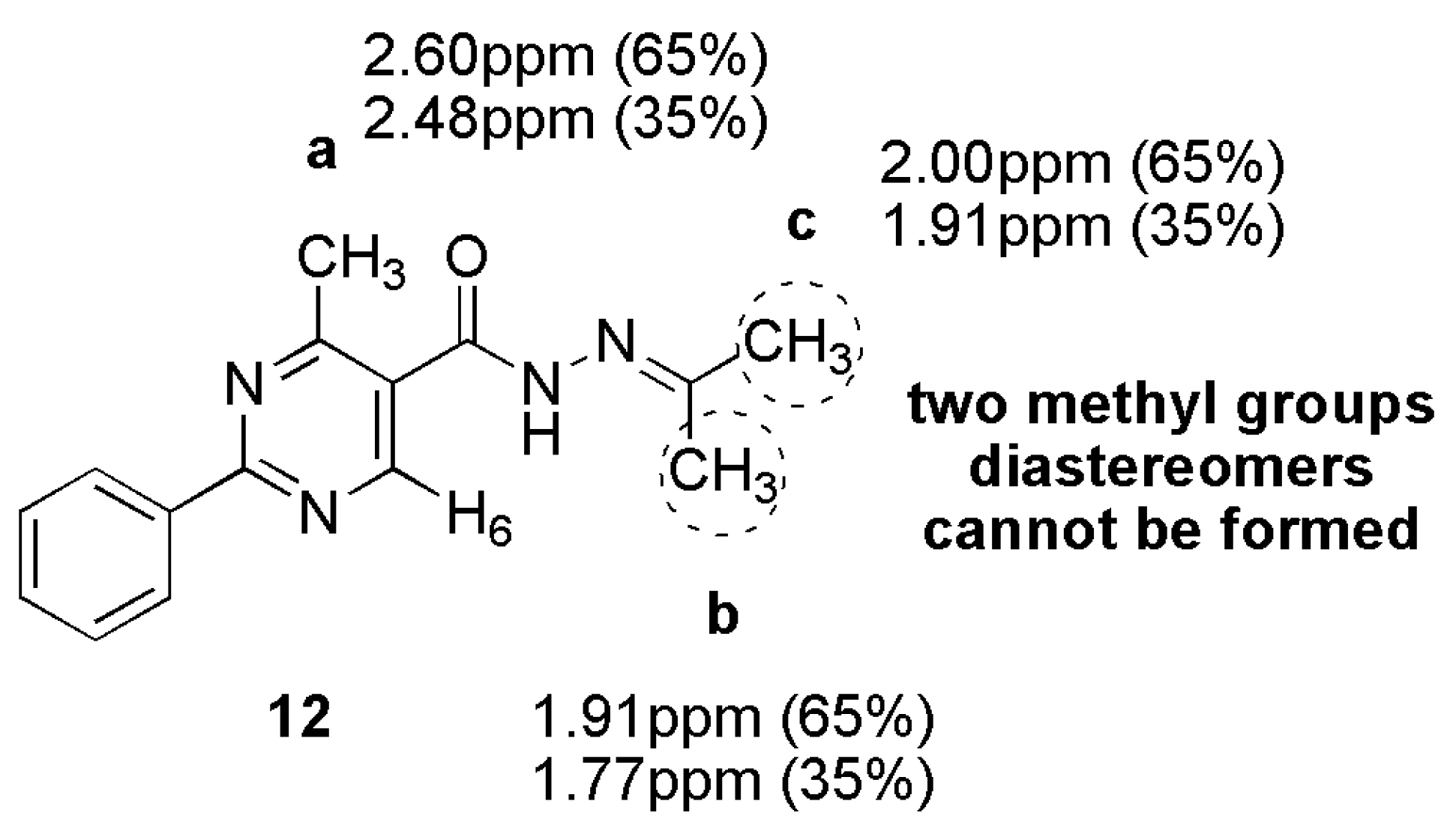
3.13. 4-Methyl-2-Phenyl-N’-(Propan-2-Ylidene)Pyrimidine-5-Carbohydrazide (12)
3.14. Molecular Modeling
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Duarte, C.D.; Barreiro, E.J.; Fraga, C.A.M. Privileged structures: A useful concept for the rational design of new lead drug candidates. Mini Rev. Med. Chem. 2007, 7, 1108–1119. [Google Scholar] [CrossRef]
- Diaz, P.; Phatak, S.S.; Xu, J.; Astruc-Diaz, F.; Cavasotto, C.N.; Naguib, M. 6-Methoxy-N-alkyl isatin acylhydrazone derivatives as a novel series of potent selective cannabinoid receptor 2 inverse agonists: Design, synthesis, and binding mode prediction. J. Med. Chem. 2009, 52, 433–444. [Google Scholar] [CrossRef]
- Tributino, J.L.M.; Santos, M.L.H.; Mesquita, C.M.; Lima, C.K.F.; Silva, L.L.; Maia, R.C.; Duarte, C.D.; Barreiro, E.J.; Fraga, C.A.M.; Castro, N.G.; et al. LASSBio-881: An N-acylhydrazone transient receptor potential vanilloid subfamily type 1 antagonist orally effective against the hypernociception induced by capsaicin or partial sciatic ligation. Br. J. Pharmacol. 2010, 159, 1716–1723. [Google Scholar] [CrossRef]
- Zapata-Sudo, G.; Pereira, S.L.; Beiral, H.J.V.; Kummerle, A.E.; Raimundo, J.M.; Antunes, F.; Sudo, R.T.; Barreiro, E.J.; Fraga, C.A.M. Pharmacological characterization of (3-thienylidene)-3,4-methylenedioxybenzoylhydrazide: A novel muscarinic agonist with antihypertensive profile. Am. J. Hypertens. 2010, 28, 135–141. [Google Scholar] [CrossRef]
- Palla, G.; Predieri, G.; Domiano, P. Conformational behaviour and E/Z isomerization of N-acyl and N-aroylhydrazones. Tetrahedron 1986, 42, 3649–3654. [Google Scholar] [CrossRef]
- Collier, H.; Dinneen, L.; Johnson, C.; Schneider, C. The abdominal constriction response and its suppression by analgesic drugs in the mouse. Br. J. Pharmacol. 1968, 32, 295–310. [Google Scholar]
- Palla, G.; Pelizzi, C.; Predieri, G.; Vignali, C. Conformational study on N-acylhydrazones of aromatic aldehydes by NMR spectroscopy. Gazz. Chim. Ital. 1982, 112, 339–341. [Google Scholar]
- Rahman, V.M.; Mukhtar, S.; Ansari, W.H.; Lemiere, G. Synthesis, stereochemistry and biological activity of some novel long alkyl chain substituted thiazolidin-4-ones and thiazan-4-one from 10- undecenoic acsid hydrazide. Eur. J. Med. Chem. 2005, 40, 173–184. [Google Scholar]
- Tian, B.; He, M.; Tang, S.; Hewlett, I.; Tan, Z.; Li, J.; Jin, Y.; Yang, M. Synthesis and antiviral activities of novel acylhydrazone derivatives targeting HIV-1 capsid protein. Bioorg. Med. Chem. Lett. 2009, 19, 2162–2167. [Google Scholar]
- Quattropani, A.; Dorbais, J.; Covini, D.; Pittet, P.-A.; Colovray, V.; Thomas, R.J.; Coxhead, R.; Halazy, S.; Scheer, A.; Missotten, M.; et al. Discovery and development of a new class of potent, selective, orally active oxytocin receptor antagonists. J. Med. Chem. 2005, 48, 7882–7905. [Google Scholar] [CrossRef]
- Schenone, P.; Sansebastiano, L.; Mosti, L. Reaction of 2-dimethylaminomethylene-1,3-diones with dinucleophiles. VIII. Synthesis of ethyl and methyl 2,4-disubstituted 5-pyrimidinecarboxylates. J. Heterocycl. Chem. 1990, 27, 295–305. [Google Scholar] [CrossRef]
- Golec, J.M.C.; Scrowston, R.M.; Dunleavy, M. Tricyclic heteroaromatic systems containing a bridgehead nitrogen atom. Part 3. [1,2,4]Triazolo[3′,4′:3,2]pyrazolo[3,4-d]pyrimidines, tetrazolo[1′,5′:1,5]pyrazolo[3,4-d]pyrimidines and pyrimido-[5′,4′: 4,5]pyrazolo[3,2-c][1,2,4]triazines. J. Chem. Soc. Perkin Trans. 1 1992, 2, 239–244. [Google Scholar]
- Tobe, M.; Isobe, Y.; Tomizawa, H.; Nagasaki, T.; Takahashi, H.; Fukasawa, T.; Hayashi, H. Discovery of quinazolines as a noved structural class of potent inhibitors of NFκB activation. Bioorg. Med. Chem. 2003, 11, 383–391. [Google Scholar]
- Brown, D.S.; Cumming, J.G.; Nash, I.A. Amide derivatives bearing a cyclopropylaminoacarbonyl substituent useful as cytokine inhibitors. WO/2005/061465. 2005. [Google Scholar]
- Bonnefous, C.; Vernier, J.; Hutchinson, J.H.; Gardner, M.F.; Cramer, M.; James, J.K.; Rowe, B.A.; Daggett, L.P.; Schaffhauser, H.; Kamenecka, T.M. Biphenyl-indanones: Allosteric potentiators of the metabotropic glutamate subtype 2 receptor. Bioorg. Med. Chem. Lett. 2005, 15, 4354–4358. [Google Scholar]
- Lima, P.C.; Lima, L.M.; Da Silva, K.C.M.; Léda, P.H.O.; Miranda, A.L.P.; Fraga, C.A.M.; Barreiro, E.J. Synthesis and analgesic activity of novel N-acylarylhydrazones and isosters, derived from natural safrole. Eur. J. Med. Chem. 2000, 35, 187–203. [Google Scholar]
- Kümmerle, A.E.; Raimundo, J.M.; Leal, C.M.; Da Silva, G.S.; Balliano, T.L.; Pereira, M.A.; de Simone, C.A.; Sudo, R.T.; Zapata-Sudo, G.; Fraga, C.A.M.; Barreiro, E.J. Studies towards the identification of putative bioactive conformation of potent vasodilator arylidene N-acylhydrazone derivatives. Eur. J. Med. Chem. 2009, 44, 4004–4009. [Google Scholar]
- Mspin 1.03 software, Mestrelab Research. University of Santiago de Compostela: Santiago de Compostela, Spain, 2009.
- PC Spartan Pro 1.0.5 software. Wavefunction, Inc.: Irvine, CA, USA, 2001.
- Costa, P.; Vasconcellos, M.; Pilli, R.; Pinheiro, S. Substâncias Carboniladas e Derivados; Bookman Editora: Porto Alegre, Brazil, 2003; p. 412. [Google Scholar]
- Anslyn, E.V.; Dougherty, D.A. Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, USA, 2006. [Google Scholar]
- Wiberg, K.B. The interaction of carbonyl groups with substituents. Acc. Chem. Res. 1999, 32, 922–929. [Google Scholar]
- Lauvergnat, D.; Hiberty, P.C. Role of conjugation in the stabilities and rotational barriers of formamide and thioformamide. An ab initio valence-bond study. J. Am. Chem. Soc. 1997, 119, 9478–9482. [Google Scholar] [CrossRef]
- Taha, A.N.; True, N.S. Experimental 1H NMR and computational studies of internal rotation of solvated formamide. J. Phys. Chem. A 2000, 104, 2985–2993. [Google Scholar]
- Olsen, R.A.; Liu, L.; Ghaderi, N.; Johns, A.; Hatcher, M.E.; Mueller, L.J. The amide rotational barriers in picolinamide and nicotinamide: NMR and ab Initio studies. J. Am. Chem. Soc. 2003, 125, 10125–10132. [Google Scholar]
- Kümmerle, A.E.; Schmitt, M.; Cardozo, S.V.S.; Lugnier, C.; Villa, P.; Lopes, A.B.; Romeiro, N.C.; Justiniano, H.; Martins, M.A.; Fraga, C.A.M.; et al. Design, synthesis, and pharmacological evaluation of N-acylhydrazones and novel conformationally constrained compounds as selective and potent orally active phosphodiesterase-4 inhibitors. J. Med. Chem. 2012, 55, 7525–7545. [Google Scholar]
- Wyrzykiewicz, E.; Prukala, D. New isomeric N-substituted hydrazones of 2-, 3- and 4-pyridinecarboxaldehydes. J. Heterocycl. Chem. 1998, 35, 381–387. [Google Scholar] [CrossRef]
- Barreiro, E.J.; Kümmerle, A.E.; Fraga, C.A.M. The methylation effect in medicinal chemistry. Chem. Rev. 2011, 111, 5215–5246. [Google Scholar] [CrossRef]
- Oki, M. Applications of Dynamic NMR Spectroscopy to Organic Chemistry; VCH Pub.: Weinheim, Germany, 1995; pp. 43–46. [Google Scholar]
- Marshall, J.L. Carbon-Carbon and Carbon-Proton NMR Couplings: Applications to Organic Stereochemistry and Conformational Analysis; VCH Pub.: Weinheim, Germany, 1983. [Google Scholar]
- Sandstrom, J. Dynamic NMR Spectroscopy; Academic Press: New York, NY, USA, 1983. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lopes, A.B.; Miguez, E.; Kümmerle, A.E.; Rumjanek, V.M.; Fraga, C.A.M.; Barreiro, E.J. Characterization of Amide Bond Conformers for a Novel Heterocyclic Template of N-acylhydrazone Derivatives. Molecules 2013, 18, 11683-11704. https://doi.org/10.3390/molecules181011683
Lopes AB, Miguez E, Kümmerle AE, Rumjanek VM, Fraga CAM, Barreiro EJ. Characterization of Amide Bond Conformers for a Novel Heterocyclic Template of N-acylhydrazone Derivatives. Molecules. 2013; 18(10):11683-11704. https://doi.org/10.3390/molecules181011683
Chicago/Turabian StyleLopes, Alexandra Basilio, Eduardo Miguez, Arthur Eugen Kümmerle, Victor Marcos Rumjanek, Carlos Alberto Manssour Fraga, and Eliezer J. Barreiro. 2013. "Characterization of Amide Bond Conformers for a Novel Heterocyclic Template of N-acylhydrazone Derivatives" Molecules 18, no. 10: 11683-11704. https://doi.org/10.3390/molecules181011683
APA StyleLopes, A. B., Miguez, E., Kümmerle, A. E., Rumjanek, V. M., Fraga, C. A. M., & Barreiro, E. J. (2013). Characterization of Amide Bond Conformers for a Novel Heterocyclic Template of N-acylhydrazone Derivatives. Molecules, 18(10), 11683-11704. https://doi.org/10.3390/molecules181011683