Novel Method of Synthesis of 5''-Phosphate 2'-O-ribosyl-ribonucleosides and Their 3'-Phosphoramidites


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of 5″-Phosphate 2'-O-ribosylribonucleosides

2.2. Synthesis of 3'-O-Phosphoramidites of 5″-Phosphate 2'-O-ribosylribonucleosides and Their Use in the Preparation of a Dinucleoside Monophosphate



3. Experimental
3.1. General Methods
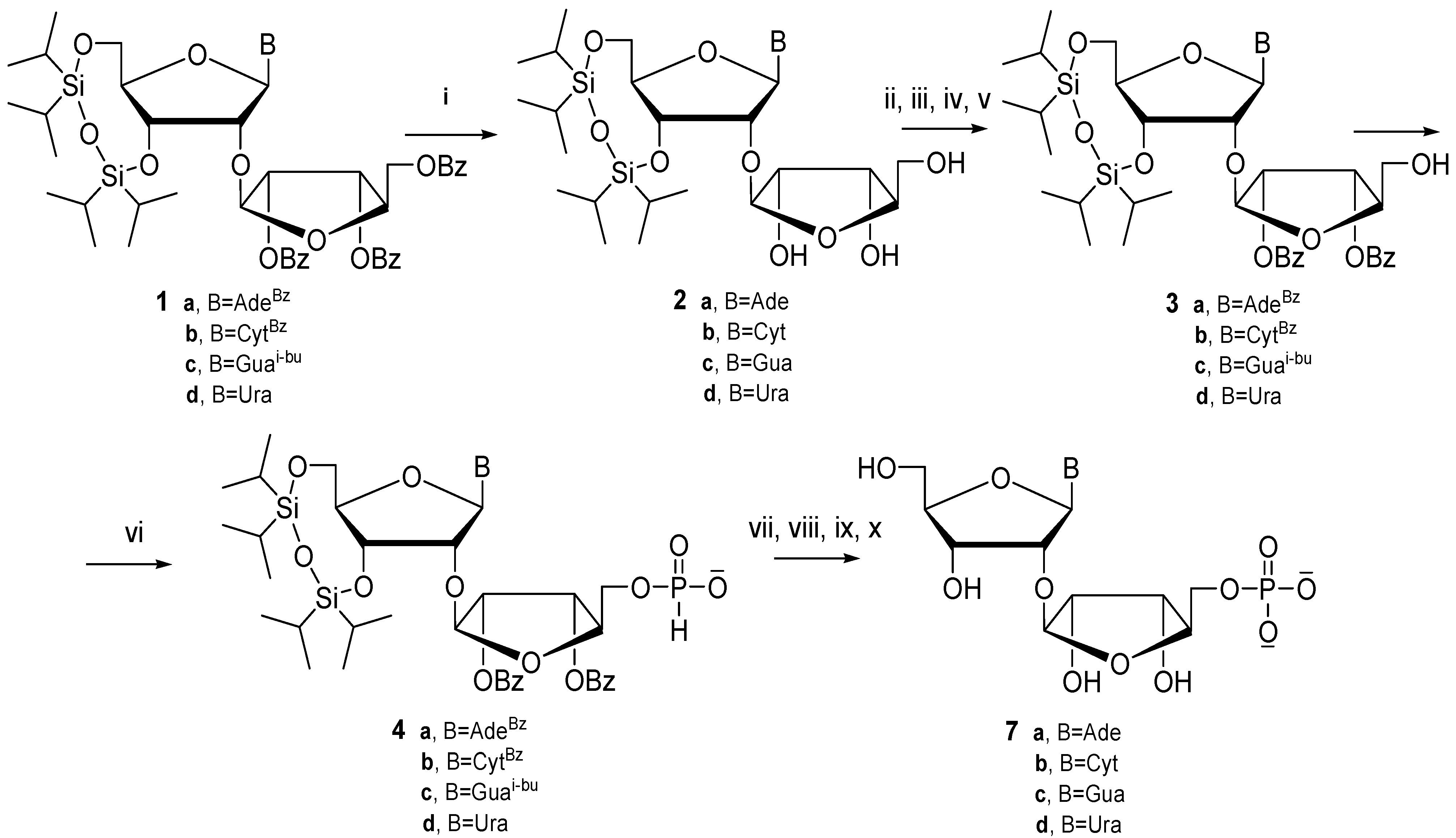
3.2. General Procedure for the Synthesis of 3',5'-O-(Tetraisopropyldisiloxane-1,3-diyl)-2'-O-β-d-ribo-furanosyl Ribonucleosides 2
- 2b: yield 89%; 1H-NMR (CDCl3): δ (ppm) 7.92 (s, 1, H-8); 5.79 (s, 1, H-1'); 5.37 (s, 1, H-1''); 4.49 (m, 2, H-3', 3''); 4.37 (d, 1, H-2'); 4.25 (d, 1, H-2''); 4.16–4.10 (m, 2, H-4', 5); 4.04–3.92 (m, 3,H-4'', 5'', 5'); 3.73 (2×d, 1, H-5''); 1.11–0.98 (m, 28, TIPDSi).
- 2c: yield 72%; 1H-NMR (CDCl3): δ (ppm) 7.84 (d, 1, H-6); 5.81 (d, 1, H-5); 5.54 (s, 1, H-1'); 5.27 (s, 1, H-1''); 4.33–4.29 (m, 1, H-2'); 4.23–4.17 (m, 3, H-2'', 3', 4'); 4.08–3.96 (m, 3, H-3'', 5', 5'); 3.89–3.87 (m, 1, H-4''); 3.79 (2×d, 1, H-5''); 3.58 (2×d, 1, H-5''); 1.12–1.01 (m, 28, TIPDSi).
- 2d: yield 87%; 1H-NMR (CDCl3): δ (ppm) 7.89 (d, 1, H-6); 5.71 (d, 1, H-5); 5.64 (s, 1, H-1'); 5.39 (s, 1, H-1''); 4.53 (m, 1, H-2'); 4.30–4.19 (m, 3, H-2'', 3', 4'); 4.12–3.94 (m, 3, H-3'', 5', 5'); 3.76–3.71 (m, 1, H-4''); 3.66 (2×d, 1, H-5''); 3.36 (2×d, 1, H-5''); 1.11–1.02 (m, 28, TIPDSi).
3.3. General Procedure for the Synthesis of N-Protected-3',5'-O-(tetraisopropyldisiloxane-1,3-diyl)-2'-O-(2″,3″-di-O-benzoyl-β-d-ribofuranosyl) Ribonucleosides 3
Transient Protecting Procedure
- 3a: yield 75%; 1H-NMR (CDCl3): δ (ppm) 8.67 (s, 1, H-2), 8.45 (s, 1, H-8), 7.98–7.84 and 7.60–7.33 (m, 15, 3×Bz), 6.22 (s, 1, H-1'), 6.05 (m, 1, H-3''), 5.85 (d, 1, H-2''), 5.63 (s, 1, H-1''), 5.03 (m, 1, H-3'), 4.60–4.49 (m, 3, H-5'', 4″, 2'), 4.32–4.25 (m, 2, H-5'', 4'), 4.11–4.03 (m, 2, H-5', 5''-OH), 3.84–3.77 (m, 1, H-5'), 1.09–0.96 (m, 28, TIPDSi).
- 3b: yield 84%; 1H-NMR (CDCl3): δ (ppm) 8.36 (d, 1, H-6), 7.88–7.25 (m, 16, 3×Bz, H-5), 5.85 (m, 1, H-3''), 5.71 (s, 1, H-1'), 5.69 (d, 1, H-2''), 5.63 (s, 1, H-1″), 4.46 (m, 2, H-2', H-4'), 4.23–4.19 (m, 2, H-3', H-4''), 3.99–3.93 (m, 2, H-5'), 3.68–3.63 (m, 2, H-5''), 1.03–0.88 (m, 28, TIPDSi).
- 3c: yield 73%; 1H-NMR (CDCl3): δ (ppm) 12.20 (s, 1, N-H), 10.21 (s, 1, N-H), 8.16 (s, 1, H-8), 7.99–7.32 (m, 10, 2×Bz), 5.92 (s, 1, H-1'), 5.89 (t, 1, H-3''), 5.87 (s, 1, H-1''), 5.83 (d, 1, H-2''), 4.62 (m, 1, H-4''), 4.53 (t, 1, H-3'), 4.50 (s, 1, 5''-OH), 4.34 (d, 1, H-2'), 4.26–4.16 (m, 3, H-4'', 5'), 4.07–3.96 (4×d, 2, H-5''), 2.66 (m, 1, CH of i-bu), 1.28 (s, 3, CH3 of i-bu), 1.27 (s, 3, CH3 of i-bu), 1.10–0.95 (m, 28, TIPDSi).
- 3d: yield 81%; 1H-NMR (CDCl3): δ (ppm) 8.85 (s, 1, N-H), 7.77 (d, 1, H-6), 7.9–7.26 (m, 10, 2×Bz), 5.88 (m, 1, H-3''), 5.79 (d, 1, H-2''), 5.74 (s, H-1'), 5.72 (d, 1, H-5), 5.67 (s, 1, H-1''), 4.54 (m, 1, H-4''), 4.39 (d, 1, H-2'), 4.3–4.23 (m, 3, H-3', H-4', H-5'), 4.07–3.97 (m, 2, H-5', H-5''), 3.80 (2×d, 1, H-5''), 1.09–0.96 (m, 28, TIPDSi).
3.4. General Procedure for the Synthesis of N-Protected 3',5'-O-(tetraisopropyldisiloxane-1,3-diyl)-2'-O-(5″-H-phosphonate-2″,3″-di-O-benzoyl-β-d-ribofuranosyl) Ribonucleosides 4
- 4a: yield 96%; 31P-NMR (D2O): δ = 3.01 ppm JP-H = 610 Hz
- 4b: yield 98%; 31P-NMR (D2O): δ = 4.37 ppm JP-H = 658 Hz
- 4c: yield 95%; 31P-NMR (D2O): δ = 4.87 ppm JP-H = 640 Hz
- 4d: yield 96%; 31P-NMR (D2O): δ = 5.06 ppm JP-H = 674 Hz
3.5. General Procedure for the Synthesis of 5″Phosphate 2'-O-β-d-ribofuranosyl Ribonucleosides 5
- 5a: 31P-NMR (D2O): δ = 9.46 and 9.03 ppm JP-H = 717 Hz and 7.19 Hz
- 5b: 31P-NMR (D2O): δ = 10.10 and 9.45 ppm JP-H = 734 Hz
- 5c: 31P-NMR (D2O): δ = 11.66 and 9.56 ppm JP-H = 742 Hz and 726 Hz
- 5d: 31P-NMR (D2O): δ = 9.49 and 9.43 ppm JP-H = 720 Hz
- 6a: 31P-NMR (D2O): δ = −0.63 ppm
- 6b: 31P-NMR (D2O): δ = −1.47 ppm
- 6c: 31P-NMR (D2O): δ = −1.34 ppm
- 6d: 31P-NMR (D2O): δ = −0.58 ppm
- 7a: yield 89%; FAB-MS: m/z [M−H]−; Calcd for C15H21N5O11P−, 478.10; found m/z 477.9; Calcd for C15H20N5O11PNa−, 500.08; found m/z 499.8; 31P-NMR (D2O): δ =1.481ppm; 1H-NMR (D2O): δ (ppm) 8.24 (s, 1, H-2); 8.08 (d, 1, H-8); 6.06 (d, 1, H-1'); 4.80 (d, 1, H-1''); 4.74 (t, 1, H-2'); 4.43 (q, 1, H-3'); 4.12 (q, 1, H-4'); 4.06 (q, 1, H-2''); 4.01 and 3.99 (2×d, 1, H-3''); 3.77 (m, 1, H-4''); 3.73 (2×d, 1, H-5'); 3.68 (2×d, 1, H-5'); 3.53 (m, 1, H-5″); 3.37 (m, 1, H-5″); 13C-NMR (D2O): δ (ppm) 156.58 (s, C-6); 153.34 (s, C-2); 149.18 (s, C-4); 142.62 (s, C-8); 120.03 (s, C-5); 108.28 (s, C-1″); 88.49 (s, C-1'); 86.80 (s, C-4'); 82.73 (d, C4″); 80.42 (s, C-2'); 75.24 (s, C-2″); 71.61 (s, C-3″); 70.56 (s, C-3'); 65.90 (d, C-5″); 62.4 (s, C-5').
- 7b: yield 83%; FAB-MS: m/z [M–H]−; Calcd for C14H21N3O12P− 454.09; found m/z 454.1; Calcd for C14H20N3O12PNa−, 476.07; found m/z 475.9; 31P-NMR (D2O): δ = 0.359 ppm; 1H-NMR (D2O): δ (ppm) 7.82 (d, 1, H-6); 6.07 (d, 1, H-5); 5.94 (d, 1, H-1'); 5.13 (s, 1, H-1″); 4.45 (t, 1, H-2'); 4.35 (t, 1, H-3'); 4.24 (q, 1, H-3''); 4.17 (d, 1, H-2''); 4.08 (m, 1, H-4''); 4.08 (m, 1, H-4'); 3.92 (m, 1, H-5''); 3.87 (2×d, 1, H-5'); 3.76 (2×d, 1, H-5'); 3.74 (m, 1, H-5''); 13C-NMR (D2O): δ (ppm) 167.02 (s, C-4); 158.07 (s, C-2); 144.19 (s, C-6); 108.31 (s, C-1''); 97.36 (s, C-5); 91.42 (s, C-1'); 85.03 (s, C-4'); 82.68 (d, C-4''); 79.95 (s, C-2'); 75.19 (s, C-2''); 71.69 (s, C-3''); 69.33 (s, C-3'); 66.57 (s, C-5''); 61.85 (s, C-5'').
- 7c: yield 82%; FAB-MS: m/z [M−H]−; Calcd for C15H21N5O12P− 494.09; found m/z 493.8; Calcd for C15H20N5O12PNa− 516.07; found m/z 515.7; 31P-NMR (D2O): δ = 3.115 ppm; 1H-NMR (D2O): δ (ppm) 7.88 (d, 1, H-8); 5.93 (d, 1, H-1'); 4.90 (d, 1, H-1''); 4.62 (t, 1, H-2'); 4.40 (q, 1, H-3'); 4.11 (q, 1, H-4'); 4.03 (q, 1, H-2''); 4.02 (d, 1, H-3''); 3.87 (m, 1, H-4''); 3.73 (2×d, 1, H-5'); 3.67 (2×d, 1, H-5'); 3.61 (m, 1, H-5''); 3.52 (m, 1, H-5''); 13C-NMR (D2O): δ (ppm) 159.86 (s, C-6); 154.62 (s, C-2); 152.06 (s, C-4); 139.71 (s, C-8); 117.46 (s, C-5); 108.53 (s, C-1''); 88.04 (s, C-1'); 85.66 (s, C-4'); 82.94 (d, C4''); 80.34 (s, C-2'); 75.28 (s, C-2''); 71.90 (s, C-3''); 70.17 (s, C-3'); 65.78 (d, C-5''); 61.98 (s, C-5').
- 7d: yield 81%; FAB-MS: m/z [M−H]−; Calcd for C14H20N2O13P− 455.07; found m/z 454.9; Calcd for C14H19N2O13PNa− 477.05; found m/z 476.8; 31P-NMR (D2O): δ = 2.176 ppm; 1H-NMR (D2O): δ (ppm) 7.73 (d, 1, H-6); 5.74 (d, 1, H-5); 5.82 (d, 1, H-1'); 5.98 (s, 1, H-1''); 4.31 (t, 1, H-2'); 4.21 (t, 1, H-3'); 4.14 (q, 1, H-3''); 4.04 (d, 1, H-2″); 3.95 (m, 1, H-4″''); 3.91 (m, 1, H-4'); 3.76 (m, 1, H-5''); 3.72 (2×d, 1, H-5'); 3.67 (2×d, 1, H-5'); 3.55 (m, 1, H-5''); 13C-NMR (D2O): δ (ppm) 167.18 (s, C-4); 152.26 (s, C-2); 144.26 (s, C-6); 108.84 (s, C-1''); 103.12 (s, C-5); 91.05 (s, C-1'); 84.83 (s, C-4'); 82.94 (d, C-4''); 80.49 (s, C-2'); 75.25 (s, C-2''); 71.70 (s, C-3''); 69.33 (s, C-3'); 66.04 (d, C-5''); 61.57 (s, C-5').
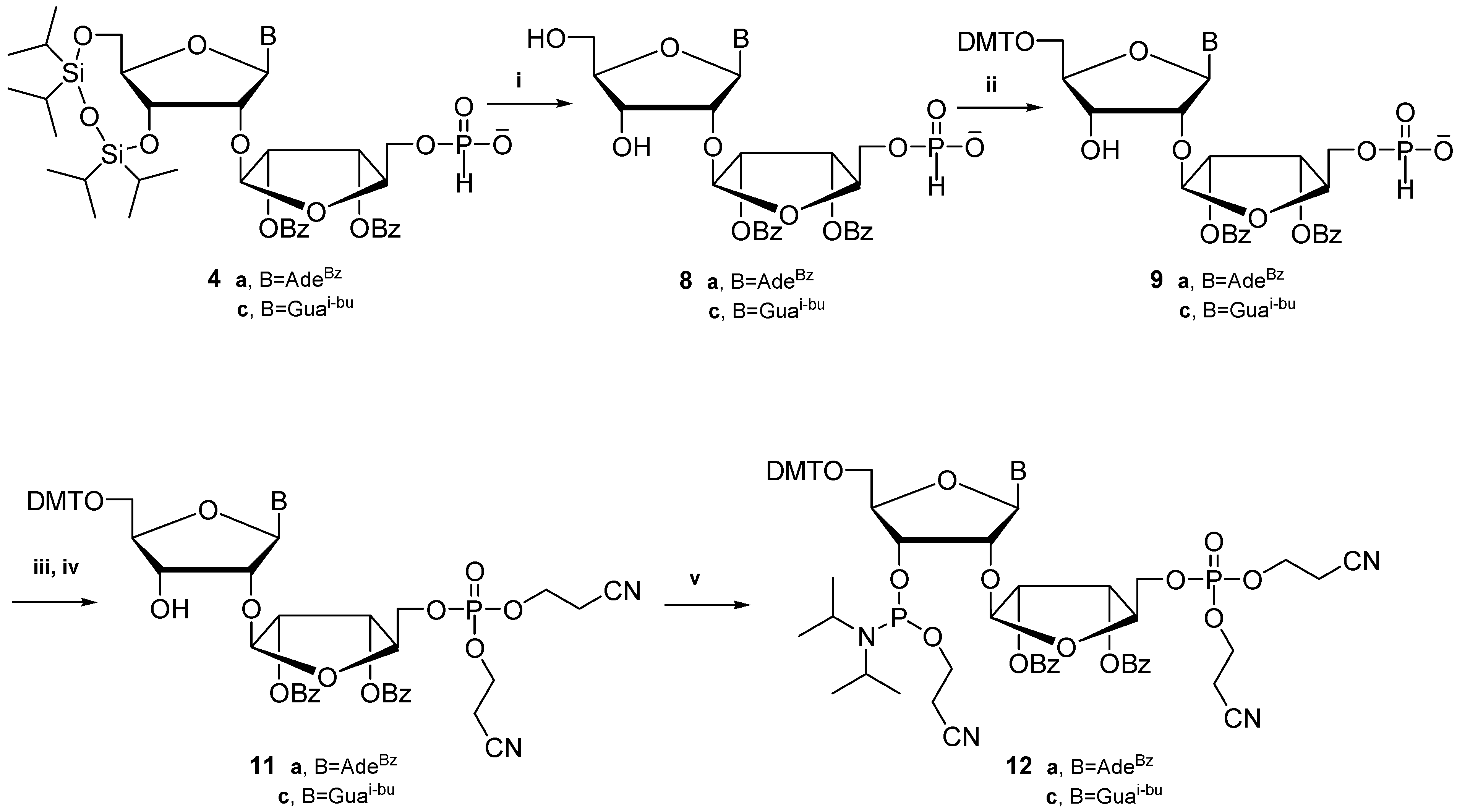
3.6. Synthesis of 6-N-Benzoyl-2'-O-(5″-H-phosphonate-2″,3″-di-O-benzoyl-β-d-ribofuranosyl)-adenosine (8a) and 2-N-Isobutyryl-2'-O-(5″-H-phosphonate-2″,3″-di-O-benzoyl-β-d-ribofuranosyl)-guanosine (8c)
- 8a: yield 89%; 31P-NMR (D2O): δ 3.01 ppm, JP-H = 611 Hz; 1H-NMR (CDCl3): δ (ppm) 8.93 (s, H-2); 8.58 (d, H-8); 7.2–7.9 (m, 15H, arom.) 6.80 (d, H-1'); 4.80 (d, H-1''); 4.74 (t, H-2'); 4.43 (q, H-3'); 4.12 (q, H-4'); 4.06 (q, H-2″); 4.01 and 3.99 (2×d, H-3''); 3.77 (m, H-4''); 3.73 (2×d, H-5'); 3.68 (2×d, H-5'); 3.53 (m, H-5''); 3.37 (m, H−5'').
- 8c: yield 84%; 31P-NMR (D2O): δ = 2.855 ppm, JP-H = 609 Hz; 1H-NMR (CDCl3): δ (ppm) 13.25 (s, 1, N-H); 12.38 (s, 1, N-H); 7.71 (s, 1, H-8); 7.92–7.87 (m, 2, arom.); 7.68–7.26 (m, 8, arom.); 5.89 (d, 1, H-1'); 5.73 (m, 1, H-2''); 5.69 (s, 1, H-1''); 5.65 (m, 1, H-3″); 4.69 (m, 1, H-4''); 4.40 (m, 1, H-3'); 4.28–4.21 (m, 2, H-2', 5''); 4.13–4.04 (m, 2, H-4', 5''); 3.56–3.49 (m, 2, H-5', 5'); 3.20 (s, 3, CH3-ibu); 3.17 (m, 1, CH of i-bu); 3.14 (s, 3, CH3 of i-bu).
3.7. Synthesis of 6-N-benzoyl-5'-O-(4,4'-dimethoxytrityl)-2'-O-(5″-H-phosphonate-2″,3″-di-O-benzoyl-β-d-ribofuranosyl)adenosine (9a) and 2-N-isobutyryl-5'-O-(4,4'-dimethoxytrityl)-2'-O-(5″-H-phosphonate-2″,3″-di-O-benzoyl-β-d-ribofuranosyl)guanosine (9c)
- 9a: yield 93%; FAB-MS: m/z [M−H]−; Calcd for C57H51N5O15P−, 1076.31; found m/z 1076.1; 31P-NMR (CDCl3): δ = 4.02 ppm JP-H = 629 Hz; 1H-NMR (CDCl3): δ (ppm) 8.75 (s,1,H-2); 8.59 (s, 1, H-8); 7.9–7.3 (m, 28, H-arom.); 5.92 (s, 1, H-1'); 5.82 (t, 1, H-3''); 5.66 (2×d, 1, H-2''); 5.12 (d, H-1″); 5.09 (m, 1, H-3'); 4.62 (m, 1, H-2') 4.39 (d, 1, H-5'); 4.29 (d, 1, H-5'); 4.22–4.05 (m, 3, H-4', 5'', 4''); 3.76 (s,6,O-CH3); 3.52 (2×d, 2, CH2); 1.31 (t, 3, CH3).
- 9c: yield 91%; 31P-NMR (CDCl3): δ = 1.458 ppm JP-H = 603 Hz; 1H-NMR (CDCl3): δ (ppm) 13.60 (s, 1, N-H); 12.38 (s, 1, N-H); 7.82 (s, 1, H-8); 7.97–7.85 (m, 4, arom); 7.53–7.15 (m, 15, H-arom.); 6.82–6.76 (m, 4, H-arom); 6.42 (d, 1, H-1'); 5.92 (s, 1, H-1''); 5.82 (t, 1, H-3''); 5.67 (m, 1, H-2''); 5.33 (m, 1, H-4''); 4.81 (m, 1, H-3'); 4.58 (d, 1, H-2'); 4.53 (m, 1, H-4'); 4.41 (2×d, 1, H-5'); 4.17–4.12 (m, 3, H-5', 5'', 5″); 3.76 (s, 6, CH3) 2.34 (m, 1, CH of ibu); 1.19 (s, 3, CH3 of ibu); 1.17 (s, 3, CH3 of ibu).
3.8. Synthesis of 6-N-Benzoyl-5'-O-(4,4'-dimethoxytrityl)-2'-O-(5″-dicyanoethyl phosphotriester 2″,3″-di-O-benzoyl-β-d-ribofuranosyl)adenosine (11a) and 2-N-isobutyryl-5'-O-(4,4'-dimethoxy-trityl)-2'-O-(5″-dicyanoethyl phosphotriester 2″,3″-di-O-benzoyl-β-d-ribofuranosyl)guanosine (11c)
- 10a: 31P-NMR (Py): δ = 9.351 ppm JP-H = 719 Hz and δ = 9.153 ppm JP-H = 720 Hz
- 10c: 31P-NMR (Py): δ = 9.748 ppm JP-H = 726 Hz and δ = 9.428 ppm JP-H = 720 Hz
- 11a: yield 42%; 31P-NMR (CDCl3): δ = −2.29 ppm; 1H-NMR (CDCl3): δ (ppm) 8.69 (s, 1, H-2); 8.36 (s, 1, H-8); 8.1–7.26 (m, 28, arom); 6.88 (2×d, 4, trytyl-arom) 6.39 (d, 1, H-1'); 5.78 (t, 1, H-3''); 5.68 (2×d, 1, H-2''); 5.42 (d, H-1''); 5.25 (t, 1, H-3'); 4.70 (t, 1, H-2') 4.47 (m, 1, H-4'); 4.37–4.25 (m, 6, H-5', 4'', CH2); 4.17 (m, 1, H-5') 3.79 (s, 6, O-CH3); 3.52 (2×d, 1, H-5''); 3.44 (2×d, 1, H-5''); 2.79 (m, 4, CH2CN).
- 11c: yield 37%; 31P-NMR (CDCl3): δ = −2.031 ppm; 1H-NMR (CDCl3): δ (ppm) 12.20 (s, 1, N-H); 10.25 (s, 1, N-H); 7.74 (s, 1, H-8); 7.96–7.85 (m, 4, arom); 7.58–7.21 (m, 15, arom); 6.85–6.79 (m, 4, arom); 6.32 (d, 1, H-1'); 5.89 (t, 1, H-3''); 5.30 (s, 1, H-1''); 5.70 (m, 1, H-2″); 4.71 (t, 1, H-3'); 4.57 (m, 1, H-4''); 4.55 (d, 1, H-2'); 4.48 (m, 1, H-4'); 4.46 (2×d, 1, CH2); 4.46–4.39 (m,3, H-5', 5', 5'', 5''); 3.77 (s, 6, CH3) 2.34 (m, 1, CH of i-bu); 1.19 (s, 3, CH3 of i-bu); 1.17 (s, 3, CH3 of i-bu).
3.9. Synthesis of 6-N-Benzoyl-3'-[(2-O-cyanoethyl)-N,N-diisopropylphosphoramidite]-5'-O-(4,4'-dimethoxytrityl)-2'-O-(5″-dicyanoethyl phosphotriester 2″,3″-di-O-benzoyl-β-d-ribofuranosyl)-adenosine (12a) and 2-N-Isobutyryl-3'-[(2-O-cyanoethyl)-N,N-diisopropylphosphoramidite]-5'-O-(4,4'-dimethoxytrityl)-2'-O-(5″-dicyanoethyl phosphotriester 2″,3″-di-O-benzoyl-β-d-ribofuranosyl)-guanosine (12c)
- 12a: yield 45%;31P-NMR (benzene): δ (ppm) = −2.48, −2.42, 149.97, 150.58; FAB-MS: m/z [M−H]−; Calcd for C72H76N9O17P2+, 1400.48; found m/z 1401.4
- 12c: yield 42%; 31P-NMR (benzene): δ (ppm) = −3.09, 149.93, 14970.
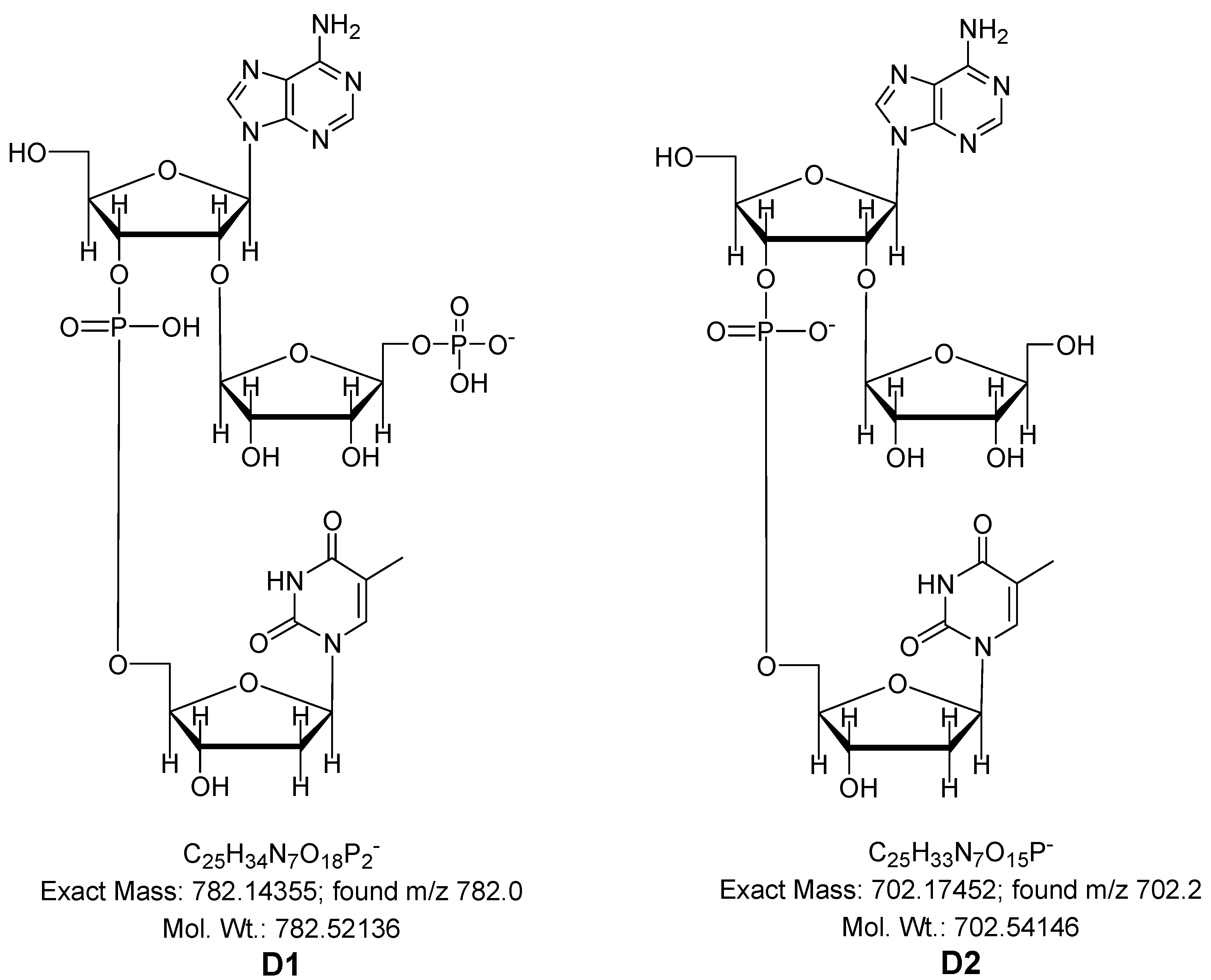
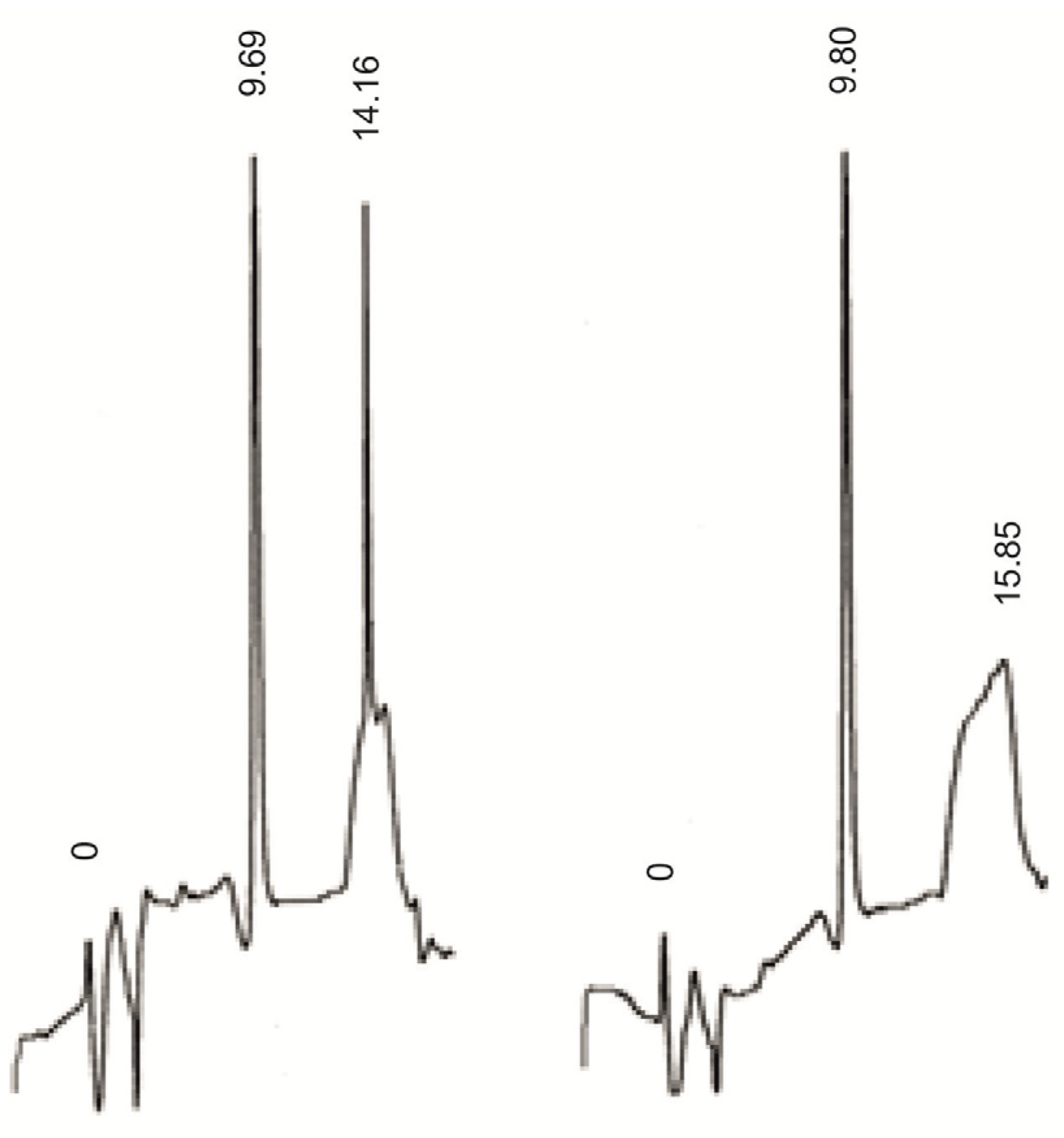
3.10. Synthesis of Dimers ArT and Ar(p)T
- Ar(p)T (D1); FAB-MS: m/z [M−H]−; Calcd for C25H34O18N7P2, 782.14; found m/z 782.0.
- ArT (D2); FAB-MS: m/z [M–H]−; Calcd for C25H33O15N7P, 702.17; found m/z 702.2.
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Kiesewetter, S.; Ott, G.; Sprinzl, M. The role of modified purine 64 in initiator/elongator discrimination of tRNA(iMet) from yeast and wheat germ. Nucl. Acid Res. 1990, 18, 4677–4682. [Google Scholar] [CrossRef]
- Glasser, A.; Desgres, J.; Heitzler, J.; Gehrke, C.W.; Keith, G. O-ribosyl-phosphate purine as a constant modified nucleotide located at position 64 in cytoplasmic initiator tRNAs(Met) of yeasts. Nucleic Acid Res. 1991, 19, 5199–5203. [Google Scholar] [CrossRef]
- Basavappa, R.; Sigler, P.B. The 3 A crystal structure of yeast initiator tRNA: Functional implications in initiator/elongator discrimination. EMBO J. 1991, 10, 3105–3011. [Google Scholar]
- Forster, C.; Chakraburtty, K.; Sprinzl, M. Discrimination between initiation and elongation of protein biosynthesis in yeast: Identity assured by a nucleotide modification in the initiator tRNA. Nucleic Acid Res. 1993, 21, 5679–5683. [Google Scholar] [CrossRef]
- Niewczyk, A.; Krzyżaniak, A.; Barciszewski, J.; Markiewicz, W.T. Studies on the synthesis of O-ribosyl-adenosine a new minor nucleoside of tRNA. Nucleos. Nucleot. 1991, 10, 635–638. [Google Scholar] [CrossRef]
- Efimtseva, E.V.; Kulikova, I.V.; Mikhailov, S.N. Disaccharide nucleosides as an important group of natural compounds. Mol. Biol. 2009, 43, 301–312. [Google Scholar] [CrossRef]
- Efimtseva, E.V.; Kulikova, I.V.; Mikhailov, S.N. Disaccharide nucleosides and their incorporation into oligonucleotides. Curr. Org. Chem. 2007, 11, 335–354. [Google Scholar]
- Markiewicz, W.T.; Niewczyk, A.; Gdaniec, Z.; Adamiak, D.; Dauter, Z.; Rypniewski, W.; Chmielewski, M. Studies on synthesis and structure of O-β-d-ribofuranosyl(1''-2')ribonucleosides and oligonucleotides. Nucleos. Nucleot. 1998, 17, 411–424. [Google Scholar] [CrossRef]
- Mikhailov, S.; Debruyn, A.; Herdewijn, P. Synthesis and properties of some 2'-O-d-ribofuranosyl-nucleosides. Nucleos. Nucleot. 1995, 14, 481–484. [Google Scholar] [CrossRef]
- Efimtseva, E.V.; Ermolinsky, B.S.; Fomitcheva, M.V.; Meshkov, S.V.; Padyukova, N.S.; Mikhailov, S.N.; Aerschot, A.V.; Rozenski, J.; Herdewijn, P. Regioselective incorporation of reactive dialdehyde groups into synthetic oligonucleotides. Collect. Czech. Chem. Commun. 1996, 61, 206–209. [Google Scholar]
- Pearson, D.; Hienzsch, A.; Wagner, M.; Globisch, D.; Reiter, V.; Özden, D.; Carell, T. LC-MS Based Quantification of 2'-Ribosylated Nucleosides Ar(p) and Gr(p) in tRNA. Chem. Commun. 2011, 47, 5196–5198. [Google Scholar] [CrossRef] [Green Version]
- Gurskaya, G.V.; Zhukhlistova, N.E.; Nekrasov, Y.V.; Bobkov, G.V.; Efimtseva, E.V.; Mikhailov, S.N. Disaccharide Nucleosides: The Crystal and Molecular Structure of 2'-O-β-d-Ribopyranosylcytidine. Crystallogr. Rep. 2005, 50, 395–399. [Google Scholar] [CrossRef]
- Rodinov, A.A.; Efimtseva, E.V.; Mikhailov, S.N.; Rozenski, J.; Luyten, I.; Herdewijn, P. Synthesis and properties of O-β-d-ribofuranosyl-(1''-2')-adenosine-5''-O-phosphate and its derivatives. Nucleos. Nucleot. Nucleic Acids 2000, 19, 1847–1859. [Google Scholar] [CrossRef]
- Rodionov, A.A.; Efimtseva, E.V.; Mikhailov, S.N. Synthesis of O-β-d-ribofuranosyl-(1''-2')-adenosine-5''-O-phosphate. Nucleos. Nucleot. 1999, 18, 623–624. [Google Scholar] [CrossRef]
- Efimtseva, E.V.; Shelkunova, A.A.; Mikhailov, S.N.; Naawelaerts, K.; Rozenski, J.; Lescrinier, E.; Herdewijn, P. Synthesis and conformational properties of O-β-d-ribofuranosyl-(1"-2')-guanosine and (adenosine)-5"-phosphate. Nucleos. Nucleot. Nucleic Acids 2003, 22, 1109–1111. [Google Scholar] [CrossRef]
- Mikhailov, S.N.; Efimtseva, E.V.; Rodionov, A.A.; Shelkunova, A.A.; Rozenski, J.; Emmerechts, G.; Schepers, G.; van Aerschot, A.; Herdewijn, P. Synthesis of RNA Containing O-β-d-Ribofuranosyl-(1″-2′)-adenosine-5″-phosphate and 1-Methyladenosine, Minor Components of tRNA. Chem. Biodivers. 2005, 2, 1153–1163. [Google Scholar] [CrossRef]
- Luyten, I.; Esnouf, R.M.; Mikhailov, S.N.; Efimtseva, E.V.; Michiels, P.; Heus, H.A.; Hilbers, C.W.; Herdewijn, P. Solution structure of a RNA decamer duplex, containing 9-[2-O-(β-d-ribofuranosyl)-β-d-ribofuranosyl]adenine, a special residue in lower eukaryotic initiator tRNAs. Helv. Chim. Acta 2000, 83, 1278–1289. [Google Scholar] [CrossRef]
- Efimtseva, E.V.; Victorova, L.S.; Rodionov, A.A.; Ermolinsky, B.S.; Fomitcheva, M.V.; Tunitskaya, V.L.; Mikhailov, S.N.; Oivanen, M.; Aerschot, A.V.; Herdewijn, P. Disaccharide nucleosides and their enzymatic and chemical incorporation into oligonucleotides. Nucleos. Nucleot. 1998, 17, 1681–1684. [Google Scholar] [CrossRef]
- Efimtseva, E.V.; Shelkunova, A.A.; Mikhailov, S.N.; Nauwelaerts, K.; Rozenski, J.; Lescrinier, E.; Herdewijn, P. Synthesis and properties of O-β-d-ribofuranosyl-(1''-2')-guanosine-5''-O-phosphate and its derivatives. Helv. Chim. Acta 2003, 86, 504–514. [Google Scholar] [CrossRef]
- Jankowska, J.; Sobkowski, M.; Stawiński, J.; Kraszewski, A. Studies on aryl H-phosphonates. 1. An efficient method for the preparation of deoxyribo- and ribonucleoside 3'-H-phosphonate monoesters by transesterification of diphenyl H-phosphonate. Tetrahedron Lett. 1994, 35, 3355–3358. [Google Scholar] [CrossRef]
- Sobkowski, M.; Stawiński, J.; Sobkowska, A.; Kraszewski, A. Studies on reactions of nucleoside H-phosphonates with bifunctional reagents. 2. Stability of nucleoside H-phosphonate diesters in the presence of amino alcohols. J. Chem. Soc. Perkin Trans. 1 1994, 13, 1803–1808. [Google Scholar]
- Garegg, P.J.; Regberg, T.; Stawiński, J.; Stromberg, R. Nucleoside phosphonates: Part 7. Studies on the oxidation of nucleoside phosphonate esters. J. Chem. Soc. Perkin Trans. 1 1987, 1269–1273. [Google Scholar]
- Chmielewski, M.K. Chemical Synthesis of Oligonucleotides with 2'-ribosyl units. Ph.D. Thesis, Polish Academy of Sciences, Poznan, Poland, 2000. [Google Scholar]
- Stawiński, J.; Stromberg, R.; Zain, R. Stereospecific oxidation and oxidative coupling of H-phosphonate and H-phosphonothioate diesters. Tetrahedron Lett. 1992, 33, 3185–3188. [Google Scholar] [CrossRef]
- Kierzek, R.; Rozek, M.; Markiewicz, W.T. Some steric aspects of synthesis of oligoribonucleotides by phosphoroamidite approach on solid support. Bull. Polish Acad. Sci. Chem. 1987, 35, 507–516. [Google Scholar]
- Markiewicz, W.T.; Biała, E.; Kierzek, R. Application of the Tetraisopropyldisiloxane-1,3-Diyl Group in the Chemical Synthesis of Oligoribonucleotides. Bull. Polish Acad. Sci. Chem. 1984, 32, 433–451. [Google Scholar]
- Sample Availability: Samples of the compounds 7 and 12 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chmielewski, M.K.; Markiewicz, W.T. Novel Method of Synthesis of 5''-Phosphate 2'-O-ribosyl-ribonucleosides and Their 3'-Phosphoramidites. Molecules 2013, 18, 14780-14796. https://doi.org/10.3390/molecules181214780
Chmielewski MK, Markiewicz WT. Novel Method of Synthesis of 5''-Phosphate 2'-O-ribosyl-ribonucleosides and Their 3'-Phosphoramidites. Molecules. 2013; 18(12):14780-14796. https://doi.org/10.3390/molecules181214780
Chicago/Turabian StyleChmielewski, Marcin K., and Wojciech T. Markiewicz. 2013. "Novel Method of Synthesis of 5''-Phosphate 2'-O-ribosyl-ribonucleosides and Their 3'-Phosphoramidites" Molecules 18, no. 12: 14780-14796. https://doi.org/10.3390/molecules181214780
APA StyleChmielewski, M. K., & Markiewicz, W. T. (2013). Novel Method of Synthesis of 5''-Phosphate 2'-O-ribosyl-ribonucleosides and Their 3'-Phosphoramidites. Molecules, 18(12), 14780-14796. https://doi.org/10.3390/molecules181214780