1. Introduction
Previously we reported the transformation of diazonium hydrogen sulphate
2 derived from 2-amino-
N-methyl-
N-(3-methyl-1-phenyl-1
H-pyrazol-5-yl)benzamide (
1). This reaction was carried out with CuSO
4 and NaCl, in the presence of ascorbic acid as a reducing agent [
1] (
Scheme 1). The above mixture was used earlier by Hanson and co-workers [
2] to perform the Sandmeyer reaction on 4-chlorobenzendiazonium chloride in a homogeneous aqueous phase. Ascorbic acid reduces Cu(II) to Cu(I) which, in turn, reduces the diazonium ion to a diazenyl radical. The latter intermediate decomposes to dinitrogen and the phenyl radical. Copper(II) ions form complexes with chloride ions which are able to transfer a chloro radical to the 4-chlorophenyl by a ligand transfer process to afford 1,4-dichlorobenzene [
2]. The diazonium hydrogen sulfate
2 afforded neither the chloro derivative
3, the product of the classical Sandmeyer reaction, nor the tricyclic derivative
9, the expected product of the competitive Pschorr ring closure, and instead we obtained epimers
7 and
8 [
1].
Scheme 1.
Transformation of the diazonium salt 2 by reaction with CuSO4/NaCl/ascorbic acid.
Scheme 1.
Transformation of the diazonium salt 2 by reaction with CuSO4/NaCl/ascorbic acid.
Recently a re-investigation of the transformation reaction of diazonium salt
2 in the presence of CuSO
4/NaCl/ascorbic acid has allowed to establish that a demethylation process also takes place to give
N-(3-methyl-1-phenyl-1
H-pyrazol-5-yl)benzamide
4 (for the mechanism see
Scheme 5, below). Moreover, trace amounts of the hydroxy spiro derivatives
5 and
6 [
1] were detected by TLC analysis. In our continuing research on this reaction [
1,
3,
4,
5], we became interested in investigating the behaviour of substrates possessing an alkyl group, such as methyl, in lieu of the pyrazole N-phenyl, as well as a substituent at the 4-position of the pyrazole nucleus, in order to verify whether such modifications are able to influence the course of the reaction. In fact, phenyl and methyl groups exert different electronic as well as steric effects on the pyrazole ring. Moreover, electronic and steric effects engendered by the substituent at the 4-position of the pyrazole nucleus could also affect the reaction outcome. Here, we describe the CuSO
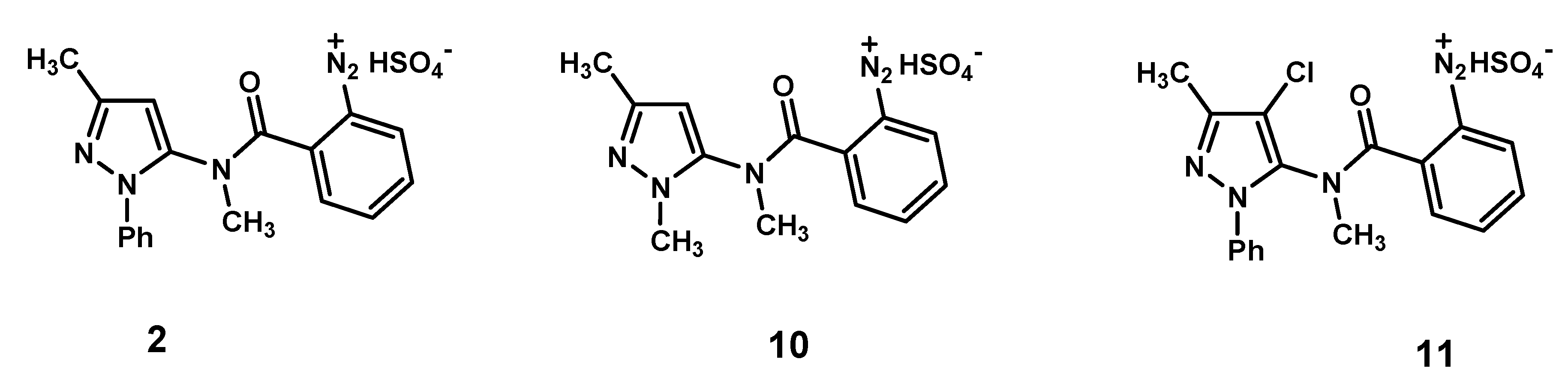
4/ascorbic acid-catalyzed decomposition in the presence of NaCl of the diazonium hydrogen sulfates
10 and
11 (
Figure 1). These intermediates bear a methyl group or a chloro atom on the pyrazole nucleus, at positions 1 or 4, respectively.
Figure 1.
Diazonium hydrogen sulfates 2, 10 and 11.
Figure 1.
Diazonium hydrogen sulfates 2, 10 and 11.
2. Results and Discussion
Benzendiazonium hydrogen sulfates derivatives
10 and
11 were prepared following
Scheme 2 and
Scheme 3.
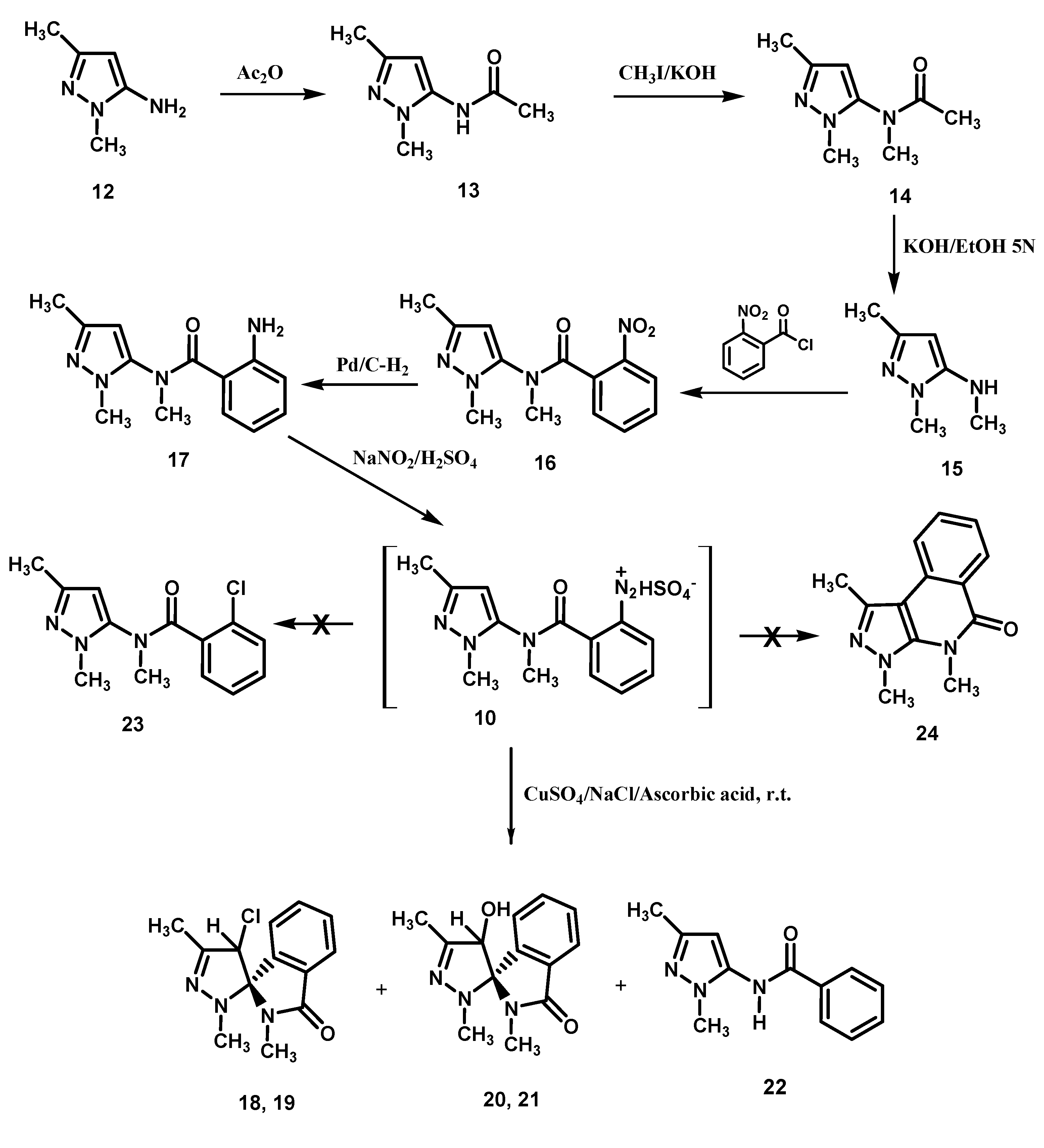
Scheme 2.
Preparation of the amino derivative 17 and its transformation.
Scheme 2.
Preparation of the amino derivative 17 and its transformation.
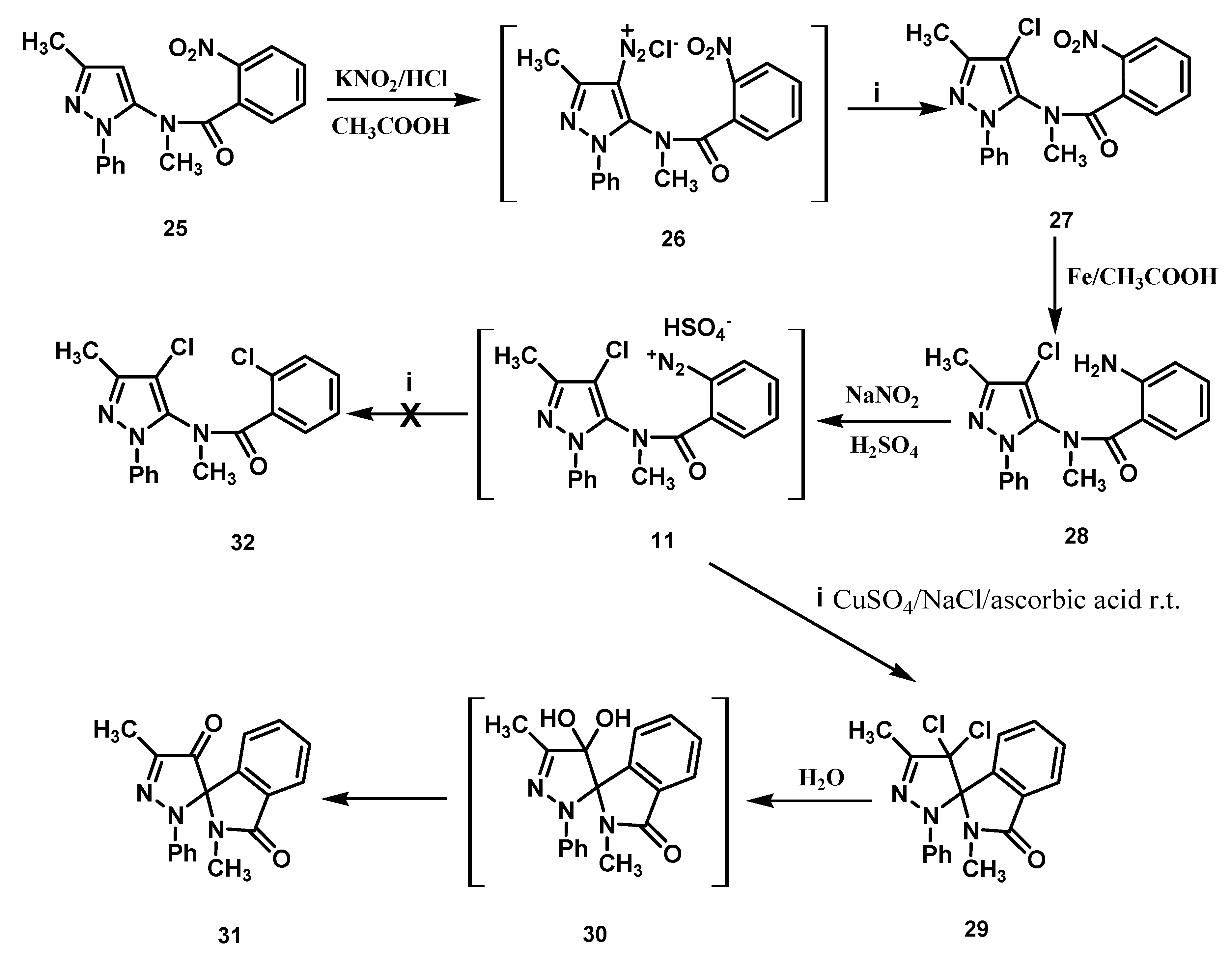
Scheme 3.
Preparation of the amino derivative 28 and its transformation.
Scheme 3.
Preparation of the amino derivative 28 and its transformation.
The amine
12 was reacted with acetic anhydride to give the acetamide derivative
13 which, in turn, was methylated with methyl iodide (
Scheme 2). The obtained di-substituted acetamide derivative
14 was hydrolyzed with a potassium hydroxide ethanol-water solution to give the pyrazol-5-amine
15.
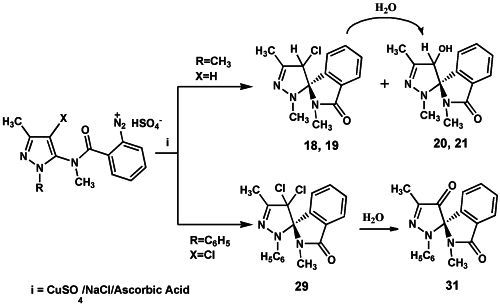
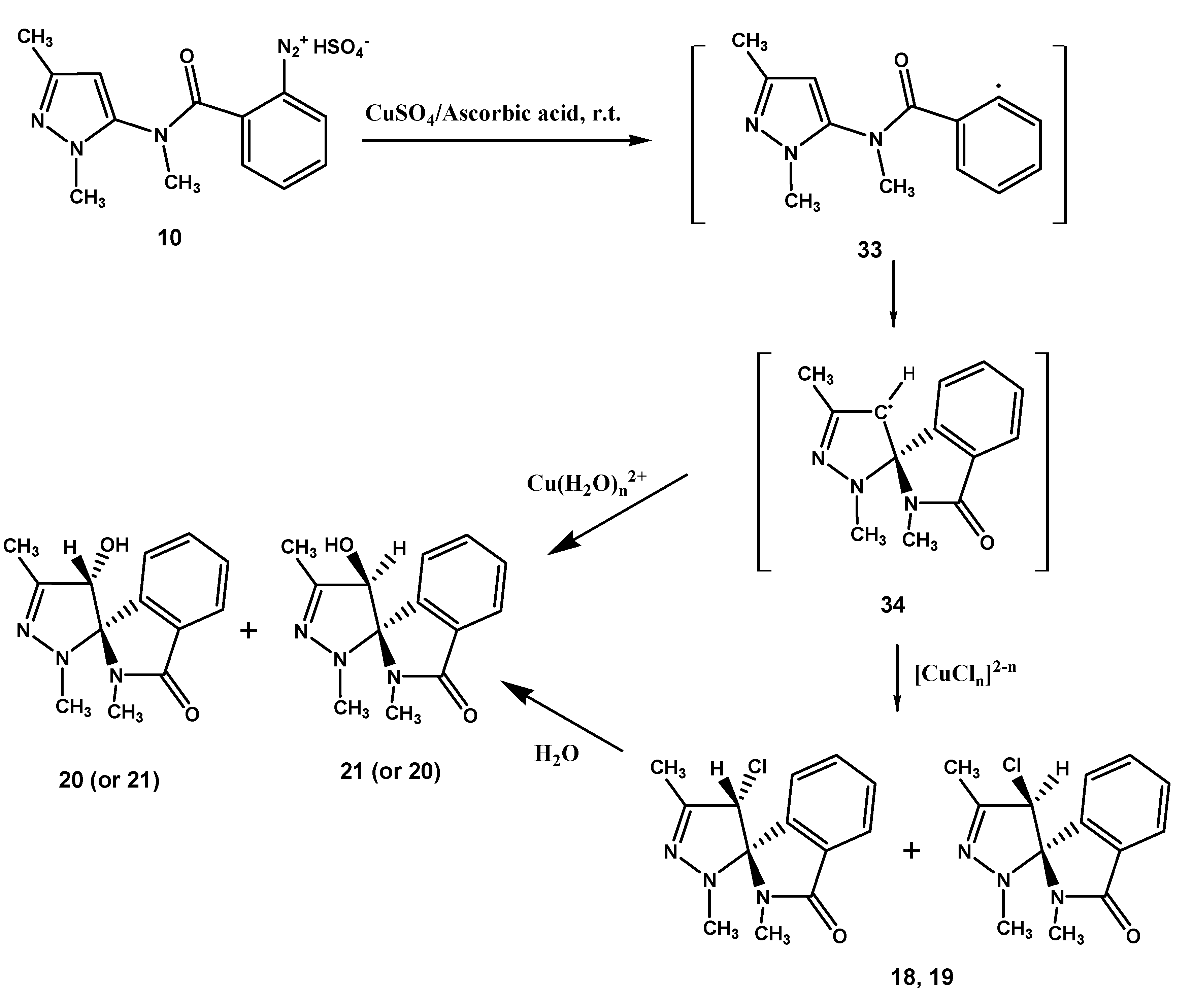
Condensation of 15 with 2-nitrobenzoyl chloride afforded the 2-nitrobenzamide derivative 16 which was reduced with hydrogen in the presence of palladium/activated charcoal catalyst to give the 2-amino-N-methylbenzamide derivative 17, diazotization of which produced the diazonium hydrogen sulfate 10. Exposure of 10 to CuSO4/NaCl/ascorbic acid, as seen earlier for 2, afforded a mixture of the chlorinated epimers (1S,4′R)- (or (1S,4′S)-) and (1S,4′S)- (or (1S,4′R)-) 18 and 19, respectively, and the hydroxyl epimers (1S,4′R)- (or (1S,4′S)-) and (1S,4′S)- (or (1S,4′R)-) 20 and 21, together obviously with the related enantiomers (not represented in the Schemes), and the benzamide derivative 22. Finally, no evidence was obtained for the formation of the chloro derivative 23, the product of the classical Sandmeyer reaction, as well as of 1,3,4-trimethylpyrazole[3,4-c]isoquinoline-5(4H)-one (24), the expected product of a possible competing Pschorr ring closure.
The preparation of substrate
11 started with the reaction of the 2-nitrobenzamide derivative
25 with a seven-fold excess of nitrous acid in acetic acid, resulting in the direct introduction of a diazo group at the 4-position of the pyrazole nucleus [
6,
7] (
Scheme 3). The transformation
in situ of the diazonium salt
26 into the chloro derivative
27 was performed with CuSO
4, NaCl and ascorbic acid [
1,
2]. Compound
27 was then reduced with iron in acetic acid, and the aniline derivative
28 thus obtained was diazotized to furnish
11.
The reaction of this diazonium salt under the same conditions employed earlier for
2 and
10 afforded the dichloro spiro derivative
29, as the sole product of reaction (
Scheme 3). Compound
29 was then converted into dione derivative
31, possibly via intermediate
30, upon refluxing in water. The new compounds were characterized by means of analytical and spectral data. The relative configuration of the pyrazoline C(4′)-atom of the spiro-compounds
18–
21 were not determined. The work up of the reaction mixture obtained by transformation of
10 did not allow us to isolate the epimers
18 and
19. The
1H-NMR spectrum of the obtained mixture showed singlets at 2.05, 2.07, 2.37, 2.44, 2.79 and 2.81 ppm, as well as at 5.54 and 5.83 ppm (ratio 1:2.25), consistent with the methyls and the pyrazoline H-4 of both epimers. On the contrary, epimers
20 and
21 were obtained as single compounds. The
1H-NMR spectrum of
20 showed three singlets at 1.94, 2.34 and 2.78 ppm, attributable to three methyls, and two doublets centered at 5.11 and 6.12 ppm for the pyrazoline H-4 and OH-4, respectively. Upon D
2O exchange the OH signal in the the spectrum disappeared, and the H-4 doublet collapsed to a singlet. The IR spectrum confirmed the presence of the pyrazoline hydroxyl by the absorption band at 3,263 cm
−1. The epimer
21 produced very similar IR and
1H-NMR spectra. The analogous compounds
22 and
4 were identified by comparison of their physical and spectroscopic data with authentic specimens (see the Experimental Section).
As regards
29, the
1H-NMR spectrum showed signals at 2.38 and 2.66 ppm for two methyls, as well as those in the range 6.70–7.96 ppm for nine aromatic protons. The assigned structure of compound
29 was confirmed by its
13C-NMR spectrum and by its chemical modification. In fact, the action of water on compound
29 afforded a product which was identical in all respects (mixed melting point, TLC, MS, IR) to dione derivative
31 [
1].
Rationalization of the formation of
18,
19,
20,
21 and
4,
22 is outlined in the
Scheme 4 and
Scheme 5. Formation of the chloro epimers
18 and
19 from
10 takes place
via the intermediates
33 and
34 (
Scheme 4). The latter transforms to
18 and
19 by a chloro transfer process from copper(II)-chloro complexes [
1,
2]. As regards the hydroxy spiro epimers
20 and
21, they might be afforded by two different mechanisms: by transfer of H
2O
+ from the hydratation shell of the aqueous copper(II)-complex [
8] to radical spiro intermediate
34, or by nucleophilic replacement of the chloro atom in
18 and
19 by attack of a molecule of water. We noted that the chloro epimers
7,
8 (
Scheme 1) and
18,
19 (
Scheme 2) were obtained as precipitates from the solutions of diazonium salts
2 and
10 respectively, being the yield for
7,
8 [
1] quite higher than that of
18,
19 (45%
versus 8%). Moreover, extraction of the mother liquors of the above reaction mixtures of
2 and
10 allowed us to obtain hydroxy spiro epimers only in the case of
10 (that is
20 and
21, yield 7%). Instead, only trace amounts of the hydroxy spiro epimers
5,
6 could be detected by TLC in the crude mixture of
7,
8. We also observed that the mixture of
18 and
19 could be transformed in
20 and
21 when it was reacted with cold (5 °C) 0.09 M sulfuric acid solution, where
18,
19 slowly dissolved, allowing the mixture to stand at r.t. for 1 h, mimicking thus the pH conditions of the reaction medium in which
10 transformed. When the same experiment was performed with the mixture of
7 and
8 no dissolution of the epimers in the acidic medium was observed. The suspension was extracted with ethyl acetate, and TLC of the extract did not reveal any transformation to give
5 and
6. At this point we realized that the low yield for
18,
19 is due to their solubility in the reaction medium, where they undergo a nucleophilic substitution by attack of a molecule of water to afford the hydroxy spiro epimers
20 and
21. Nevertheless, a radical transfer process of H
2O
+ to the spiro intermediate
34 to give
20,
21 can’t be ruled out (
Scheme 4).
Scheme 4.
Suggested mechanism for the transformation of 10.
Scheme 4.
Suggested mechanism for the transformation of 10.
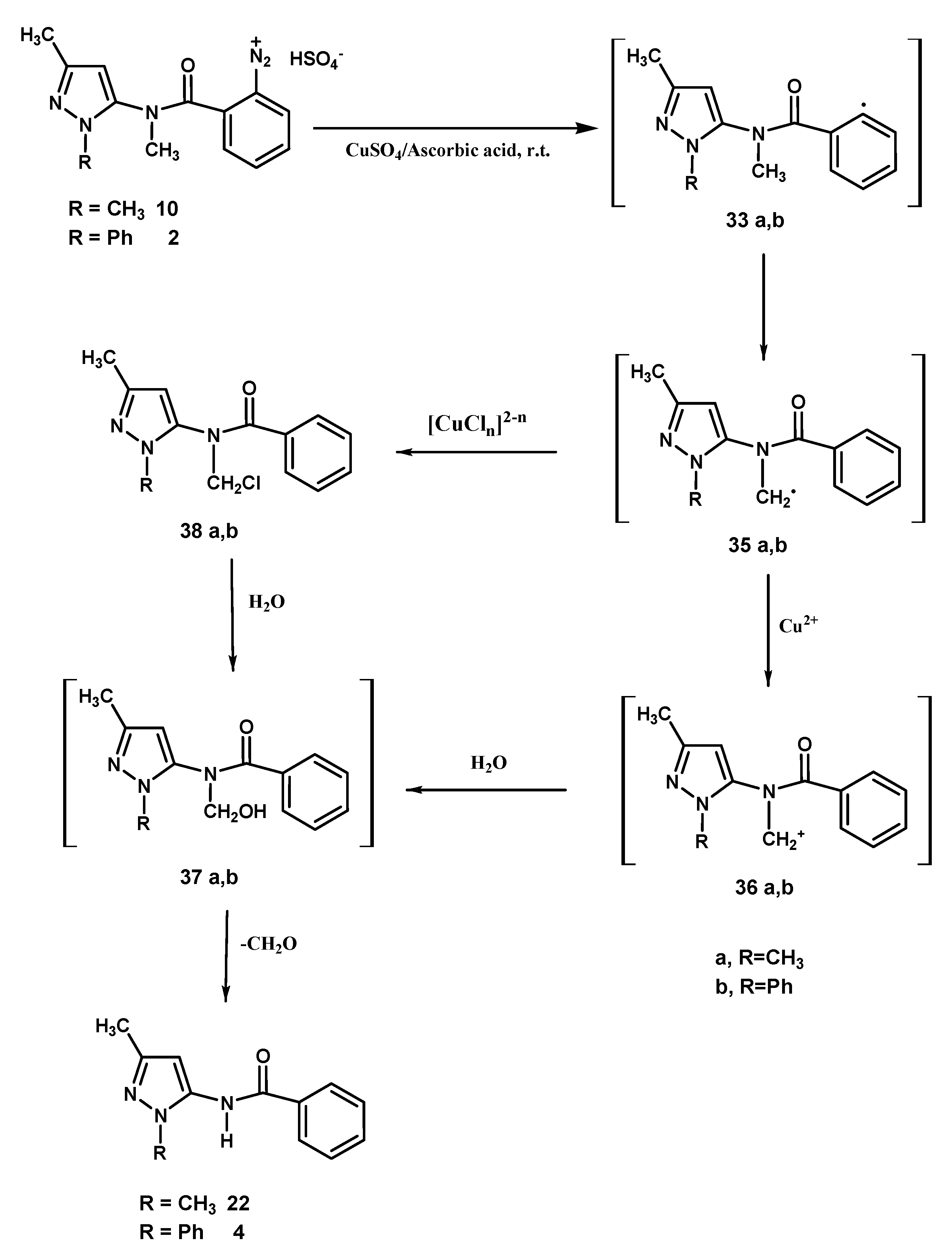
The latter process can be invoked for the formation of the trace amounts of
5 and
6. Lastly, compounds
4 and
22 (
Scheme 5) could be justified by considering a 1,5-hydrogen atom transfer process affording the radical intermediates
35a,
b which possibly undergo oxidation by cupric ions [
9] to give carbocations
36a,
b. The oxidation process would probably be favoured by the resonance stability of
36a,
b, but the direct ligand radical transfer route from
35a,
b to
38a,
b cannot be ruled out. Intermediates
36a,
b and
38a,
b react with water affording the unstable compounds
37a,
b which by loss of formaldehyde transform into benzamide derivatives
22 and
4, respectively.
Considering all the above data we concluded that the apparent different chemical behaviour between diazonium salts 2 and 10 is due to different solubilities of phenyl- and methyl-substituted chloro epimers, rather than to any electronic and steric effects of the N-phenyl- or N-methyl-pyrazole substituents.
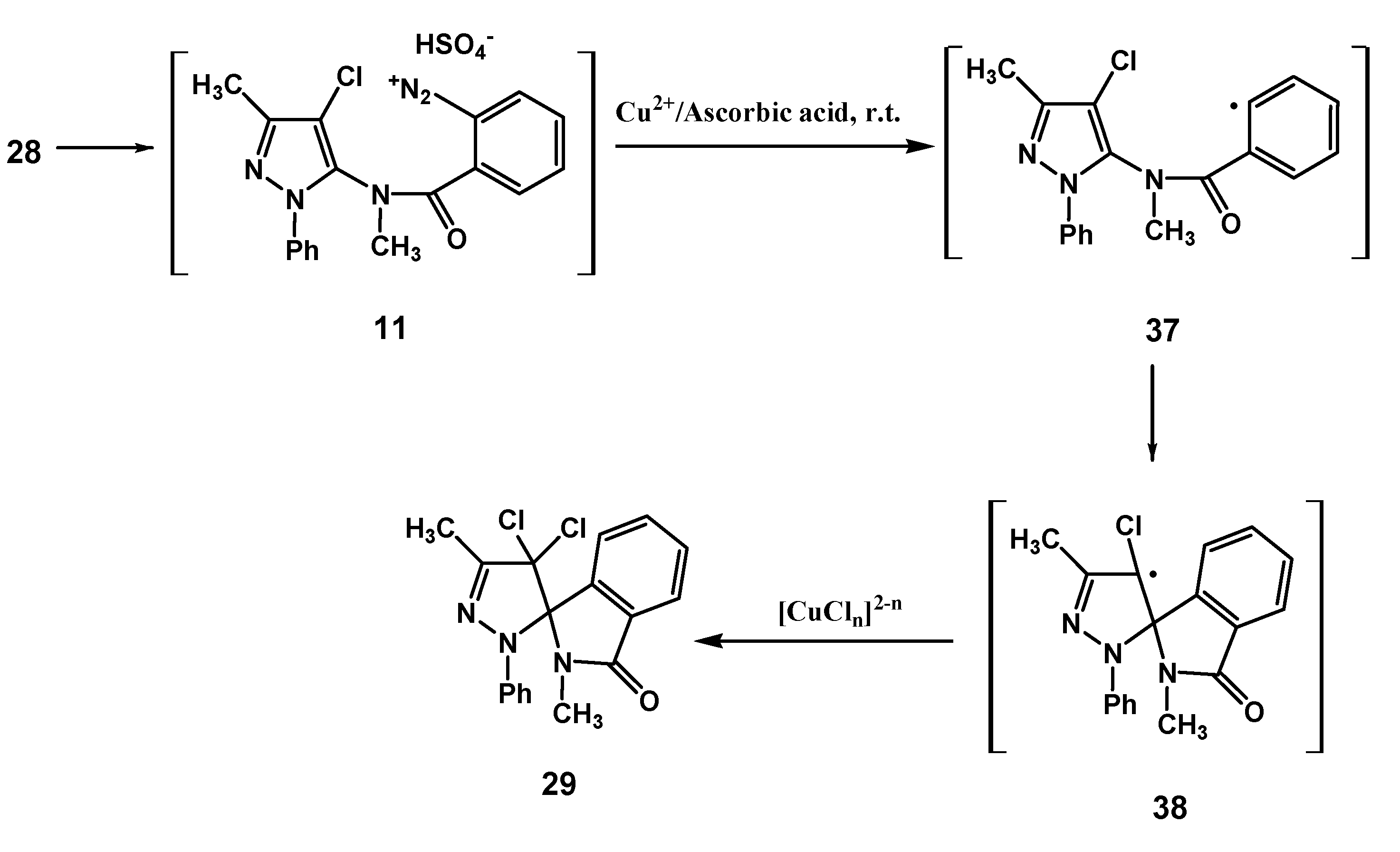
As regards the transformation of the diazonium salt
11, performed under the identical conditions followed for the chemical analogues
10 and
2, the production of the dichloro spiro compound
29 was observed, which demonstrates that substitution at the 4-position of the pyrazoline nucleus does not hinder the ligand radical transfer process from the copper(II)-chloro complex to radical species
38 (
Scheme 6).
Scheme 5.
1,5-hydrogen atom transfer by -reaction in the transformation of diazonium salts 10, 2.
Scheme 5.
1,5-hydrogen atom transfer by -reaction in the transformation of diazonium salts 10, 2.
Scheme 6.
Suggested mechanism for the transformation of 11.
Scheme 6.
Suggested mechanism for the transformation of 11.
The spiro compound
20 proved to be a useful intermediate since it was transformed by heating at 210 °C into 1,3-dimethylisochromeno[4,3-
c]pyrazol-5(1
H)-one (
39) (
Scheme 7). The latter compound has the potential as a benzodiazepine receptor ligand. In fact, this compound shows two hydrogen bond acceptor atoms at the distance of about 3.5 Å, that is the isochromene oxygen and the nitrogen at position 2 of the pyrazole nucleus, which are mandatory in the molecule for a good affinity at the benzodiazepine binding site [
10].
Scheme 7.
Transformation of 20 by fusion.
Scheme 7.
Transformation of 20 by fusion.
3. Experimental
3.1. General
Reaction progress was monitored by TLC on silica gel plates (Merck 60, F254, 0.2 mm). Organic solutions were dried over Na2SO4. Evaporation refers to the removal of solvent on a rotary evaporator under reduced pressure. All melting points were determined on a Büchi 530 capillary melting point apparatus and are uncorrected. IR spectra were recorded with a Perkin Elmer Spectrum RXI FT-IR System spectrophotometer as solid in KBr disc. 1H-NMR and 13C-NMR spectra were obtained in CDCl3 or DMSO-d6 at 300.13 and 75.47 MHz respectively, using a Bruker AC series 300 MHz spectrometer (tetramethylsilane as an internal standard): chemical shifts are expressed in ppm values. Mass spectra at 70 eV were obtained using an Autospec Ultima Ortogonal T.O.F.T. (Micromass) spectrometer. Merck silica gel (Kiesegel 60/230-400 mesh) was used for flash chromatography columns. Microanalyses data (C, H, N) were obtained by an Elemental Vario EL. III apparatus and are within ±0.4% of the theoretical values. Yields refer to products after one crystallization. The names of the compounds were obtained using the Chem Draw 9.0.1 software of Cambridge Soft (Cambridge MA, USA).
3.2. Preparation of N-(1,3-Dimethyl-1H-pyrazol-5-yl)acetamide (13)
This compound was prepared following a literature method [
11] modified by us: 1,3-dimethyl-1
H-pyrazol-5-amine (
12, 11.4 g, 0.102 mol) was reacted under stirring at r.t. with acetic anhydride (45 mL) for 24 h. After this time the solution was evaporated to give an oily residue which solidified when treated with triturated ice (20 g) and scraped. After filtration the material was air dried and crystallized from ethyl acetate/petroleum ether (b.p. 40–70 °C) to give
13 in 70% yield; mp: 43–45 °C; IR (KBr, cm
−1): 3,123 (broad, NH), 1,667 (CO);
1H-NMR (DMSO-
d6, ppm): 2.04 (3H, s, CH
3); 2.07 (3H, s, CH
3); 3.55 (3H, s, CH
3); 5.95 (1H, s, pyrazole H-4); 9.84 (1H, s, exchangeable with D
2O, NH); Anal. Calcd. for C
7H
11N
3O (153.18) : C, 54.89%; H, 7.24%; N, 27.43%. Found: C, 54.51%; H, 6.86%; N, 27.28%.
3.3. Preparation of N-(1,3-Dimehyl-1H-pyrazol-5-yl)-N-methylacetamide (14)
To a solution of N-(1,3-dimethyl-1H-pyrazol-5-yl)acetamide (13, 1 g, 6.5 mmol) in hot acetone (26 mL) was added KOH (1.58 g), and the mixture was refluxed for 10 min. After this time CH3I (0.67 mL, 10 mmol) in acetone (4 mL) was added and reflux was continued for one hour. The mixture was filtered, and evaporation of the filtrate afforded a residue which was treated with water (20 mL) and extracted with diethyl ether (4 × 20 mL). The combined ether extracts were evaporated to give a solid residue which was crystallized from ethyl acetate affording compound 14 as colorless crystals in 46% yield, mp: 128–130 °C. MS (m/z): 167 (M+); IR (KBr, cm−1): 1,681 (CO); 1H-NMR (DMSO-d6, ppm): 1.75 (3H, s, CH3); 2.12 (3H, s, CH3); 3.03 (3H, s, CH3); 3.57 (3H, s, CH3); 6.04 (1H, s, pyrazole H-4). 13C-NMR (DMSO-d6) (ppm): 13.79 (CH3), 21.39 (CH3), 34.54 (CH3), 35.39 (CH3), 101.50 (CH), 141.83 (C), 146.30 (C), 169.82 (CO); Anal. Calcd. for C8H13N3O (167.21) : C, 57.46%; H, 7.84%; N, 25.13%. Found: C, 57.21%; H, 7.90%; N, 24.76%.
3.4. Preparation of N,1,3-Trimethyl-1H-pyrazol-5-amine (15) [12]
To a solution of N-(1,3-dimethyl-1
H-pyrazol-5-yl)-
N-methylacetamide (
14, 7.16 g, 42 mmol) in ethanol (69 mL) was added a 5 N aqueous solution of KOH (40 mL), and the mixture was refluxed for 10 h. After this time the reaction mixture was first filtered and then evaporated under vacuum to give a residue which was extracted with diethyl ether (4 × 90 mL). The combined extracts were evaporated under reduced pressure and the obtained residue was processed by flash chromatography [
13]: column external diameter 4 cm, silica gel 0.040–0.063 mm, ethyl acetate/acetone (1:1 v/v ) as eluent (1.5 L), fractions each 50 mL. The first ten fractions were discarded, and fractions 11-25 were evaporated affording pure
15 as a pale yellow oil in 70% yield
. IR (KBr, cm
−1): 3,228 (NH);
1H-NMR (CDCl
3, ppm): 2.18 (3H, s, CH
3), 2.80 (3H, s, CH
3), 3.53 (4H, s, CH
3, NH; 3H after exchange with D
2O), 5.27 (1H, s, pyrazole H-4); Anal. Calcd. for C
6H
11N
3 (125.17) : C, 57.57%; H, 8.86%; N, 33.57%. Found: C, 57.21%; H, 8.86%; N, 33.53%.
3.5. Preparation of N-(1,3-dimethyl-1H-pyrazol-5-yl)-N-methyl-2-nitrobenzamide (16)
A solution containing equimolar amounts (6.16 mmol) of N,1,3-trimethyl-1H-pyrazol-5-amine (15) and 2-nitrobenzoyl chloride in dry chloroform (29 mL) was refluxed for 5 h. After the first hour triethylamine (0.86 mL) was added in four portions (0.43 mL; 0.21 mL; 2 × 0.11 mL respectively, with intervals of 1 h between additions). The solution was evaporated under reduced pressure, the residue washed with cold water and extracted with dichloromethane (3 × 50 mL). The combined extracts were dried (sodium sulfate) and the solid residue afforded by evaporation was crystallized from ethyl acetate to give 16, as colorless crystals, in 64% yield, mp 132–133 °C; MS (m/z) 274 (M+); IR (KBr, cm−1): 1,671 (CO); 1H-NMR (CDCl3, ppm): 2.03 (3H, s, CH3), 3.40 (3H, s, CH3), 3.63 (3H, s, CH3), 5.70 (1H, s, pyrazole H-4), 7.29–8.30 (4H, a set of signals, C6H4); 13C-NMR (DMSO-d6, ppm): 13.92 (CH3), 35.35 (CH3), 36.36 (CH3), 102.44 (CH), 124.80 (CH), 128.67 (CH), 131.09 (CH), 132.34 (C), 134.96 (CH), 140.62 (C), 145.49 (C), 146.50 (C), 167.31 (CO); Anal. Calcd. for C13H14N4O3 (274.28) : C, 56.93%; H, 5.14%; N, 20.43%. Found: C, 56.75%; H, 5.13%; N, 20.62%.
3.6. Preparation of 2-Amino-N-(1,3-Dimethyl-1H-pyrazol-5-yl)-N-methylbenzamide (17)
To a solution of
N-(1,3-dimethyl-1
H-pyrazol-5-yl)-
N-methyl-2-nitrobenzamide (
16, 3.84 g, 14 mmol) in ethanol (150 mL) was added 10% palladium on activated charcoal catalyst (380 mg). The mixture was reacted with hydrogen in a Parr apparatus at 50 psi for 20 h. After this time the reaction mixture was filtered, and the filtrate was evaporated under vacuum to give an oily residue which was processed by Flash chromatography [
13]: column external diameter 4.5 cm, silica gel (0.040-0.063 mm), ethyl acetate as eluent (1.8 L), fractions each 50 mL. The first 17 fractions were discarded. Fractions 18–21 were collected and evaporated under vacuum to afford
17 as a pale yellow pure oil in 44% yield; MS (
m/z): 244 (M
+); IR (KBr) (cm
−1): 3490–3200 (multiple bands, NH
2), 1638 (CO);
1H-NMR (CDCl
3, ppm): 2.16 (3H, s, CH
3), 3.34 (3H, s, CH
3), 3.44 (3H, s, CH
3), 4.81 (2H, s, NH
2, exchangeable with D
2O), 5.87 (1H, s, pyrazole H-4), 6.40–7.07 (4H, a set of signals, C
6H
4); Anal. Calcd. for C
13H
16N
4O (244.29) : C, 63.91%; H, 6.60%; N, 22.93%. Found: C, 64.18%; H, 6.63%; N, 22.67%.
3.7. Preparation of 2-((1,3-Dimethyl-1H-pyrazol-5-yl)(methyl)carbamoyl)benzendiazonium hydrogen sulfate (10)
The pulverized amine 17 (2.07 g, 8.48 mmol) was dissolved in cooled (0–5 °C) aqueous sulfuric acid (5 N) (17 mL), and aqueous sodium nitrite (2.5 M) (3.5 mL) was added dropwise to the stirred solution. The solution was stirred for a further 15 min in the ice bath and was then checked for excess nitrous acid with potassium iodide starch paper; the eventual excess can be destroyed by addition of urea. This solution was utilized in the next step.
3.8. Transformation of the Diazonium Hydrogen Sulphate (10)
To a cold (0–5 °C) solution. (400 mL) of CuSO4·5 H2O (0.3 M) and NaCl (0.75 M), first the soln. of 10, obtained from the previous procedure, and then ascorbic acid (370 mg, 2.11 mmol) were added under stirring. The mixture was stirred for 1 h at r.t. and then filtered. The solid product obtained was washed with cold water, dried in desiccator (anhydrous CaCl2, 24 h) and then crystallized from diethyl ether to give the epimeric mixture of (1S,4′R)- (or (1S,4′S)-) and (1S,4′S)- (or (1S,4′R)-)4′-chloro-2,2′,5′-trimethy-2′,4′-dihydrospiro[isoindoline-1,3′-pyrazol]-3-one (18) and 19, respectively in 8% overall yield; GLC-MS: two peaks, 228 (m/z) for each, (M+-Cl); IR (KBr, cm−1): 1,710 (CO); 1H-NMR (DMSO-d6, ppm): 2.05 (3H, s, CH3), 2.07 (3H, s, CH3), 2.37 (3H, s, CH3), 2.44 (3H, s, CH3), 2.79 (3H, s, CH3), 2.81 (3H, s, CH3), 5.54 (1H, s, pyrazoline H-4); 5.83 (1H, s, pyrazoline H-4); 7.59–7.77 (2 × 4H, a set of signals, 2 × C6H4); Anal. Calcd. for C13H14ClN3O (263.72) : C, 59.21%; H, 5.35%; N, 15.93%. Found: C, 59.28%; H, 5.33%; N, 15.93%.
The mother liquors were saturated with NaCl and extracted with diethyl ether (3 × 100 mL). The combined extracts were dried (Na
2SO
4) and evaporated under reduced pressure. The oily residue (850 mg) was chromatographed following the flash procedure [
13]: external diameter of the column 4.5 cm, silica gel (0.040–0.063 mm), ethyl acetate as eluent (2L), fractions each 50 mL. The initial seven fractions were discarded. Fractions 8 and 9 were evaporated under vacuum to give 130 mg (7%) of pure (1
S,4′
R)- (or (1
S,4′
S)-)4′-hydroxy-2,2′,5′-trimethyl-2′,4′-dihydrospiro[isoindole-1,3′-pyrazol]-3(2
H)-one (
20) as a colourless solid, mp 160–163 °C (ethyl acetate); MS (
m/z): 245 (M
+), 214 (M
+-CH
3NH
2); IR(KBr, cm
−1): 3,263 (OH), 1,671 (CO);
1H-NMR (DMSO-
d6, ppm): 1.94 (3H, s, CH
3), 2.34 (3H, s, CH
3), 2.78 (3H, s, CH
3), 5.11 (1H, d,
J = 6.3 Hz, pyrazoline H-4), 6.12 (1H, d,
J = 6.3 Hz, OH, exchangeable with D
2O), 7.52-7.70 (4H, a set of signals, C
6H
4); Anal. Calcd. for C
13H
15N
3O
2 (245.28): C, 63.66%; H, 6.16%; N, 17.13%. Found: C, 63.42%; H, 6.55%; N, 17.02%.
The combined fractions 11–14 when evaporated gave a residue (170 mg) which was chromatographed by preparative TLC on silica gel (20 × 20 cm, thickness 2 mm; ethyl acetate/chloroform 1:1 as eluent). The plate showed three bands (U.V. light at 254 nm) among which the intermediate one underwent extraction with ethyl acetate. The extract was evaporated under vacuum and the residue obtained was crystallized to give a compound which was identical in all respects (mp, mixed mp, Rf, IR,
1H-NMR) with an authentic specimen of
N-(1,3-dimethyl-1
H-pyrazol-5-yl)benzamide (
22) [
7], 2% yield. The mother liquors from the crystallization of
22 were evaporated under vacuum, and the residue was processed by preparative TLC: 20 × 20 cm, thickness 2 mm, ethyl acetate/chloroform 1:1 as eluent. The crude product obtained underwent the same procedure to give 10 mg of pure (1
S,4′
S)- (or (1
S,4′
R)-)4′-hydroxy-2,2′,5′-trimethy-2′,4′-dihydrospiro[isoindoline-1,3′-pyrazol]-3-one (
21), as a colorless solid, mp 177–180 °C; MS (
m/z): 245 (M
+), 214 (M
+-CH
3NH
2);
1H-NMR (DMSO-
d6, ppm): 1.97 (3H, s, CH
3), 2.22 (3H, s, CH
3), 2.85 (3H, s, CH
3), 4.92 (1H, d,
J =5.7 Hz, pyrazoline H-4), 5.85 (1H, d,
J = 6.3 Hz, pyrazoline OH-4, exchangeable with D
2O), 7.43-7.70 (4H, a set of signals, C
6H
4);
13C-NMR (DMSO-
d6, ppm): 12.75 (CH
3), 24.74 (CH
3), 34.02 (CH
3), 77.76 (CH), 91.86 (C), 111.21 (C), 122.10 (CH), 124.65 (CH), 128.83 (CH), 130.45 (CH), 131.52 (C), 139.47 (C), 152.38 (C), 166.02 (CO); Anal. Calcd. for C
13H
15N
3O
2 (245.28) : C, 63.66%; H, 6.16%; N, 17.13%. Found: C, 63.45%; H, 6.17%; N, 17.20%.
3.9. Preparation of 1,3-Dimethylisochromeno[4,3-c]pyrazol-5(1H)-one (39)
Compound 20 (30 mg) was melted at 210 °C for 10 min. The obtained material was crystallized from ethanol to give 39, as colorless solid, in 69% yield, mp 237–239 °C; MS (m/z): 214 (M+); IR (KBr, cm−1): 1715 (CO); 1H-NMR (CDCl3, ppm): 2.37 (3H, s, CH3), 4.23 (3H, s, CH3), 7.55–8.46 (4H, a set of signals, C6H4); 13C-NMR (CDCl3, ppm): 9.96 (CH3), 40.07 (CH3), 119.86 (C), 120.47 (CH), 122.41 (C), 128.10 (CH), 128.58 (C), 132.47 (CH), 132.91 (C), 134.97 (CH), 136.95 (C), 162.08 (CO); Anal. Calcd. for C12H10N2O2 (214.22) : C, 67.28%; H, 4.71%; N, 13.08%. Found: C, 67.56%; H, 4.87%; N, 13.09%.
3.10. Preparation of N-(3-Methyl-1-phenyl-1H-pyrazol-5-yl)benzamide (4)
From the transformation of the diazonium salt
2 by the CuSO
4/NaCl/ascorbic acid reagent combination: the diazonium hydrogen sulfate
2 derived from compound
1 [
1] (5.9 mmol) was decomposed following the procedure used for
10. The suspension obtained was filtered, and the solid was crystallized from ethanol (95% V/V) to give epimers
7,
8. The mother liquors when evaporated leave a residue (350 mg) which was processed by preparative TLC (two plates, 20 × 20 cm, thickness 2 mm, ethyl acetate/petroleum ether 3:7 as eluent). Work up allowed us to obtain a product (140 mg) which was identical in all respects (mp, mixed mp, IR,
1H-NMR) to an authentic specimen of compound
4 [
7].The aqueous mother liquors were saturated with sodium chloride and extracted with ethyl acetate (4 × 150 mL). The combined extracts were evaporated to give a residue (70 mg) which was processed by preparative TLC. More of compound
4 (20 mg) was obtained following the above procedure, overall yield 10%.
3.11. Preparation of N-(4-Chloro-3-methyl-1-phenyl-1H-pyrazol-5-yl)-N-methyl-2-nitrobenzamide (27)
This compound was prepared by modifying the procedure reported in reference [
14]. Compound
25 [
15] (2 g, 5.95 mmol) was dissolved in acetic acid (57 mL) and then concentrated hydrochloric acid (36.5% w/w, 4.3 mL) was added. To the stirred solution a potassium nitrite aqueous solution (3.5 g of KNO
2 in 1.8 mL of H
2O) was added dropwise and stirring was continued for a further 24 h. The obtained suspension was filtered and the filtrate was treated first with an aqueous solution (300 mL) of hydrochloric acid (0.24 M), copper sulphate pentahydrate (0.3 M) and sodium chloride (0.75 M) and then with ascorbic acid (260 mg, 1.48 mmol). The mixture was stirred for 1 h at r.t. and then filtered. The solid product obtained was crystallized from ethanol (95% V/V) to give
27 (yield 60%) identical in all respects (mixed melting point, TLC, MS, IR) to an authentic specimen of compound
27 [
14].
3.12. Preparation of 2-Amino-N-(4-chloro-3-methyl-1-phenyl-1H-pyrazol-5-yl)-N-methylbenzamide (28)
A suspension of Fe filings (12.41 g) in 5% (V/V) aqueous AcOH (16 mL) was heated at about 100 °C under stirring on the water bath of a rotavapor until H2 evolution ceased. The nitro derivative 27 (6.9 g, 18.5 mmol) was added in four portions, with intervals of 20 min. between additions. After the last addition, stirring was continued at 100 °C for 1 h, then the mixture was cooled to r.t. The pH of the suspension was adjusted to 7–8 with a saturated NaHCO3 solution. The solid was separated by filtration, air dried overnight, and then extracted with boiling CHCl3 (3 × 30 mL). The combined extracts were evaporated under vacuum, leaving an residue which was crystallized from ethyl acetate to give 28 in 80% yield, mp 165–166 °C; MS (m/z): 342 (M+); IR (cm−1): 3485, 3358 (NH2), 1640 (CO); 1H-NMR (ppm): 2.26 (3H, s, Me), 3.40 (3H, s, Me); 4.33 (2H, s, exchangeable with D2O, NH2), 6.40–7.35 (9H, m, C6H5 and C6H4); Anal. Calcd. for C18H17ClN4O (340.81) : C, 63.44%; H, 5.03%; N, 16.44%. Found: C, 63.35%; H, 5.21%; N, 16.56%.
3.13. Preparation of 2-((4-Chloro-3-methyl-1-phenyl-1H-pyrazol-5-yl)(methyl)carbamoyl)benzendiazonium Hydrogen sulphate (11)
The pulverized amine 28 (1.5 g, 4.41 mmol) was dissolved in cooled (0–5 °C) aqueous sulfuric acid (10 M) (17.6 mL) and aqueous sodium nitrite (2.5 M) (1.82 mL) was added drop-wise to the stirred solution. The solution was stirred for a further 15 min in the ice bath and was then checked for excess nitrous acid with potassium iodide starch paper. The eventual excess can be destroyed by addition of urea.
3.14. Decomposition of the Diazonium Hydrogen Sulphate 11
To a cold (0–5 °C) soln. (220 mL) of CuSO4·5H2O (0.3 M) and NaCl (0.75M), first the soln. of 11 obtained from the previous procedure, and then ascorbic acid (195 mg, 1.11 mmol) were added under stirring. The mixture was stirred for 1 h at r.t. and then filtered. The solid product obtained was dried in desiccator (anhydrous CaCl2) for 24 h and then crystallized from ethyl acetate to give compound 29 in 80% yield, mp 202–203 °C; MS (m/z): 359 (M+); IR (cm−1):1713 (CO); 1H-NMR (CDCl3, ppm): 2.38 (3H, s, Me), 2.66 (3H, s, Me), 6.70–7.96 (9H, m, C6H5 and C6H4); 13C-NMR (CDCl3, ppm): 11.13 (CH3), 26.64 (CH3), 96.61 (C), 93.23 (C), 115.59 (2xCH), 122.45 (CH), 124.30 (CH), 127.21 (CH), 129.05 (2xCH), 130.98 (CH), 131.62 (CH), 132,66 (C), 139.08 (C), 142.21 (C), 147 (C), 167 (CO); Anal. Calcd. for C18H15Cl2N3O (359.06) : C, 60.01%; H, 4.20%; N, 11.66%. Found: C, 60.20%; H, 4.58%; N, 11.69%.
3.15. Reaction of 4′,4′-Dichloro-2,5′-dimethyl-2′-phenyl-2′,4′-dihydrospiro[isoindoline-1,3′-pyrazol]-3-one (29) with Water
To a solution of
29 (10 mg) in acetonitrile (1 mL) was added water (1 mL) and the mixture was refluxed for 30 min. The solution was evaporated under vacuum to give a pure residue which was identical in all respects to an authentic specimen of compound
31 [
1].



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







