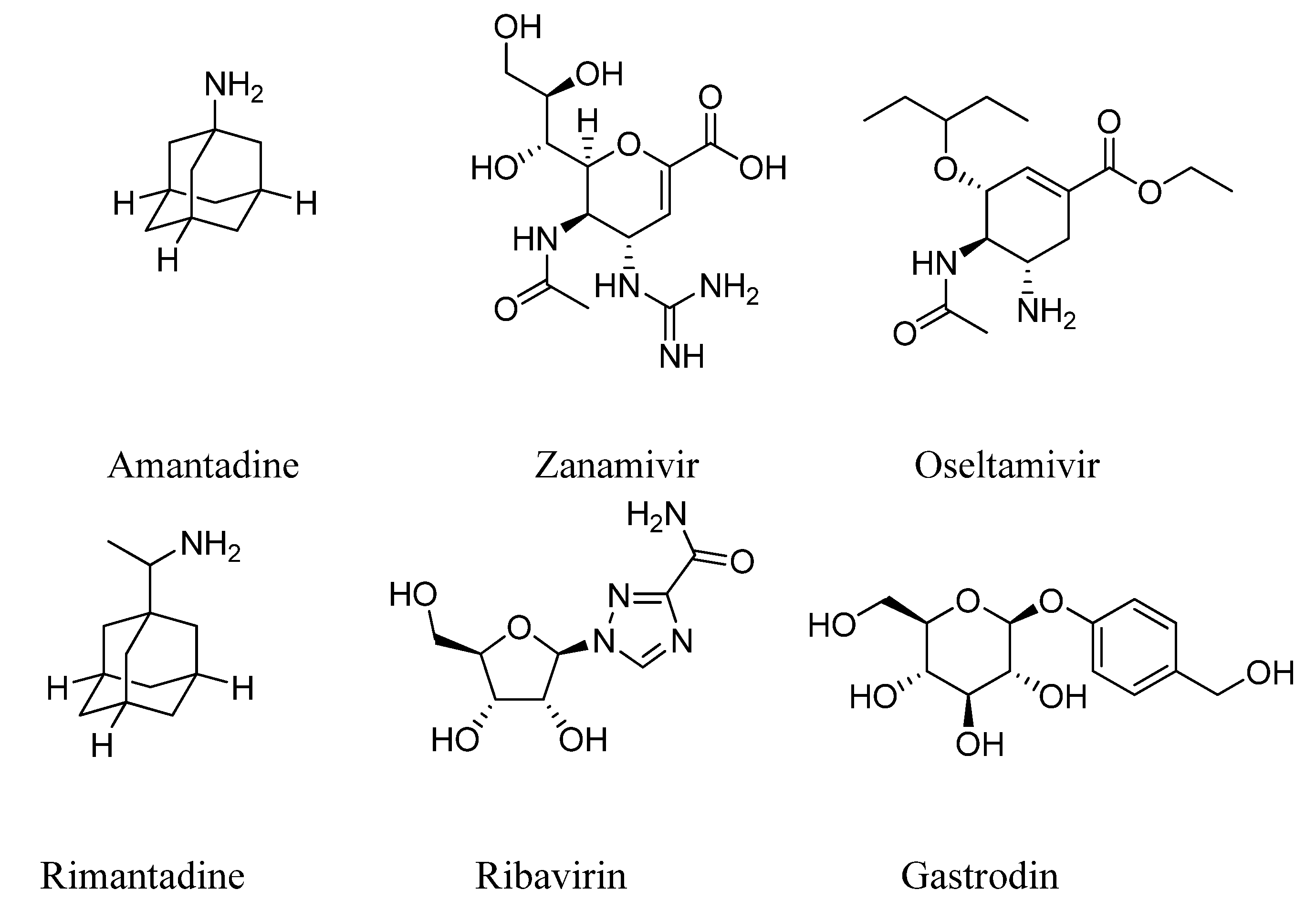
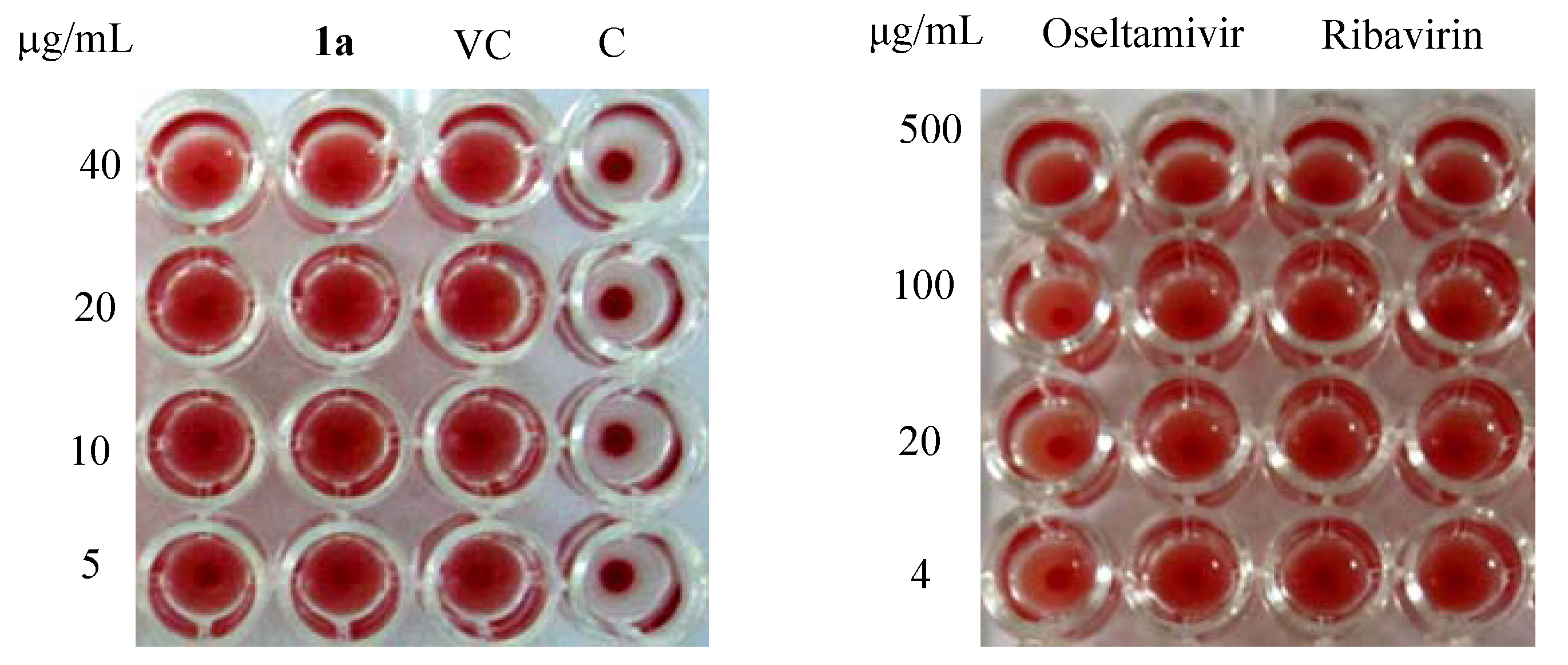
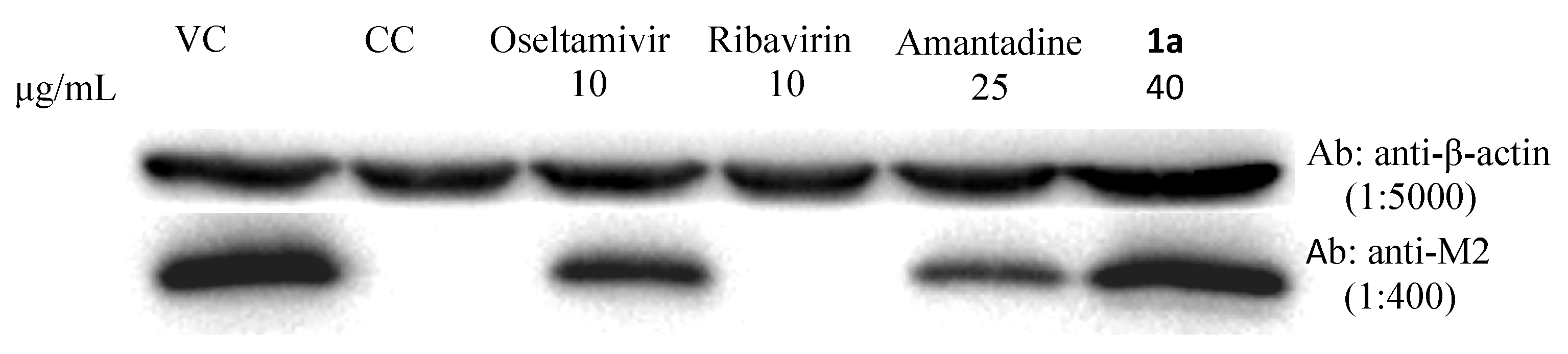
3.2. Synthesis
Methyl 4-fluoro-3-((2S,3R,4S,5R,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)-tetrahydro-2H-pyran-2-yloxy)benzoate (1a). Methyl 4-fluoro-3-hydroxybenzoate (1, 1.7 g, 10 mmol), 2,3,4,6-tetra-O-acetyl-α-d-glucopyranosyl bromide (5 g, 12 mmol) and K2CO3 (1.7 g, 12 mmol) were added to anhydrous acetone (75 mL), and the resulting mixture was stirred at 56 °C for 16 h before being cooled to ambient temperature and filtered. The solvent was then evaporated in vacuo to give a sticky residue, which was partitioned between dichloromethane (60 mL) and a 0.1 N NaOH solution (20 mL). The organic phase was collected and concentrated, and the resulting solid was purified by column chromatography on silica gel to yield compound 1a (1.65 g, 36%). White solid; m.p.: 144–146 °C; 1H-NMR (400 MHz, CDCl3) δ (ppm): 7.86 (1H, d, J = 7.6 Hz, PhH), 7.79 (1H, m, PhH), 7.14 (1H, t, J = 8.0 Hz, PhH), 5.15 (4H, m, CH), 4.28 (2H, m, CH), 3.93 (1H, d, J = 4.0 Hz, CH), 3.90 (3H, s, OCH3), 2.10 (3H, s, CH3), 2.07 (3H, s, CH3), 2.02 (6H, s, CH3); ESI-MS (m/z): 523 [M+Na]+; HRMS-ESI (m/z): calculated for C22H26FO12 [M+H]+: 501.14083; measured: 501.14078.
Methyl 2,6-dimethoxy-4-((2S,3R,4S,5R,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)-tetrahydro-2H-pyran-2-yloxy)benzoate (2a). Compound 2a was synthesized using a method similar to that described above for 1a. Yield: 35%, white solid; m.p.: 121–123 °C; 1H-NMR (400 MHz, CDCl3) δ (ppm): 6.50 (2H, s, PhH), 5.28 (3H, m, CH), 5.15 (1H, d, J = 7.2 Hz, CH), 4.22 (1H, m, CH), 4.09 (1H, d, J = 8.4 Hz, CH), 3.89 (3H, s, OCH3), 3.85 (6H, s, OCH3), 3.69 (1H, m, CH), 2.04 (3H, s, CH3), 2.01 (9H, s, CH3); ESI-MS (m/z): 565 [M+Na]+; HRMS-ESI (m/z): calculated for C24H31O14 [M+H]+: 543.17138; measured: 543.17148.
Ethyl 3,5-dichloro-4-((2S,3R,4S,5R,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)-tetrahydro-2H-pyran-2-yloxy)benzoate (3a). Compound 3a was synthesized using a method similar to that described above for 1a. Yield: 32%, white solid; m.p.: 112–114 °C; 1H-NMR (400 MHz, CDCl3) δ (ppm): 7.93 (2H, s, PhH), 6.39 (1H, s, CH), 5.30 (3H, m, CH), 4.33 (3H, m, CH), 4.11 (2H, m, CH), 2.06 (3H, s, CH3), 2.03 (9H, s, CH3), 1.33 (3H, s, CH3); ESI-MS (m/z): 587 [M+Na]+; HRMS-ESI (m/z): calculated for C22H25Cl2O12 [M+H]+: 551.07231; measured: 551.07226.
Methyl 4-amino-3-((2S,3R,4S,5R,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)-tetrahydro-2H-pyran-2-yloxy)benzoate (4a). Compound 4a was synthesized using a method similar to that described above for 1a. Yield: 29%; brown solid; m.p.: 126–128 °C; 1H-NMR (400 MHz, CDCl3) δ (ppm): 7.84 (1H, d, J = 6.0 Hz, PhH), 7.77 (1H, m, PhH), 7.10 (1H, t, J = 8.8. Hz, PhH), 5.54 (1H, t, J = 8.4 Hz, CH), 5.44 (1H, d, J = 3.2 Hz, CH), 5.14 (1H, dd, J = 8.8 Hz, J = 3.2 Hz, CH), 5.02 (1H, d, J = 8.4 Hz, CH), 4.21 (2H, m, CH), 4.05 (1H, t, J = 6.4 Hz, CH), 3.90 (3H, s, OCH3), 3.74 (2H, s, NH2), 2.19 (3H, s, CH3), 2.10 (3H, s, CH3), 2.07 (3H, s, CH3), 2.00 (3H, s, CH3); ESI-MS (m/z): 520 [M+Na]+; HRMS-ESI (m/z): calculated for C22H28NO12 [M+H]+: 498.16115; measured: 498.16102.
Methyl 4-((2S,3R,4S,5R,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)-tetrahydro-2H-pyran-2-yloxy)benzoate (5a). Compound 5a was synthesized using a method similar to that described above for 1a. Yield: 30%; white solid; m.p.: 147–149 °C; 1H-NMR (400 MHz, CDCl3) δ (ppm): 8.00 (2H, d, J = 8.4 Hz, PhH), 7.00 (2H, d, J = 8.4 Hz, PhH), 5.53 (1H, d, J = 7.6 Hz, CH), 5.27 (2H, m, CH), 5.14 (1H, t, J = 6.4 Hz, CH), 5.02 (1H, d, J = 7.2 Hz, CH), 4.25 (4H, m, CH), 2.08 (3H, s, CH3), 2.05 (6H, s, CH3), 2.04 (3H, s, CH3), 1.38 (3H, s, CH3); ESI-MS (m/z): 519 [M+Na]+; HRMS-ESI (m/z): calculated for C23H29O12 [M+H]+: 497.16590; measured: 497.16594.
Methyl 3-fluoro-4-((2S,3R,4S,5R,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)-tetrahydro-2H-pyran-2-yloxy)benzoate (7a). Compound 7a was synthesized using a method similar to that described above for 1a. Yield: 31%; white solid; m.p.: 121–123 °C; 1H-NMR (500 MHz, CDCl3) δ (ppm): 7.83 (1H, t, J = 6.5 Hz, PhH), 6.90 (1H, dd, J = 10.5 Hz, J = 2.5 Hz, PhH), 6.83 (1H, t, J = 8.0 Hz, PhH), 5.57 (1H, d, J = 4.0 Hz, CH), 5.21 (4H, m, CH), 4.37 (2H, m, CH), 3.90 (3H, s, OCH3), 2.10 (6H, s, CH3), 2.06 (6H, s, CH3); ESI-MS (m/z): 523 [M+Na]+; HRMS-ESI (m/z): calculated for C22H26FO12 [M+H]+: 501.14083; measured: 501.14091.
Ethyl 4-fluoro-2-((2S,3R,4S,5R,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)-tetrahydro-2H-pyran-2-yloxy)-benzoate (8a). Compound 8a was synthesized using a method similar to that described above for 1a. Yield: 30%; white solid; m.p.: 136–138 °C; 1H-NMR (500 MHz, CDCl3) δ (ppm): 7.80 (1H, t, J = 7.5 Hz, PhH), 6.95 (1H, dd, J = 8.0 Hz , J = 2.0 Hz, PhH), 6.84 (1H, m, PhH), 5.92 (1H, t, J = 9.0 Hz, CH), 5.48 (1H, d, J = 3.5 Hz, CH), 5.08 (2H, m, CH), 4.17 (3H, m, CH), 3.85 (3H, s, OCH3), 2.16 (3H, s, CH3), 2.09 (3H, s, CH3), 2.05 (3H, s, CH3), 2.01 (3H, s, CH3); ESI-MS (m/z): 523 [M+Na]+; HRMS-ESI (m/z): calculated for C22H26FO12 [M+H]+: 501.14083; measured: 501.14082.
Methyl 2-fluoro-6-((2S,3R,4S,5R,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)-tetrahydro-2H-pyran-2-yloxy)benzoate (9a). Compound 9a was synthesized using a method similar to that described above for 1a. Yield: 35%; white solid; m.p.: 130–132 °C; 1H-NMR (500 MHz, CDCl3) δ (ppm): 7.31 (1H, q, J = 8.5 Hz, PhH), 6.90 (1H, d, J = 8.5 Hz, PhH), 6.86 (1H, t, J = 8.0 Hz, PhH), 5.27 (2H, m, CH), 5.18 (1H, t, J = 6.5 Hz, CH), 5.05 (1H, d, J = 7.5 Hz, CH), 4.25 (3H, m, CH), 3.89 (3H, s, OCH3), 2.11 (3H, s, CH3), 2.08 (3H, s, CH3), 2.05 (3H, s, CH3), 2.02 (3H, s, CH3); ESI-MS (m/z): 523 [M+Na]+; HRMS-ESI (m/z): calculated for C22H26FO12 [M+H]+: 501.14083; measured: 501.14077.
(2R,3R,4S,5R,6S)-2-(Acetoxymethyl)-6-(3-fluoro-2-methylphenoxy)-tetrahydro-2H-pyran-3,4,5-triyl triacetate (10a). Compound 10a was synthesized using a method similar to that described above for 1a. Yield: 33%; white solid; m.p.: 127–129 °C; 1H-NMR (500 MHz, CDCl3) δ (ppm): 7.62 (1H, m, PhH), 7.21 (1H, m, PhH), 6.90 (1H, dd, J = 9.5 Hz, J = 3.5 Hz, PhH), 6.33 (1H, d, J = 3.5 Hz, CH), 5.72 (1H, d, J = 8.0 Hz, CH), 5.50 (2H, m, CH), 4.57 (1H, t, J = 7.0 Hz, CH), 4.22 (2H, m, CH), 2.40 (3H, s, CH3), 2.07 (6H, s, CH3), 2.02 (6H, s, CH3); ESI-MS (m/z): 479 [M+Na]+; HRMS-ESI (m/z): calculated for C21H26FO10 [M+H]+: 457.15100; measured: 457.15095.
(2R,3R,4S,5R,6S)-2-(Acetoxymethyl)-6-(pyridin-4-yloxy)-tetrahydro-2H-pyran-3,4,5-triyl triacetate (11a). Compound 11a was synthesized using a method similar to that described above for 1a. Yield: 27%; colorless solid; m.p.: 132–134 °C; 1H-NMR (500 MHz, CDCl3) δ (ppm): 8.30 (2H, d, J = 9.0 Hz, PhH), 6.93 (2H, d, J = 9.0 Hz, PhH), 5.50 (1H, dd, J = 10.5 Hz, J = 3.0 Hz, CH), 5.18 (1H, d, J = 3.0 Hz, CH), 5.05 (1H, t, J = 7.0 Hz, CH), 4.15 (4H, m, CH), 2.08 (3H, s, CH3), 2.04 (3H, s, CH3), 2.02 (6H, s, CH3); ESI-MS (m/z): 448 [M+Na]+; HRMS-ESI (m/z): calculated for C19H24NO10 [M+H]+: 426.14002; measured: 426.14006.
(2R,3R,4S,5R,6S)-2-(Acetoxymethyl)-6-(3-fluoropyridin-2-yloxy)-tetrahydro-2H-pyran-3,4,5-triyl triacetate(12a). Compound 12a was synthesized using a method similar to that described above for 1a. Yield: 31%; pale yellow solid; m.p.: 140–142 °C; 1H-NMR (500 MHz, CDCl3) δ (ppm): 7.92 (1H, d, J = 5.0 Hz, PhH), 7.39 (1H, t, J = 8.5 Hz, PhH), 6.99 (1H, m, PhH), 6.18 (1H, d, J = 7.5 Hz, CH), 5.35 (2H, m, CH), 5.24 (1H, t, J = 9.5 Hz, CH), 4.30 (1H, dd, J = 12.5 Hz, J = 4.5 Hz, CH), 4.14 (1H, d, J = 12.5 Hz, CH), 3.94 (1H, m, CH), 2.10 (3H, s, CH3), 2.07 (3H, s, CH3), 2.04 (3H, s, CH3), 2.02 (3H, s, CH3); ESI-MS (m/z): 466 [M+Na]+; HRMS-ESI (m/z): calculated for C19H23FNO10 [M+H]+: 444.13060; measured: 444.13051.
(2R,3R,4S,5R,6S)-2-(Acetoxymethyl)-6-(5-(acetoxymethyl)-2-fluorophenoxy)-tetrahydro-2H-pyran-3,4,5-triyl triacetate (13a). Compound 1a (500 mg, 1 mmol) was added to THF (25 mL), and solid LiAlH4 was added in a portion-wise manner to the resulting solution. Upon completion of the addition, the mixture was stirred at 66 °C for 3 h before being cooled to ambient temperature and quenched by the dropwise addition of CH3OH (10 mL). The reaction mixture was then stirred at room temperature for 10 min and concentrated to dryness under vacuum. The resulting residue was then dissolved in a mixture of acetic anhydride (20 mL) and pyridine (1 mL). The reaction mixture was stirred at 80 °C for 4 h and then concentrated to dryness in vacuo. The residue was added to ice water and stirred until precipitation occurred. The mixture was then filtered, and the filter-cake was collected and purified by flash chromatography over silica gel, affording the title compound (401 mg, 78%). White solid; m.p.: 125–127 °C; 1H-NMR (500 MHz, CDCl3) δ (ppm): 7.80 (1H, d, J = 8.0 Hz, PhH), 7.74 (1H, m, PhH), 7.10 (1H, t, J = 8.5 Hz, PhH), 5.89 (1H, d, J = 6.5 Hz, CH), 5.30 (1H, dd, J = 10.5,4.0 Hz, CH), 5.07 (1H, t, J = 11.0 Hz, CH), 5.19 (2H, s, CH), 4.36 (1H, m, CH), 4.14 (3H, m, CH), 2.11 (3H, s, CH3), 2.08 (6H, s, CH3), 2.06 (3H, s, CH3), 2.04 (3H, s, CH3); ESI-MS (m/z): 537 [M+Na]+; HRMS-ESI (m/z): calculated for C23H28FO12 [M+H]+: 515.15648; measured: 515.15652.
(2S,3R,4S,5S,6R)-2-(2-Fluoro-5-(hydroxymethyl)phenoxy)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (14a). To a solution of 13a (510 mg, 1 mmol) in CH3OH (10 mL) was added a 0.2 N solution of CH3ONa in CH3OH, and the resulting mixture was stirred at room temperature for 1 h and then allowed to stand at ambient temperature overnight. The reaction mixture was filtered, and the filter cake was collected and recrystallized from anhydrous ethanol to afford the title compound (280 mg, 90%). White solid; m.p.: 134–136 °C; 1H-NMR (500 MHz, CDCl3) δ (ppm): 7.15 (1H, t, J = 9.5 Hz, PhH), 6.95 (1H, dd, J = 9.5 Hz, J = 4.0 Hz, PhH), 6.91 (1H, m, PhH), 5.60 (1H, s, CH), 4.66 (2H, s, CH), 4.20 (1H, s, CH), 3.47 (5H, m, CH), 2.76 (1H, s, OH), 2.16 (1H, s, OH), 1.78 (2H, s, OH), 0.99 (1H, s, OH); ESI-MS (m/z): 327 [M+Na]+; HRMS-ESI (m/z): calculated for C13H18FO7 [M+H]+: 305.10366; measured: 305.10362.
Methyl 4-fluoro-3-((2S,3R,4S,5S,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)-tetrahydro-2H-pyran-2-yloxy)benzoate (1b). Compound 1b was synthesized using a method similar to that described above for 1a. Yield: 38%; white solid; m.p.: 143–145 °C; 1H-NMR (500 MHz, CDCl3) δ (ppm): 7.88 (1H, d, J = 6.5 Hz, PhH), 7.78 (1H, m, PhH), 7.14 (1H, t, J = 9.0 Hz, PhH), 5.53 (1H, t, J = 8.5 Hz, CH), 5.46 (1H, d, J = 3.0 Hz, CH), 5.11 (1H, dd, J = 10.0, 3.0 Hz, CH), 5.01 (1H, d, J = 8.0 Hz, CH), 4.20 (2H, m, CH), 4.07 (1H, t, J = 6.5 Hz, CH), 3.90 (3H, s, OCH3), 2.19 (3H, s, CH3), 2.10 (3H, s, CH3), 2.07 (3H, s, CH3), 2.00 (3H, s, CH3); ESI-MS (m/z): 523 [M+Na]+; HRMS-ESI (m/z): calculated for C22H26FO12 [M+H]+: 501.14083; measured: 501.14086.
Methyl 3-((2S,3R,4S,5S,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)-tetrahydro-2H-pyran-2-yloxy)benzoate (6b). Compound 6b was synthesized using a method similar to that described above for 1a. Yield: 36%; white solid; m.p.: 154–156 °C; 1H-NMR (500 MHz, CDCl3) δ (ppm): 7.75 (1H, d, J = 8.0 Hz, PhH), 7.67 (1H, s, PhH), 7.37 (1H, t, J = 8.0 Hz, PhH), 7.20 (1H, dd, J = 8.0, 2.5 Hz, CH), 5.50 (2H, m, CH), 5.12 (2H, m, CH), 4.16 (3H, m, CH), 3.91 (3H, s, OCH3), 2.19 (3H, s, CH3), 2.07 (6H, s, CH3), 2.00 (3H, s, CH3); ESI-MS (m/z): 505 [M+Na]+; HRMS-ESI (m/z): calculated for C22H27O12 [M+H]+: 483.15025; measured: 483.15030.
Methyl 3-fluoro-4-((2S,3R,4S,5S,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)-tetrahydro-2H-pyran-2-yloxy)benzoate (7b). Compound 7b was synthesized using a method similar to that described above for 1a. Yield: 32%; white solid; m.p.: 121–123 °C; 1H-NMR (500 MHz, CDCl3) δ (ppm): 7.77 (2H, m, PhH), 7.21 (1H, t, J = 8.5 Hz, PhH), 5.52 (1H, t, J = 8.0 Hz, CH), 5.45 (1H, d, J = 3.0 Hz, CH), 5.11 (1H, dd, J = 10.5, 3.0 Hz, CH), 5.01 (1H, d, J = 7.5 Hz, CH), 4.20 (2H, m, CH), 4.05 (1H, m, CH), 3.90 (3H, s, OCH3), 2.15 (3H, s, CH3), 2.08 (3H, s, CH3), 2.03 (3H, s, CH3), 1.99 (3H, s, CH3); ESI-MS (m/z): 523 [M+Na]+; HRMS-ESI (m/z): calculated for C22H26FO12 [M+H]+: 501.14083; measured: 501.14076.
Methyl 4-fluoro-2-((2S,3R,4S,5S,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)-tetrahydro-2H-pyran-2-yloxy)benzoate (8b). Compound 8b was synthesized using a method similar to that described above for 1a. Yield: 30%; white solid; m.p.: 136–138 °C; 1H-NMR (500 MHz, CDCl3) δ (ppm): 7.83 (1H, t, J = 7.0 Hz, PhH), 6.92 (1H, dd, J = 8.5, 2.0 Hz, PhH), 6.83 (1H, m, PhH), 5.60 (1H, t, J = 8.0 Hz, CH), 5.47 (1H, d, J = 3.0 Hz, CH), 5.11 (1H, dd, J = 10.5, 3.5 Hz, CH), 5.04 (1H, d, J = 8.0 Hz, CH), 4.17 (3H, m, CH), 3.85 (3H, s, OCH3), 2.18 (3H, s, CH3), 2.08 (3H, s, CH3), 2.06 (3H, s, CH3), 2.02 (3H, s, CH3); ESI-MS (m/z): 523 [M+Na]+; HRMS-ESI (m/z): calculated for C22H26FO12 [M+H]+: 501.14083; measured: 501.14074.
Methyl 2-fluoro-6-((2S,3R,4S,5S,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)-tetrahydro-2H-pyran-2-yloxy)benzoate (9b). Compound 9b was synthesized using a method similar to that described above for 1a. Yield: 35%; white solid; m.p.: 130–132 °C; 1H-NMR (500 MHz, CDCl3) δ (ppm): 7.31 (1H, q, J = 8.5 Hz, PhH), 6.92 (1H, d, J = 8.0 Hz, PhH), 6.84 (1H, t, J = 8.5 Hz, PhH), 5.47 (2H, m, CH), 5.07 (1H, dd, J = 10.5 Hz, J = 3.0 Hz, CH), 5.00 (1H, d, J = 8.0 Hz, CH), 4.16 (3H, m, CH), 3.90 (3H, s, OCH3), 2.21 (3H, s, CH3), 2.12 (3H, s, CH3), 2.06 (3H, s, CH3), 2.03 (3H, s, CH3); ESI-MS (m/z): 523 [M+Na]+; HRMS-ESI (m/z): calculated for C22H26FO12 [M+H]+: 501.14083; measured: 501.14077.
(2R,3S,4S,5R,6S)-2-(Acetoxymethyl)-6-(3-fluoro-2-methylphenoxy)-tetrahydro-2H-pyran-3,4,5-triyl triacetate (10b). Compound 10b was synthesized using a method similar to that described above for 1a. Yield: 36%; white solid; m.p.: 127–129 °C; 1H-NMR (500 MHz, CDCl3) δ (ppm): 7.60 (1H, m, PhH), 7.20 (1H, m, PhH), 6.93 (1H, dd, J = 9.5, 3.5 Hz, PhH), 6.32 (1H, d, J = 3.5 Hz, CH), 5.74 (1H, d, J = 8.0 Hz, CH), 5.50 (2H, m, CH), 4.54 (1H, t, J = 7.0 Hz, CH), 4.24 (2H, m, CH), 2.40 (3H, s, CH3), 2.09 (3H, s, CH3), 2.07 (3H, s, CH3), 2.04 (3H, s, CH3), 2.02 (3H, s, CH3); ESI-MS (m/z): 479 [M+Na]+; HRMS-ESI (m/z): calculated for C21H26FO10 [M+H]+: 457.15100; measured: 457.15098.




{kind=link}
{kind=link}
{kind=link}
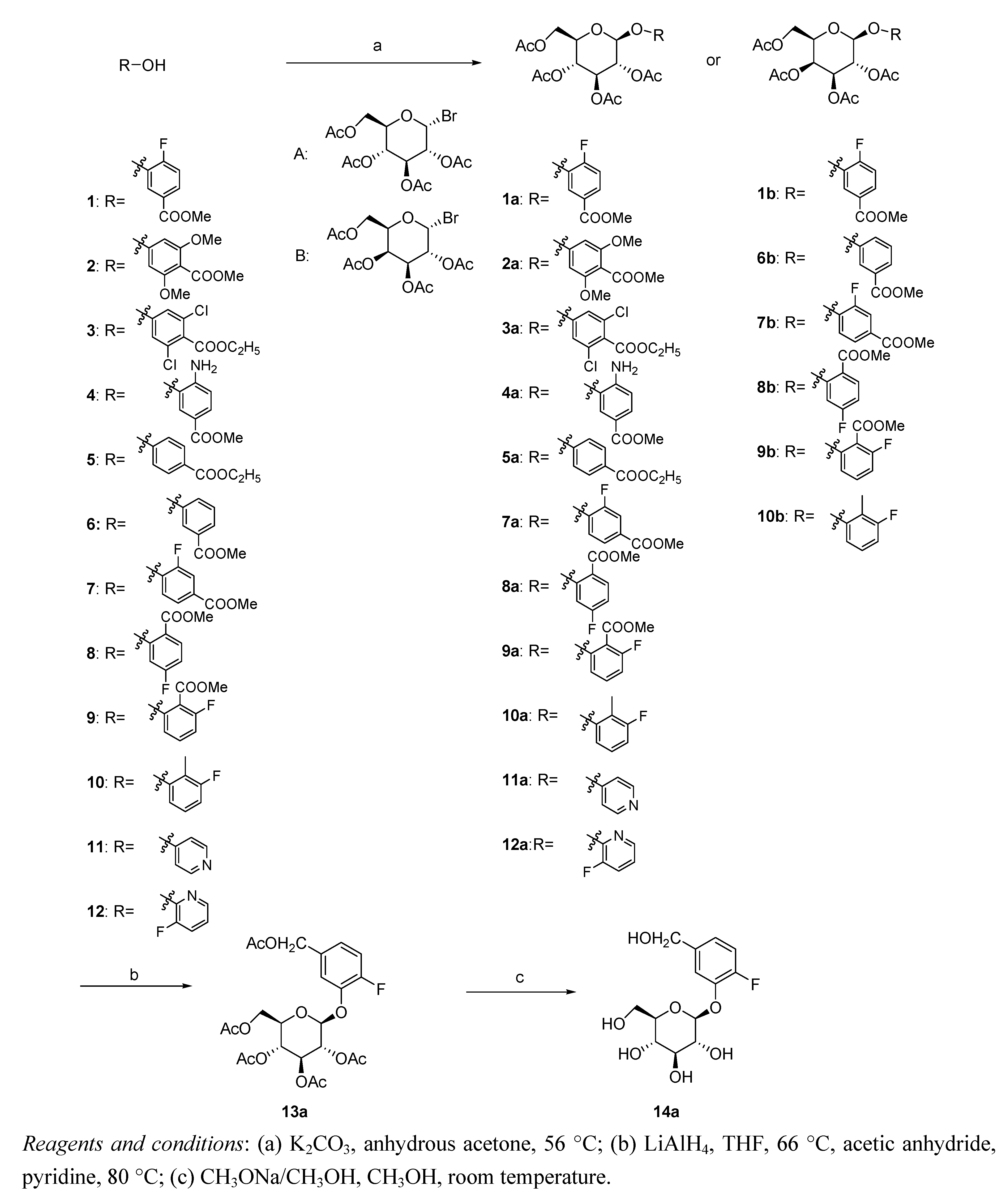{kind=link}

