Synthesis and Biological Evaluation of Thieno[3,2-d]- pyrimidinones, Thieno[3,2-d]pyrimidines and Quinazolinones: Conformationally Restricted 17b-Hydroxysteroid Dehydrogenase Type 2 (17b-HSD2) Inhibitors

Abstract
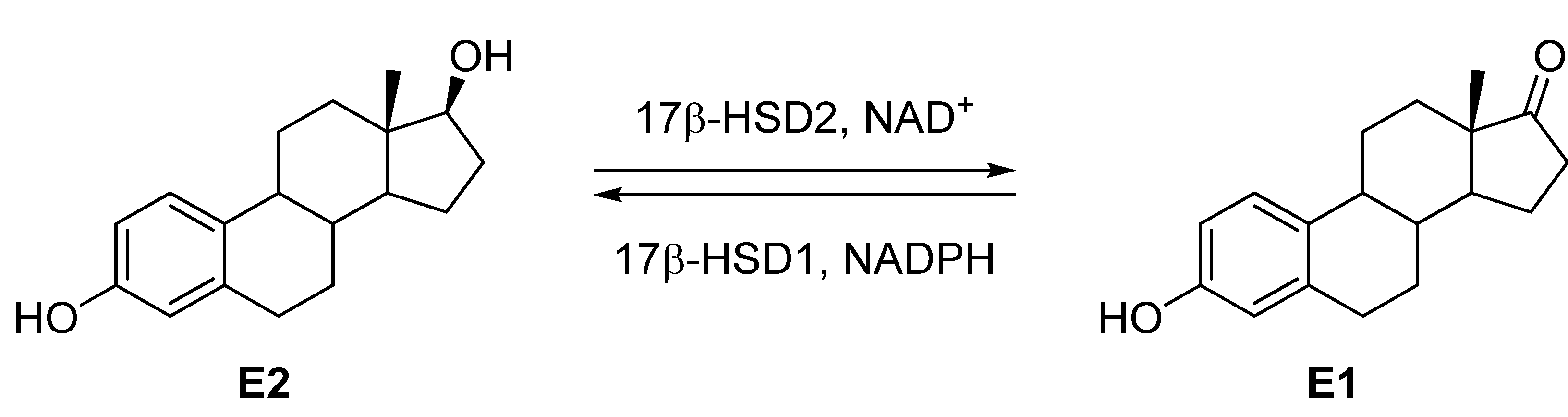
:1. Introduction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
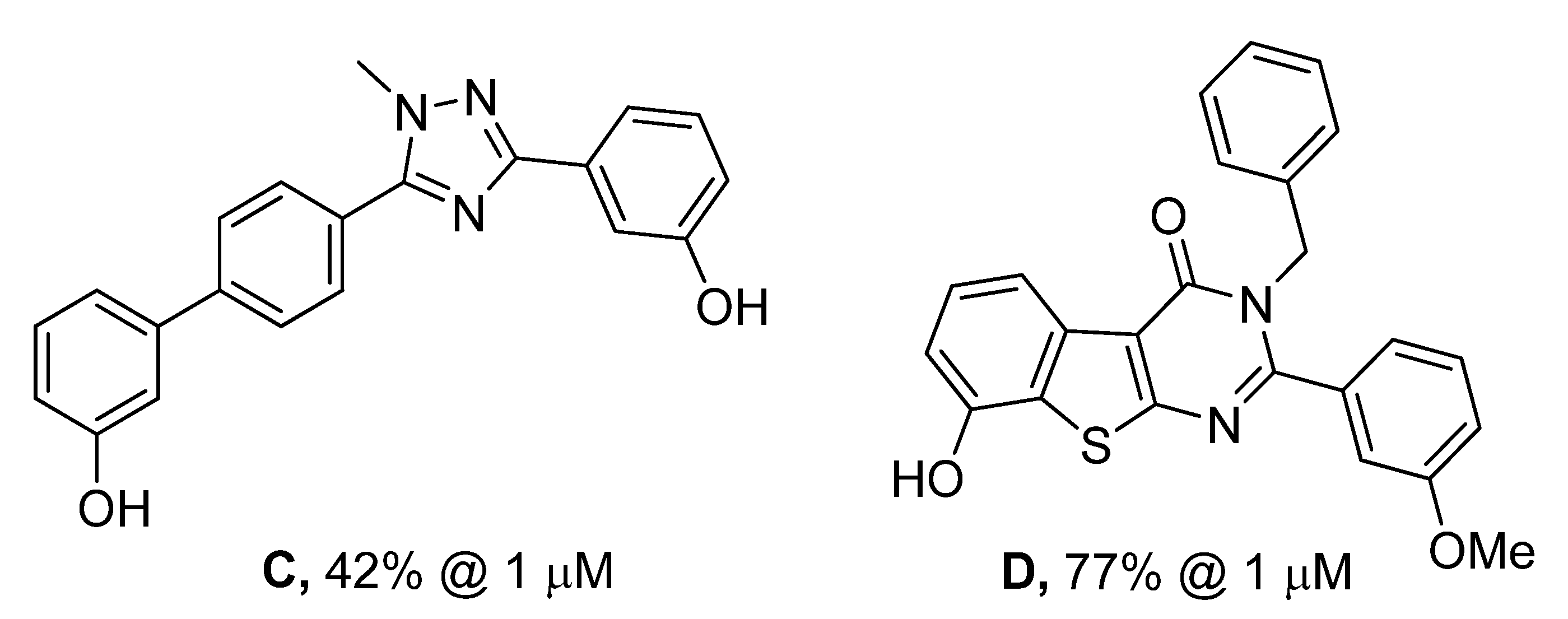{kind=link}
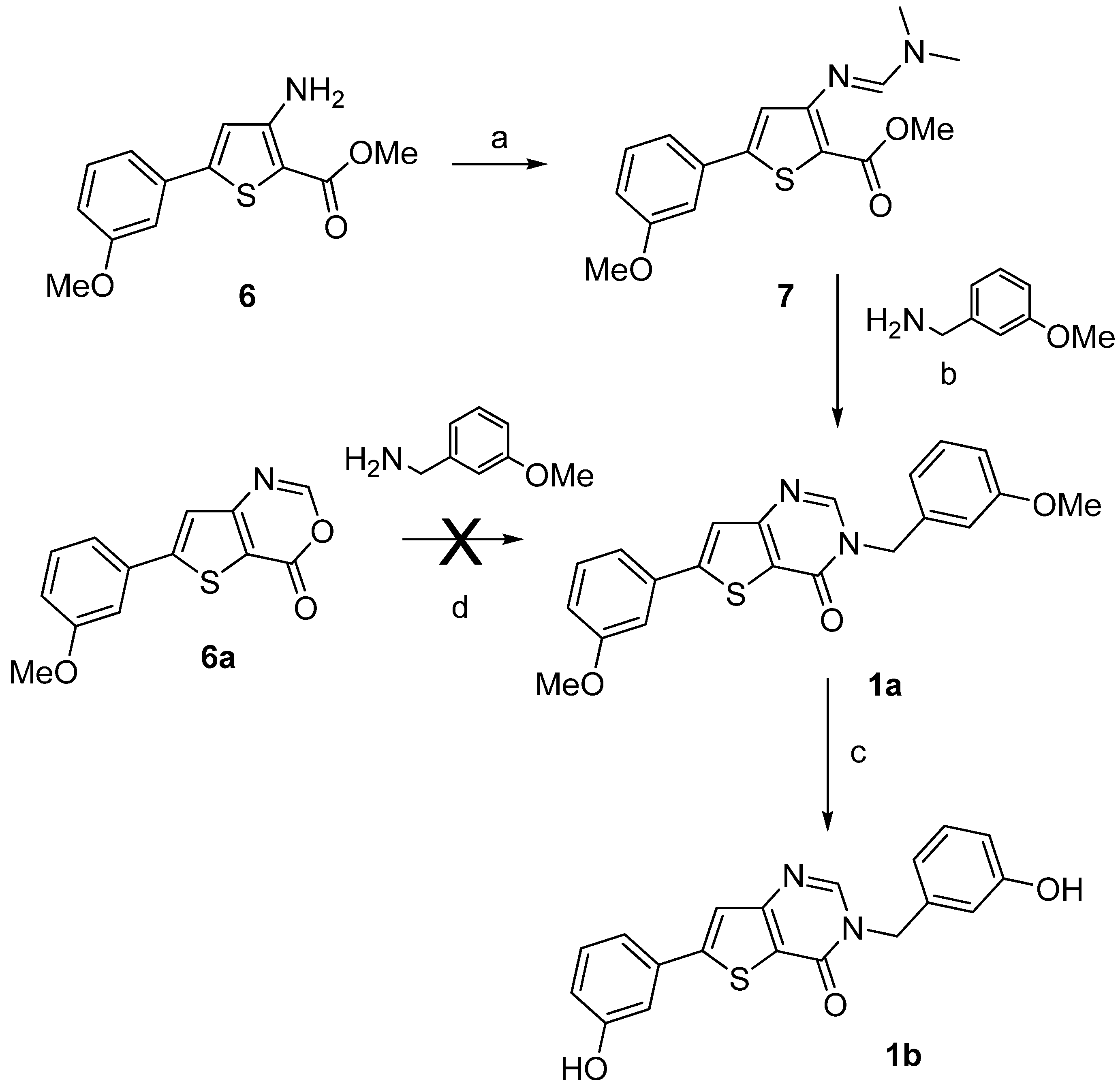{kind=link}
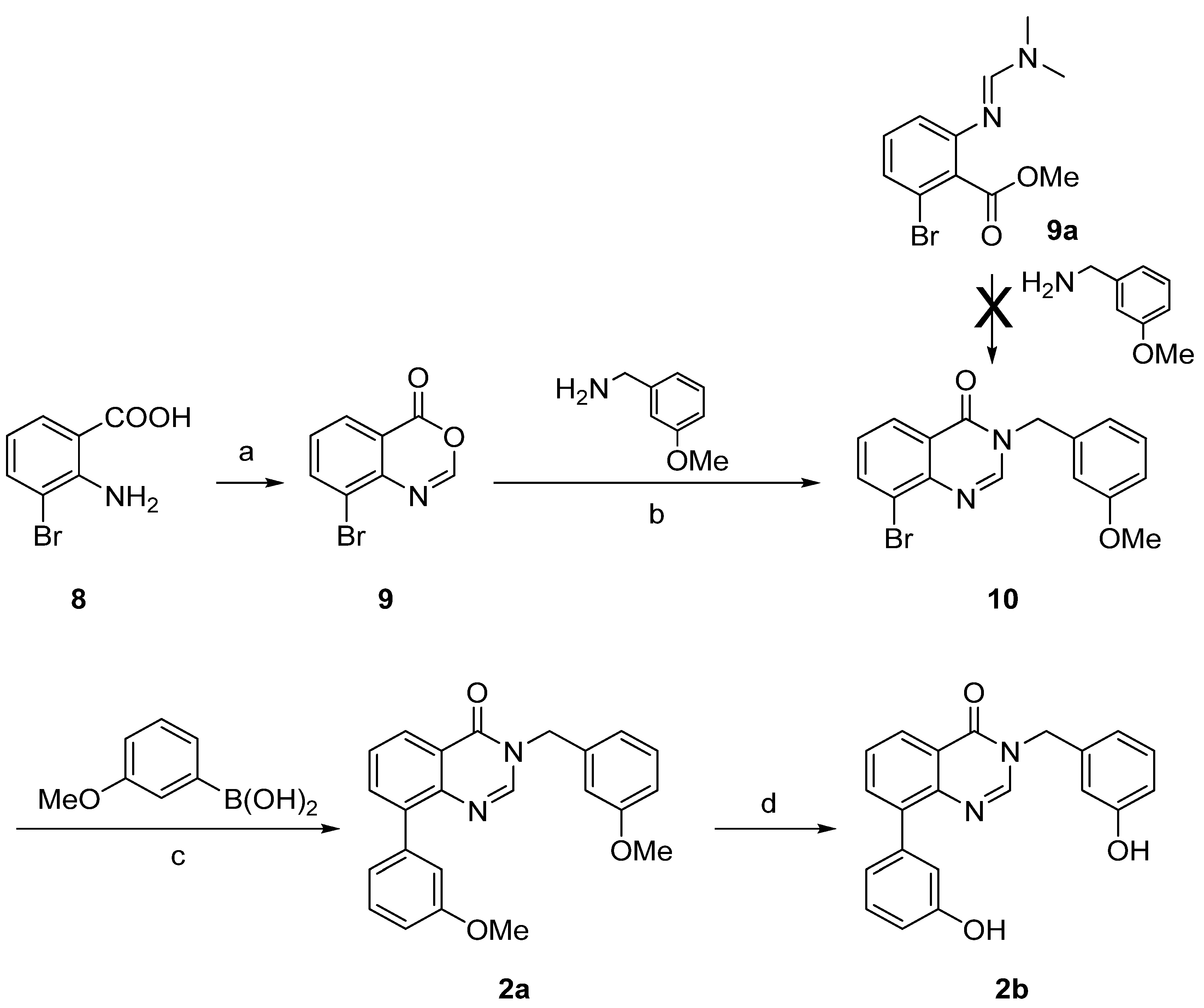{kind=link}
{kind=link}
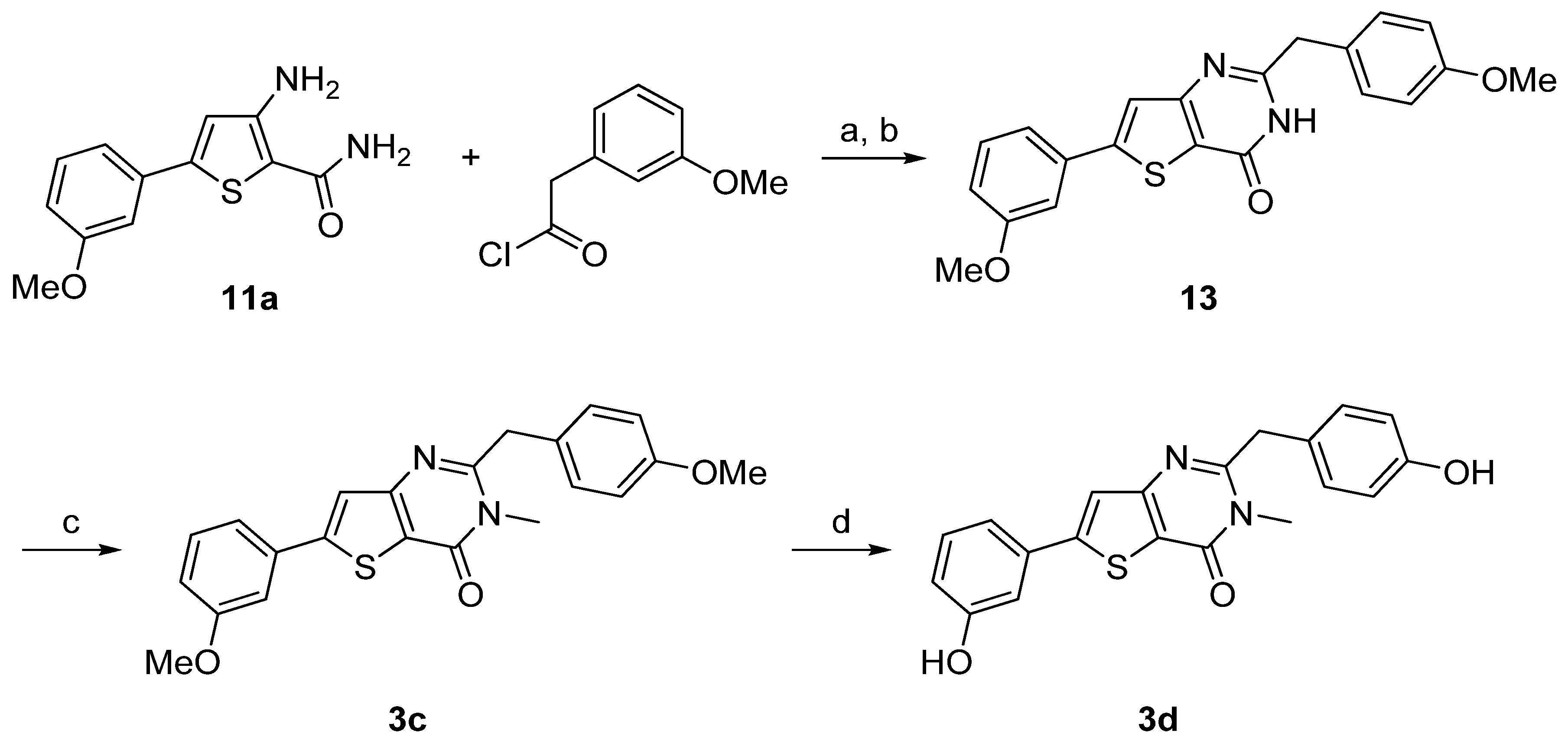{kind=link}
{kind=link}
{kind=link}

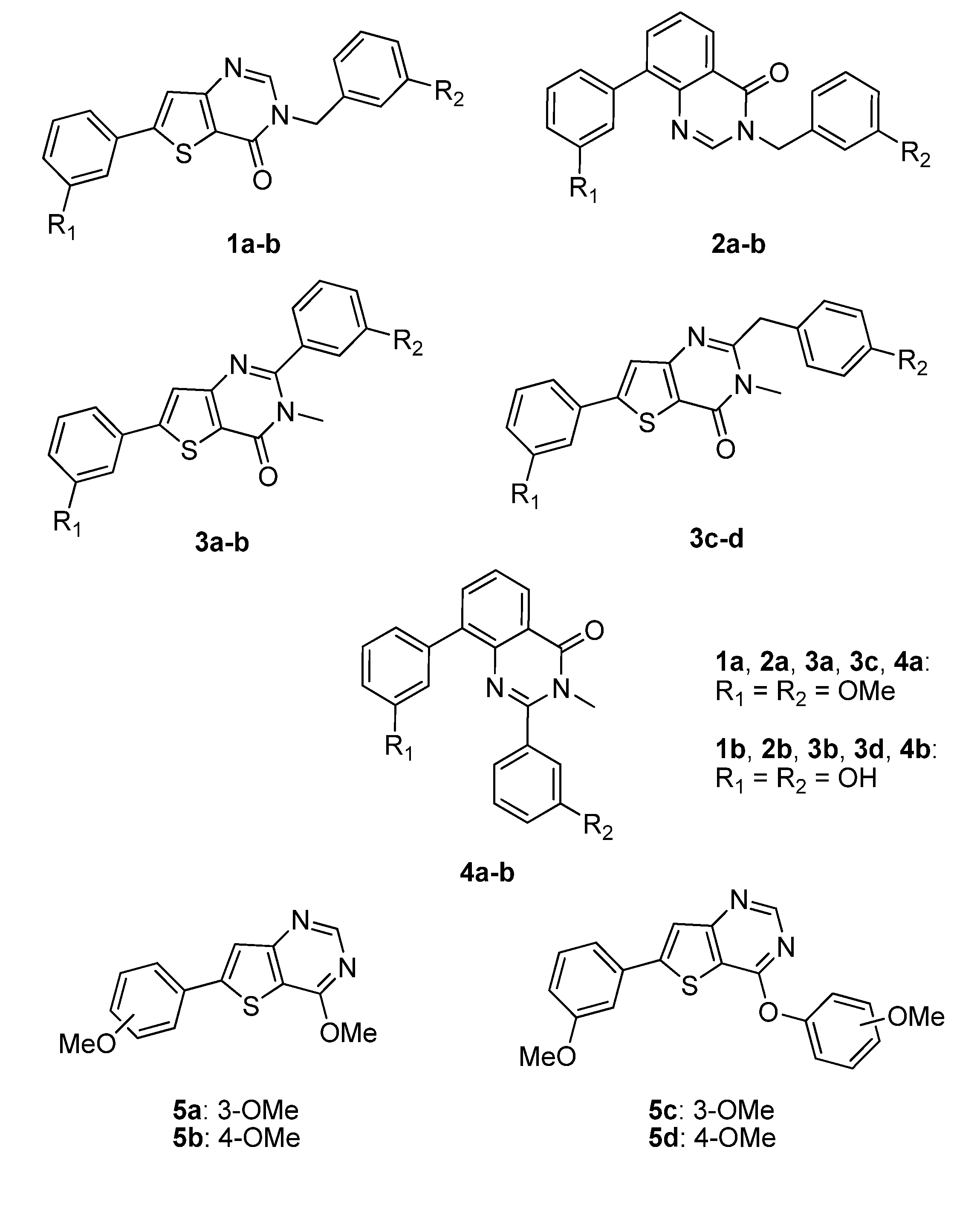
| Cmpd | R1 | R2 | Percentage of inhibition at 1µM a | |
|---|---|---|---|---|
| 17β-HSD2 b | 17β-HSD1 c | |||
| A | OMe | OMe | 63% | 0% |
| B | OH | OH | 70% | 21% |
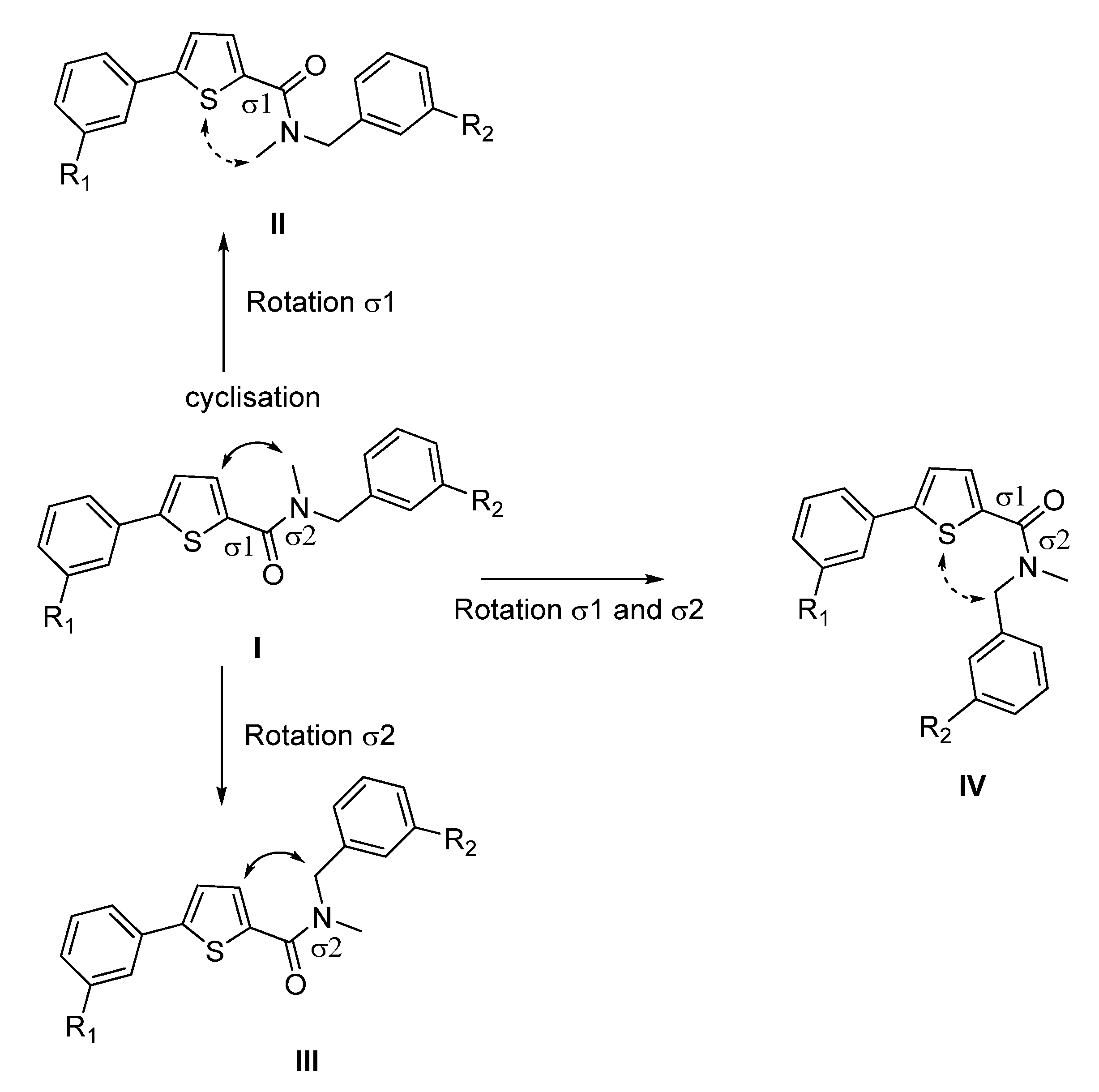
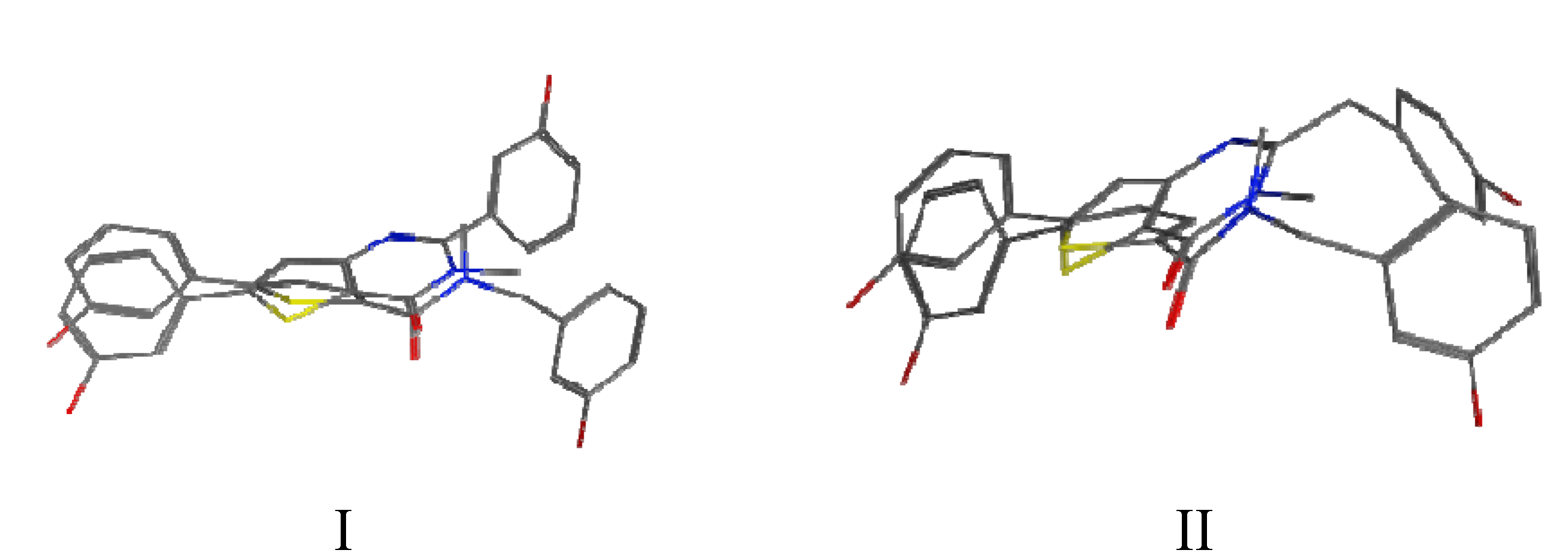
2. Results and Discussion








| Compound | Percentage of inhibition at 1 µM a | |
|---|---|---|
| 17β-HSD2 b | 17β-HSD1 c | |
| 1a | n.i. | n.i. |
| 1b | n.i. | n.i. |
| 2a | n.i. | n.i. |
| 2b | 11% | n.i. |
| 3a | n.i. | n.i. |
| 3b | 36% | 50% |
| 3c | n.i. | n.i. |
| 3d | 25% | 48% |
| 4a | n.i. | n.i. |
| 4b | n.i. | n.i. |
| 5a | n.i. | n.i. |
| 5b | n.i. | n.i. |
| 5c | n.i. | n.i. |
| 5d | n.i. | n.i. |
| 13 | n.i. | n.i. |
| 12 | n.i. | n.i. |


3. Experimental
3.1. General Procedure for Suzuki — Miyaura Coupling (2a, 17) — Method A
3.2. General Procedure for Cleavage of Ethers 1b–4b — Method B
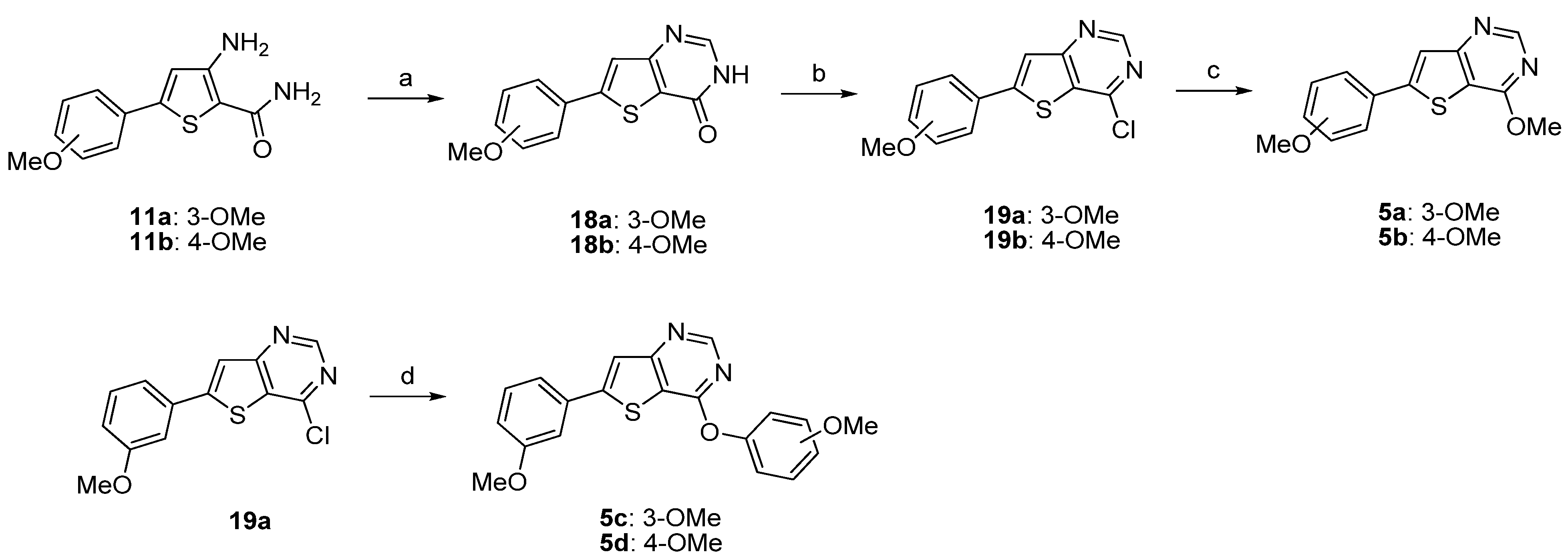
3.3. General Procedure for the Preparation of Compounds 18a–b — Method C
3.4. General Procedure for the Preparation of Compounds 19a–b — Method D
3.5. General Procedure for the Preparation of Compounds 5a–b – Method E
3.6. General Procedure for the Preparation of Compounds 5c–d – Method F
4. Conclusions
Acknowledgments
References
- Cummings, S.R.; Melton, L.J. Epidemiology and outcomes of osteoporotic fractures. Lancet 2002, 359, 1761–1767. [Google Scholar] [CrossRef]
- Bagi, C.M.; Wood, J.; Wilkie, D.; Dixon, B. Effect of 17beta-hydroxysteroid dehydrogenase type 2 inhibitor on bone strength in ovariectomized cynomolgus monkeys. J. Musculoskelet. Neuronal Interact. 2008, 8, 267–280. [Google Scholar]
- Wu, L.; Einstein, M.; Geissler, W.M.; Chan, H.K.; Elliston, K.O.; Andersson, S. Expression cloning and characterization of human 17 beta-hydroxysteroid dehydrogenase type 2, a microsomal enzyme possessing 20 alpha-hydroxysteroid dehydrogenase activity. J. Biol. Chem. 1993, 268, 12964–12969. [Google Scholar]
- Poirier, D.; Bydal, P.; Tremblay, M.R.; Sam, K.M.; Luu-The, V. Inhibitors of type ii 17beta-hydroxysteroid dehydrogenase. Mol. Cell. Endocrinol. 2001, 171, 119–128. [Google Scholar] [CrossRef]
- Bydal, P.; Auger, S.; Poirier, D. Inhibition of type 2 17beta-hydroxysteroid dehydrogenase by estradiol derivatives bearing a lactone on the d-ring: Structure-activity relationships. Steroids 2004, 69, 325–342. [Google Scholar] [CrossRef]
- Cook, J.H.; Baryza, J.; Brennan, C.; Lowe, D.; Wang, M.; Redman, A.; Scott, W.J.; Wood, J.E. Disubstituted cis-pyrrolidinones as inhibitors of 17β-hydroxysteroid dehydrogenase ii. Part 1: Synthetic approach. Tetrahedron Lett. 2005, 46, 1525–1528. [Google Scholar] [CrossRef]
- Gunn, D.; Akuche, C.; Baryza, J.; Blue, M.L.; Brennan, C.; Campbell, A.M.; Choi, S.; Cook, J.; Conrad, P.; Dixon, B.; et al. 4,5-disubstituted cis-pyrrolidinones as inhibitors of type ii 17beta-hydroxysteroid dehydrogenase. Part 2. Sar. Bioorg. Med. Chem. Lett. 2005, 15, 3053–3057. [Google Scholar] [CrossRef]
- Wood, J.; Bagi, C.M.; Akuche, C.; Bacchiocchi, A.; Baryza, J.; Blue, M.L.; Brennan, C.; Campbell, A.M.; Choi, S.; Cook, J.H.; et al. 4,5-disubstituted cis-pyrrolidinones as inhibitors of type ii 17beta-hydroxysteroid dehydrogenase. Part 3. Identification of lead candidate. Bioorg. Med. Chem. Lett. 2006, 16, 4965–4968. [Google Scholar] [CrossRef]
- Picard, F.; Baston, E.; Reichert, W.; Hartmann, R.W. Synthesis of n-substituted piperidine-4-(benzylidene-4-carboxylic acids) and evaluation as inhibitors of steroid-5alpha-reductase type 1 and 2. Bioorg. Med. Chem. 2000, 8, 1479–1487. [Google Scholar] [CrossRef]
- Leze, M.P.; Le Borgne, M.; Pinson, P.; Palusczak, A.; Duflos, M.; Le Baut, G.; Hartmann, R.W. Synthesis and biological evaluation of 5-[(aryl)(1h-imidazol-1-yl)methyl]-1h-indoles: Potent and selective aromatase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 1134–1137. [Google Scholar]
- Gobbi, S.; Cavalli, A.; Negri, M.; Schewe, K.E.; Belluti, F.; Piazzi, L.; Hartmann, R.W.; Recanatini, M.; Bisi, A. Imidazolylmethylbenzophenones as highly potent aromatase inhibitors. J. Med. Chem. 2007, 50, 3420–3422. [Google Scholar] [CrossRef]
- Hu, Q.; Negri, M.; Jahn-Hoffmann, K.; Zhuang, Y.; Olgen, S.; Bartels, M.; Muller-Vieira, U.; Lauterbach, T.; Hartmann, R.W. Synthesis, Biological evaluation, and molecular modeling studies of methylene imidazole substituted biaryls as inhibitors of human 17alpha-hydroxylase-17,20-lyase (cyp17)—part ii: Core rigidification and influence of substituents at the methylene bridge. Bioorg. Med. Chem. 2008, 16, 7715–7727. [Google Scholar] [CrossRef]
- Hille, U.E.; Hu, Q.; Vock, C.; Negri, M.; Bartels, M.; Muller-Vieira, U.; Lauterbach, T.; Hartmann, R.W. Novel cyp17 inhibitors: Synthesis, biological evaluation, structure-activity relationships and modelling of methoxy- and hydroxy-substituted methyleneimidazolyl biphenyls. Eur. J. Med. Chem. 2009, 44, 2765–2775. [Google Scholar] [CrossRef]
- Voets, M.; Antes, I.; Scherer, C.; Muller-Vieira, U.; Biemel, K.; Marchais-Oberwinkler, S.; Hartmann, R.W. Synthesis and evaluation of heteroaryl-substituted dihydronaphthalenes and indenes: Potent and selective inhibitors of aldosterone synthase (cyp11b2) for the treatment of congestive heart failure and myocardial fibrosis. J. Med. Chem. 2006, 49, 2222–2231. [Google Scholar] [CrossRef]
- Lucas, S.; Heim, R.; Negri, M.; Antes, I.; Ries, C.; Schewe, K.E.; Bisi, A.; Gobbi, S.; Hartmann, R.W. Novel aldosterone synthase inhibitors with extended carbocyclic skeleton by a combined ligand-based and structure-based drug design approach. J. Med. Chem. 2008, 51, 6138–6149. [Google Scholar] [CrossRef]
- Lucas, S.; Heim, R.; Ries, C.; Schewe, K.E.; Birk, B.; Hartmann, R.W. In vivo active aldosterone synthase inhibitors with improved selectivity: Lead optimization providing a series of pyridine substituted 3,4-dihydro-1h-quinolin-2-one derivatives. J. Med. Chem. 2008, 51, 8077–8087. [Google Scholar] [CrossRef]
- Heim, R.; Lucas, S.; Grombein, C.M.; Ries, C.; Schewe, K.E.; Negri, M.; Muller-Vieira, U.; Birk, B.; Hartmann, R.W. Overcoming undesirable cyp1a2 inhibition of pyridylnaphthalene-type aldosterone synthase inhibitors: Influence of heteroaryl derivatization on potency and selectivity. J. Med. Chem. 2008, 51, 5064–5074. [Google Scholar] [CrossRef]
- Hille, U.E.; Zimmer, C.; Haupenthal, J.; Hartmann, R.W. Optimization of the first selective steroid-11β-hydroxylase (cyp11b1) inhibitors for the treatment of cortisol dependent diseases. Med. Chem. Lett. 2011, 2, 559–564. [Google Scholar] [CrossRef]
- Hille, U.E.; Zimmer, C.; Vock, C.; Hartmann, R.W. First selective cyp11b inhibitors for the treatment of cortisol dependent diseases. ACS Med. Chem. Lett. 2011, 2, 2–6. [Google Scholar] [CrossRef]
- Marchais-Oberwinkler, S.; Wetzel, M.; Ziegler, E.; Kruchten, P.; Werth, R.; Henn, C.; Hartmann, R.W.; Frotscher, M. New drug-like hydroxyphenylnaphthol steroidomimetics as potent and selective 17beta-hydroxysteroid dehydrogenase type 1 inhibitors for the treatment of estrogen-dependent diseases. J. Med. Chem. 2011, 54, 534–547. [Google Scholar] [CrossRef]
- Oster, A.; Klein, T.; Henn, C.; Werth, R.; Marchais-Oberwinkler, S.; Frotscher, M.; Hartmann, R.W. Bicyclic substituted hydroxyphenylmethanone type inhibitors of 17 beta-hydroxysteroid dehydrogenase type 1 (17 beta-hsd1): The role of the bicyclic moiety. ChemMedChem 2011, 6, 476–487. [Google Scholar] [CrossRef]
- Spadaro, A.; Negri, M.; Marchais-Oberwinkler, S.; Bey, E.; Frotscher, M. Hydroxybenzothiazoles as new nonsteroidal inhibitors of 17beta-hydroxysteroid dehydrogenase type 1 (17beta-hsd1). PLoS One 2012, 7, e29252. [Google Scholar]
- Henn, C.; Einspanier, A.; Marchais-Oberwinkler, S.; Frotscher, M.; Hartmann, R.W. Lead optimization of 17beta-hsd1 inhibitors of the (hydroxyphenyl)naphthol sulfonamide type for the treatment of endometriosis. J. Med. Chem. 2012, 55, 3307–3318. [Google Scholar] [CrossRef]
- Bey, E.; Marchais-Oberwinkler, S.; Kruchten, P.; Frotscher, M.; Werth, R.; Oster, A.; Algul, O.; Neugebauer, A.; Hartmann, R.W. Design, synthesis and biological evaluation of bis(hydroxyphenyl) azoles as potent and selective non-steroidal inhibitors of 17beta-hydroxysteroid dehydrogenase type 1 (17beta-hsd1) for the treatment of estrogen-dependent diseases. Bioorg. Med. Chem. 2008, 16, 6423–6435. [Google Scholar] [CrossRef]
- Bey, E.; Marchais-Oberwinkler, S.; Negri, M.; Kruchten, P.; Oster, A.; Klein, T.; Spadaro, A.; Werth, R.; Frotscher, M.; Birk, B.; et al. New insights into the sar and binding modes of bis(hydroxyphenyl)thiophenes and -benzenes: Influence of additional substituents on 17beta-hydroxysteroid dehydrogenase type 1 (17beta-hsd1) inhibitory activity and selectivity. J. Med. Chem. 2009, 52, 6724–6743. [Google Scholar] [CrossRef]
- Wetzel, M.; Marchais-Oberwinkler, S.; Hartmann, R.W. 17beta-hsd2 inhibitors for the treatment of osteoporosis: Identification of a promising scaffold. Bioorg. Med. Chem. 2011, 19, 807–815. [Google Scholar] [CrossRef]
- Xu, K.; Al-Soud, Y.A.; Wetzel, M.; Hartmann, R.W.; Marchais-Oberwinkler, S. Triazole ring-opening leads to the discovery of potent nonsteroidal 17beta-hydroxysteroid dehydrogenase type 2 inhibitors. Eur. J. Med. Chem. 2011, 46, 5978–5990. [Google Scholar] [CrossRef]
- Wetzel, M.; Marchais-Oberwinkler, S.; Perspicace, E.; Moller, G.; Adamski, J.; Hartmann, R.W. Introduction of an electron withdrawing group on the hydroxyphenylnaphthol scaffold improves the potency of 17beta-hydroxysteroid dehydrogenase type 2 (17beta-hsd2) inhibitors. J. Med. Chem. 2011, 54, 7547–7557. [Google Scholar] [CrossRef]
- Xu, K.; Wetzel, M.; Hartmann, R.W.; Marchais-Oberwinkler, S. Synthesis and biological evaluation of spiro-δ-lactones as inhibitors of 17β-hydroxysteroid dehydrogenase type 2 (17β-hsd2). Lett. Drug Des. Discov. 2011, 8, 406–421. [Google Scholar] [CrossRef]
- Wetzel, M.; Gargano, E.M.; Hinsberger, S.; Marchais-Oberwinkler, S.; Hartmann, R.W. Discovery of a new class of bicyclic substituted hydroxyphenylmethanones as 17beta-hydroxysteroid dehydrogenase type 2 (17beta-hsd2) inhibitors for the treatment of osteoporosis. Eur. J. Med. Chem. 2012, 47, 1–17. [Google Scholar] [CrossRef]
- Al-Soud, Y.A.; Marchais-Oberwinkler, S.; Frotscher, M.; Hartmann, R.W. Synthesis and biological evaluation of phenyl substituted 1h-1,2,4-triazoles as non-steroidal inhibitors of 17beta-hydroxysteroid dehydrogenase type 2. Arch. Pharm. (Weinheim) 2012, 345, 610–621. [Google Scholar] [CrossRef]
- Marchais-Oberwinkler, S.; Xu, K.; Wetzel, M.; Perspicace, E.; Negri, M.; Meyer, A.; Odermatt, A.; Moller, G.; Adamski, J.; Hartmann, R.W. Structural optimization of 2,5-thiophene amides as highly potent and selective 17beta-hydroxysteroid dehydrogenase type 2 inhibitors for the treatment of osteoporosis. J. Med. Chem. 2013, 56, 167–181. [Google Scholar] [CrossRef]
- Migianu, E.; Kirsch, G. Synthesis of new thieno[b]azepinediones from α-methylene ketones. Synthesis 2002, volume, 1096–1100. [Google Scholar] [CrossRef]
- Hertzog, D.L.; Al-Barazanji, K.A.; Bigham, E.C.; Bishop, M.J.; Britt, C.S.; Carlton, D.L.; Cooper, J.P.; Daniels, A.J.; Garrido, D.M.; Goetz, A.S.; et al. The discovery and optimization of pyrimidinone-containing mch r1 antagonists. Bioorg. Med. Chem. Lett. 2006, 16, 4723–4727. [Google Scholar] [CrossRef]
- Perrissin, M.; Favre, M.; Cuong, L.D.; Huguet, F.; Gaultier, C.; Narcisse, G. Synthesis and pharmacological activities of some substituted thienopyrimidine-4-ones. Eur. J. Med. Chem. 1988, 23, 453–456. [Google Scholar] [CrossRef]
- Boehm, N.; Krasselt, U.; Leistner, S.; Wagner, G. Reaction of 4-oxo-4h-pyrido[3'2':4,5]thieno[3,2-d]-1,3-oxazines with amines. Pharmazie 1992, 47, 897–901. [Google Scholar]
- Peinador, C.; Ojea, V.; Quintela, J.M. A convenient synthesis of some new pyrido[3',2':4,5]thieno[3,2-d]pyrimidine derivatives with potential biological activity. J. Heterocycl. Chem. 1992, 29, 1693–1702. [Google Scholar] [CrossRef]
- Lessene, G.; Baell, J. Preparation of amino acid derivatives as alpha-helical mimetics. PCT Int. Appl. WO 2006002474 A1 20060112, 2006. [Google Scholar]
- Hesse, S.; Perspicace, E. Microwave-assisted synthesis of 2-aminothiophene-3-carboxylic acid derivatives, 3h-thieno[2,3-d]pyrimidin-4-one and 4-chlorothieno[2,3-d]pyrimidine. Tetrahedron Lett. 2007, 48, 5261–5264. [Google Scholar] [CrossRef]
- Kruchten, P.; Werth, R.; Marchais-Oberwinkler, S.; Frotscher, M.; Hartmann, R.W. Development of a biological screening system for the evaluation of highly active and selective 17beta-hsd1-inhibitors as potential therapeutic agents. Mol. Cell. Endocrinol. 2009, 301, 154–157. [Google Scholar] [CrossRef]
- Messinger, J.; Hirvela, L.; Husen, B.; Kangas, L.; Koskimies, P.; Pentikainen, O.; Saarenketo, P.; Thole, H. New inhibitors of 17beta-hydroxysteroid dehydrogenase type 1. Mol. Cell. Endocrinol. 2006, 248, 192–198. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1a–b, 2a–b, 3a–d, 4a–b, 5a–d are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Perspicace, E.; Marchais-Oberwinkler, S.; Hartmann, R.W. Synthesis and Biological Evaluation of Thieno[3,2-d]- pyrimidinones, Thieno[3,2-d]pyrimidines and Quinazolinones: Conformationally Restricted 17b-Hydroxysteroid Dehydrogenase Type 2 (17b-HSD2) Inhibitors. Molecules 2013, 18, 4487-4509. https://doi.org/10.3390/molecules18044487
Perspicace E, Marchais-Oberwinkler S, Hartmann RW. Synthesis and Biological Evaluation of Thieno[3,2-d]- pyrimidinones, Thieno[3,2-d]pyrimidines and Quinazolinones: Conformationally Restricted 17b-Hydroxysteroid Dehydrogenase Type 2 (17b-HSD2) Inhibitors. Molecules. 2013; 18(4):4487-4509. https://doi.org/10.3390/molecules18044487
Chicago/Turabian StylePerspicace, Enrico, Sandrine Marchais-Oberwinkler, and Rolf W. Hartmann. 2013. "Synthesis and Biological Evaluation of Thieno[3,2-d]- pyrimidinones, Thieno[3,2-d]pyrimidines and Quinazolinones: Conformationally Restricted 17b-Hydroxysteroid Dehydrogenase Type 2 (17b-HSD2) Inhibitors" Molecules 18, no. 4: 4487-4509. https://doi.org/10.3390/molecules18044487
APA StylePerspicace, E., Marchais-Oberwinkler, S., & Hartmann, R. W. (2013). Synthesis and Biological Evaluation of Thieno[3,2-d]- pyrimidinones, Thieno[3,2-d]pyrimidines and Quinazolinones: Conformationally Restricted 17b-Hydroxysteroid Dehydrogenase Type 2 (17b-HSD2) Inhibitors. Molecules, 18(4), 4487-4509. https://doi.org/10.3390/molecules18044487