Factors Influencing the Antifolate Activity of Synthetic Tea-Derived Catechins

Abstract
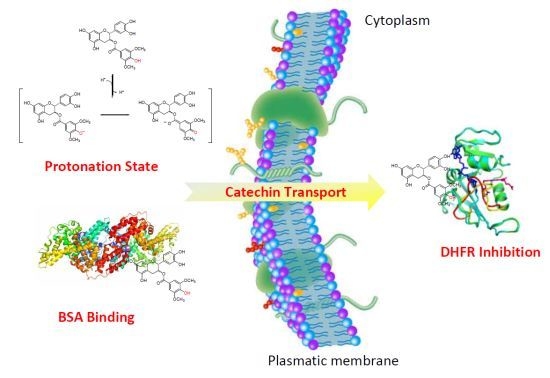
:
1. Introduction

2. Results and Discussion
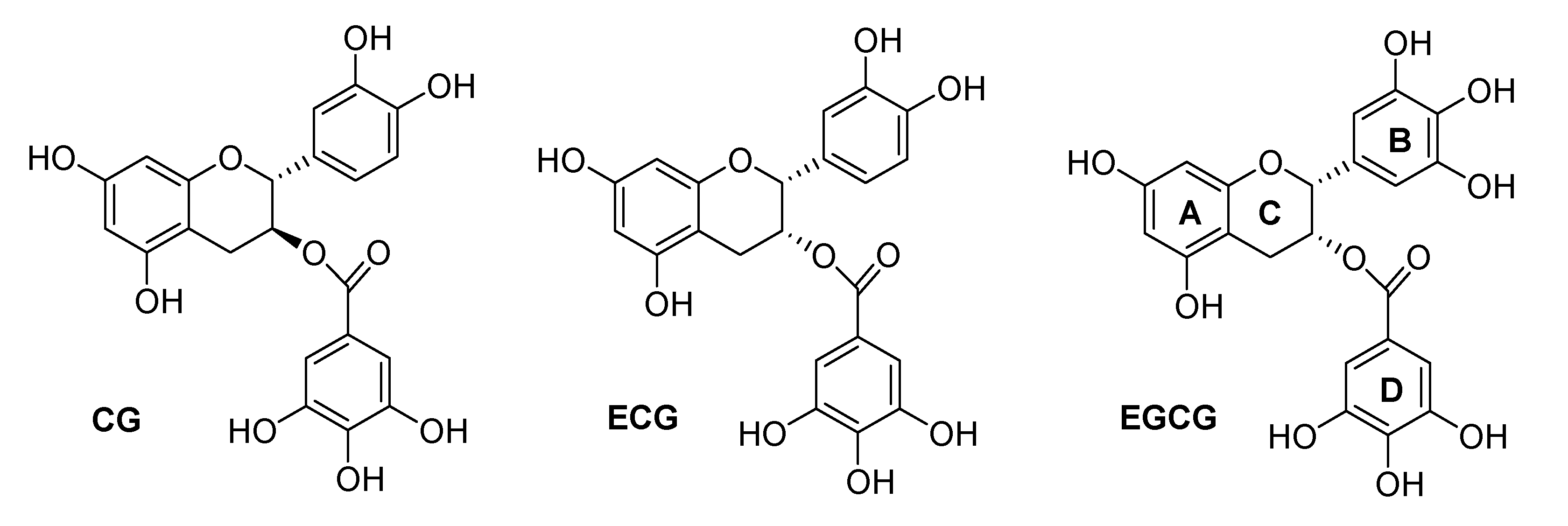
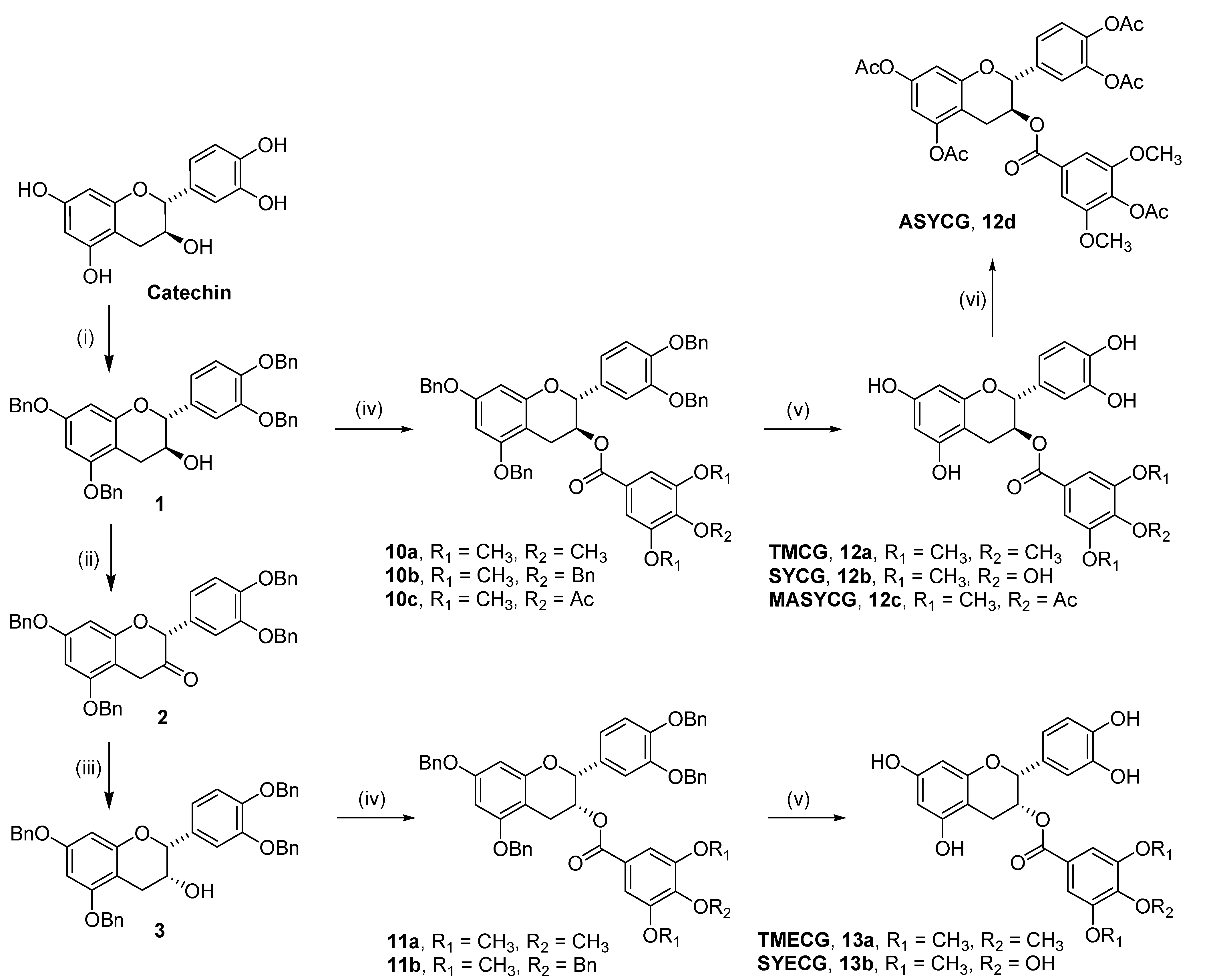
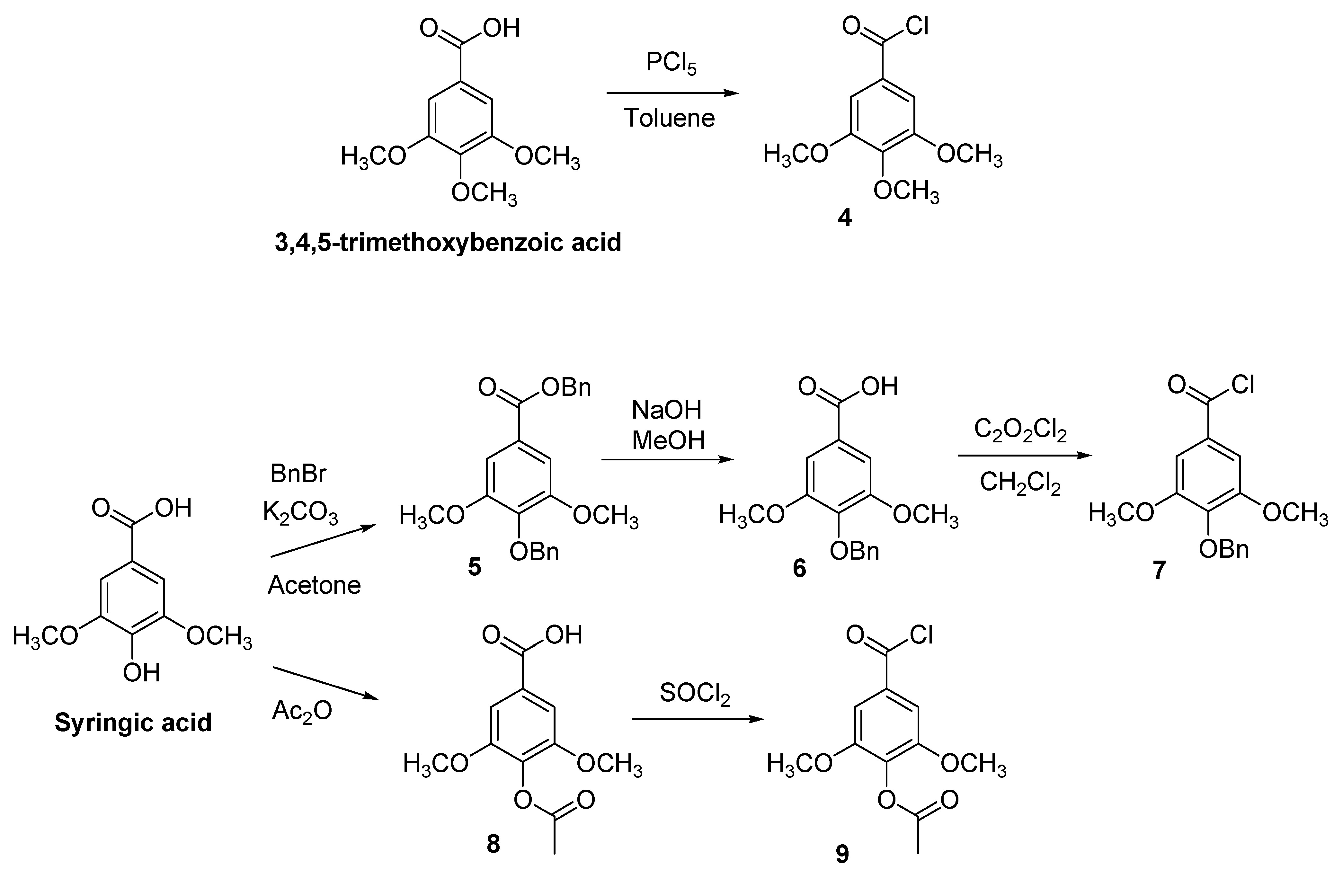
2.1. Synthesis


2.2. Biological Activities
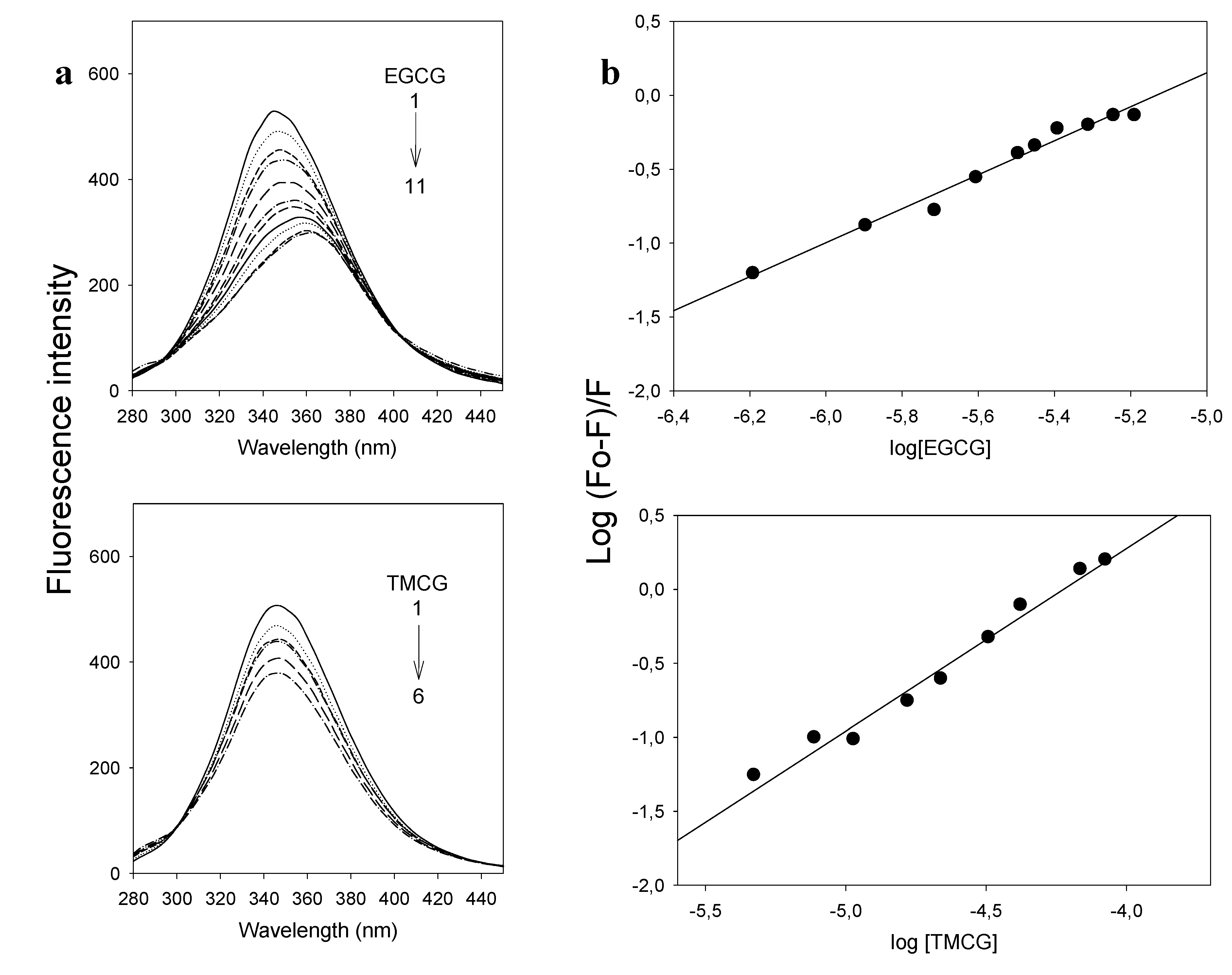
2.2.1. Binding of polyphenols to DHFR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | KD (μM) |
|---|---|
| CG | 0.56 ± 0.07 |
| ECG | 0.81 ± 0.09 * |
| TMCG | 0.9 ± 0.1 |
| TMECG | 2.1 ± 0.2 * |
| SYCG | 0.72 ± 0.10 |
| SYECG | 0.77 ± 0.07 |

| (KD, μM) | |||
|---|---|---|---|
| Catechin | 0.56 | 0.72 | 0.90 * |
| Epicatechin | 0.81 | 0.77 | 2.10 * |
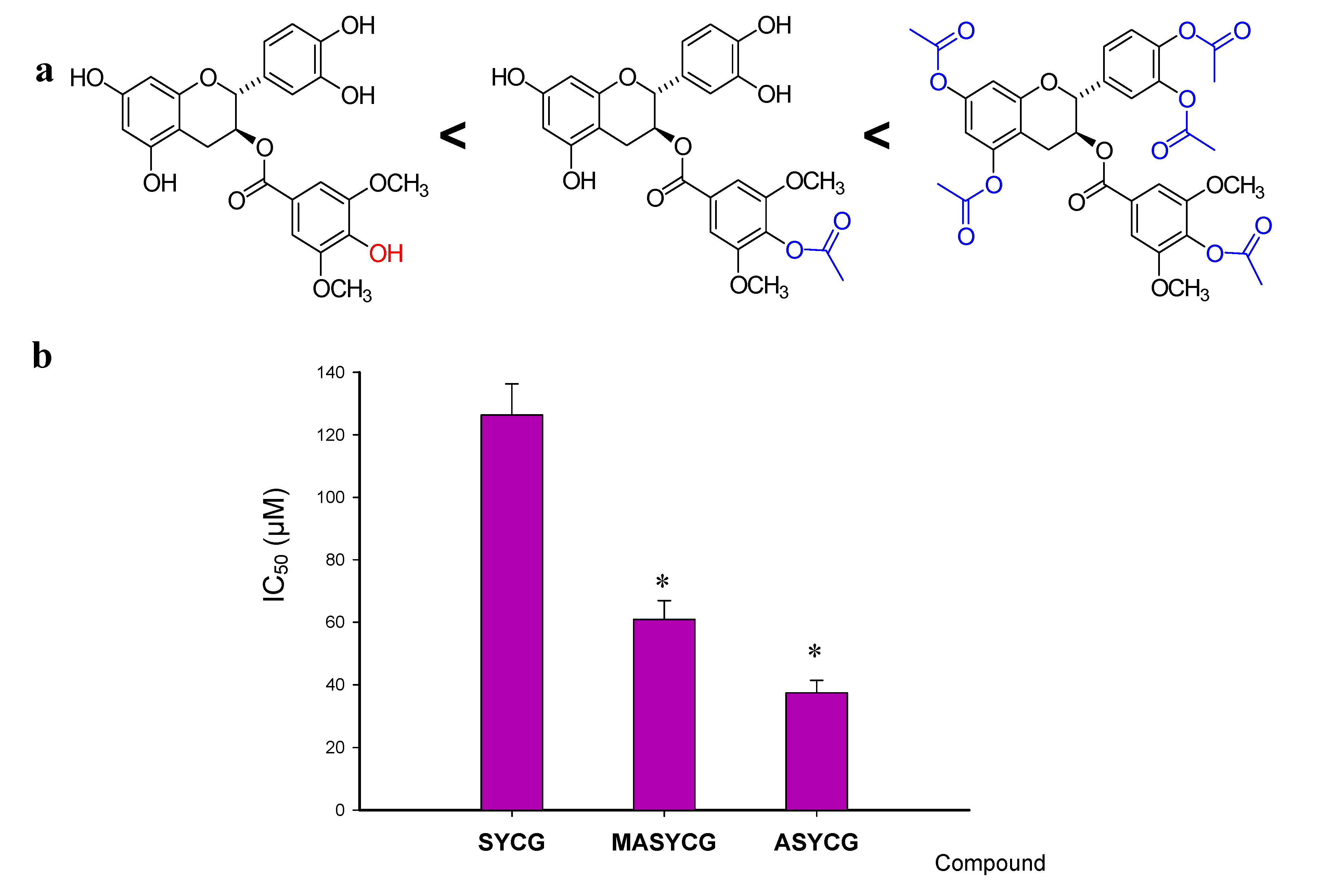
2.2.2. Cancer Cell Antiproliferative Activity
| Compound | IC50 (μM) |
|---|---|
| CG | 82.2 ± 8.0 |
| ECG | > 100 |
| TMCG | 1.5 ± 0.4 |
| TMECG | 2.5 ± 0.6 |
| SYCG | 126.3 ± 10.0 * |
| SYECG | 198.9 ± 12.0 * |
| MASYCG | 60.9 ± 6.0 ** |
| ASYCG | 37.4 ± 4.0 ** |
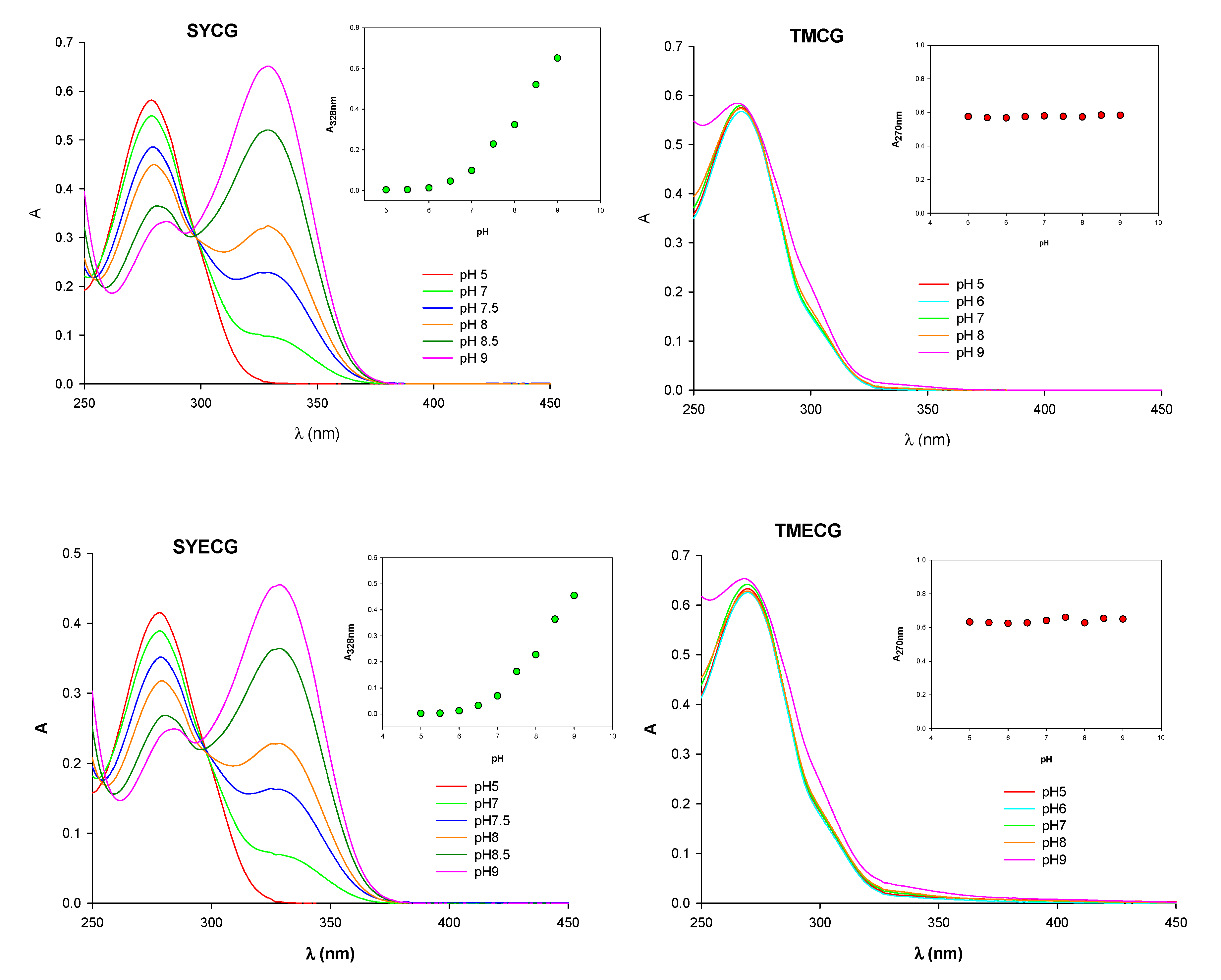
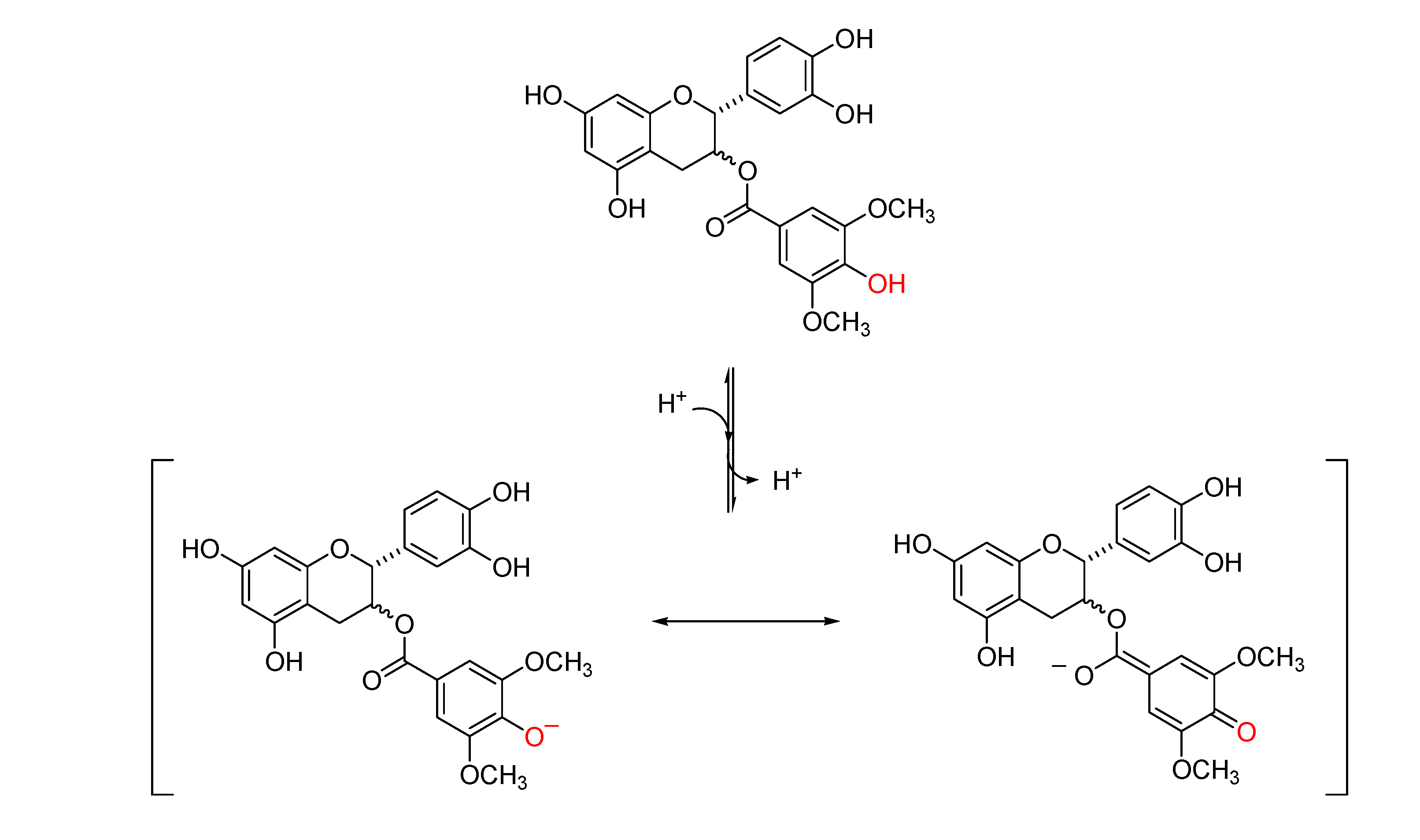
2.2.3. pH-Related Effects on Catechin Membrane Transport



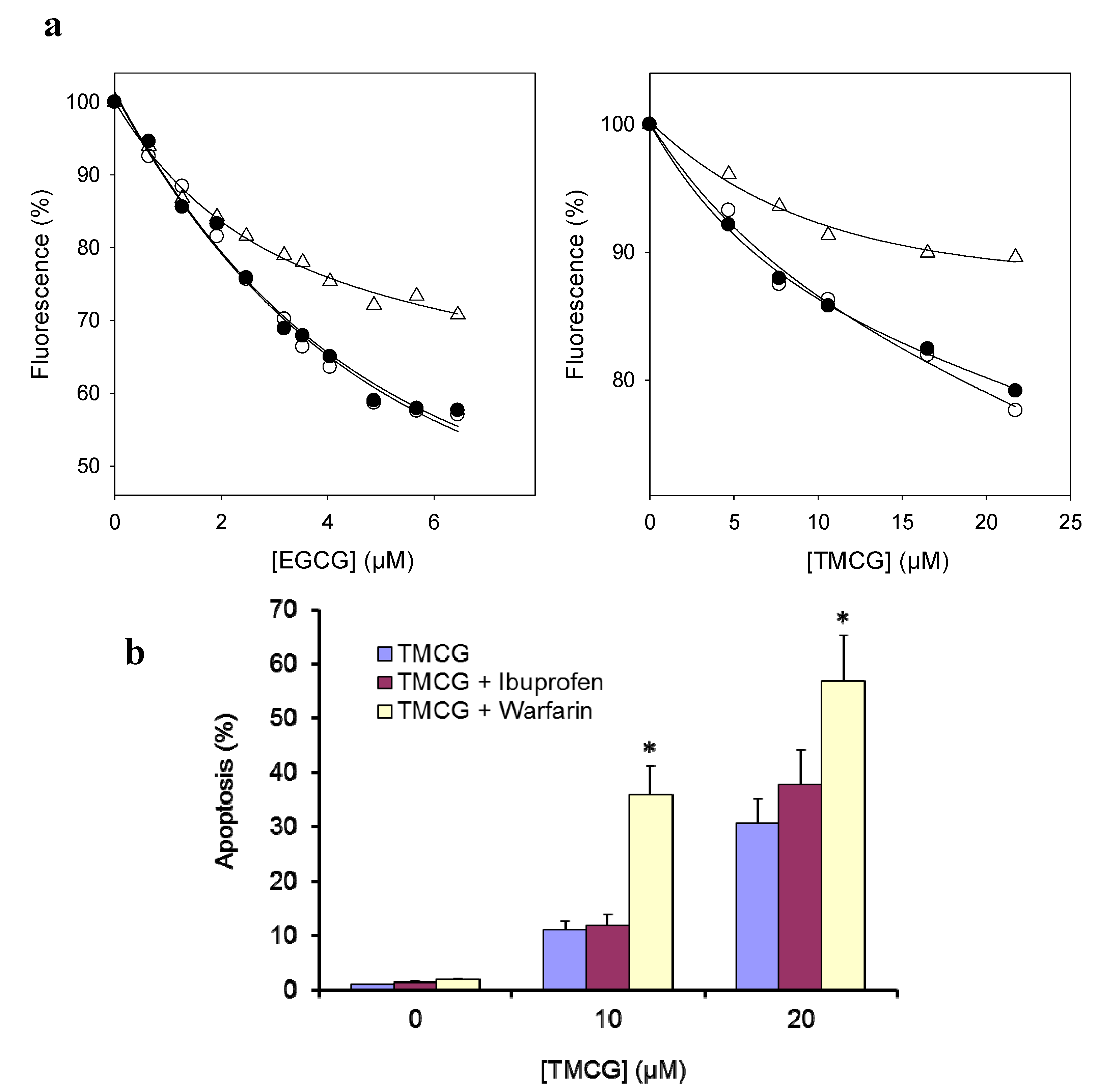
2.2.4. Influence of Catechin-Serum Albumin Interactions on Polyphenol Activity

| Catechin | Site Marker | Binding constant (KA) (×106 M−1) |
|---|---|---|
| EGCG | Blank | 15.40 |
| Warfarin | 0.13 | |
| Ibuprofen | 12.20 | |
| TMCG | Blank | 0.19 |
| Warfarin | 0.05 | |
| Ibuprofen | 0.11 |

3. Experimental
3.1. Synthesis
3.1.1. Synthesis of Acid Chlorides 7 and 9
3.1.2. General Procedure for the Synthesis of Compounds 10b, 10c, and 11b
3.1.3. General Procedure for the Synthesis of Compounds 12b, 12c, and 13b
3.1.4. Synthesis of Compound 12d
3.2. Biological Activities
3.2.1. Materials
3.2.2. Cell lines, Proliferation, and Apoptosis Assays
3.2.3. DHFR Binding Studies
3.2.4. BSA Binding Studies
3.2.5. HSA Binding Studies
3.2.6. Spectrophotometric Assays
3.2.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Mukhtar, H.; Ahmad, N. Cancer chemoprevention: future holds in multiple agents. Toxicol. Appl. Pharmacol. 1999, 158, 207–210. [Google Scholar] [CrossRef]
- Yang, C.S.; Chung, J.Y.; Yang, G.Y.; Li, C.; Meng, X.; Lee, M.J. Mechanisms of inhibition of carcinogenesis by tea. Biofactors 2000, 13, 73–79. [Google Scholar] [CrossRef]
- Shankar, S.; Ganapathy, S.; Srivastava, R.K. Green tea polyphenols: biology and therapeutic implications in cancer. Front. Biosci. 2007, 12, 4881–4899. [Google Scholar] [CrossRef]
- Nam, S.; Smith, D.M.; Dou, Q.P. Ester bond-containing tea polyphenols potently inhibit proteasome activity in vitro and in vivo. J. Biol. Chem. 2001, 276, 13322–13330. [Google Scholar] [CrossRef]
- Fang, M.Z.; Wang, Y.; Ai, N.; Hou, Z.; Sun, Y.; Lu, H.; Welsh, W.; Yang, C.S. Tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003, 63, 7563–7570. [Google Scholar]
- Li, C.; Allen, A.; Kwagh, J.; Doliba, N.M.; Qin, W.; Najafi, H.; Collins, H.W.; Matschinsky, F.M.; Stanley, C.A.; Smith, T.J. Green tea polyphenols modulate insulin secretion by inhibiting glutamate dehydrogenase. J. Biol. Chem. 2006, 281, 10214–10221. [Google Scholar] [CrossRef]
- Navarro-Peran, E.; Cabezas-Herrera, J.; Garcia-Canovas, F.; Durrant, M.C.; Thorneley, R.N.; Rodriguez-Lopez, J.N. The antifolate activity of tea catechins. Cancer Res. 2005, 65, 2059–2064. [Google Scholar] [CrossRef]
- Alemdaroglu, N.C.; Wolffram, S.; Boissel, J.P.; Closs, E.; Spahn-Langguth, H.; Langguth, P. Inhibition of folic acid uptake by catechins and tea extracts in Caco-2 cells. Planta Med. 2007, 73, 27–32. [Google Scholar] [CrossRef]
- Navarro-Peran, E.; Cabezas-Herrera, J.; Campo, L.S.; Rodriguez-Lopez, J.N. Effects of folate cycle disruption by the green tea polyphenol epigallocatechin-3-gallate. Int. J. Biochem. Cell. Biol. 2007, 39, 2215–2225. [Google Scholar] [CrossRef]
- Navarro-Martinez, M.D.; Navarro-Peran, E.; Cabezas-Herrera, J.; Ruiz-Gomez, J.; Garcia-Canovas, F.; Rodriguez-Lopez, J.N. Antifolate activity of epigallocatechin gallate against Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 2005, 49, 2914–2920. [Google Scholar] [CrossRef]
- Navarro-Peran, E.; Cabezas-Herrera, J.; Hiner, A.N.; Sadunishvili, T.; Garcia-Canovas, F.; Rodriguez-Lopez, J.N. Kinetics of the inhibition of bovine liver dihydrofolate reductase by tea catechins: origin of slow-binding inhibition and pH studies. Biochemistry 2005, 44, 7512–7525. [Google Scholar] [CrossRef]
- Spina, M.; Cuccioloni, M.; Mozzicafreddo, M.; Montecchia, F.; Pucciarelli, S.; Eleuteri, A.M.; Fioretti, E.; Angeletti, M. Mechanism of inhibition of wt-dihydrofolate reductase from E. coli by tea epigallocatechin-gallate. Proteins 2008, 72, 240–251. [Google Scholar] [CrossRef]
- Kao, T.T.; Wang, K.C.; Chang, W.N.; Lin, C.Y.; Chen, B.H.; Wu, H.L.; Shi, G.Y.; Tsai, J.N.; Fu, T.F. Characterization and comparative studies of zebrafish and human recombinant dihydrofolate reductases--inhibition by folic acid and polyphenols. Drug Metab. Dispos. 2008, 36, 508–516. [Google Scholar]
- Hannewald, P.; Maunit, B.; Muller, J.F. Screening of DHFR-binding drugs by MALDI-TOFMS. Anal. Bioanal. Chem. 2008, 392, 1335–1344. [Google Scholar] [CrossRef]
- Hong, J.; Lu, H.; Meng, X.; Ryu, J.H.; Hara, Y.; Yang, C.S. Stability, cellular uptake, biotransformation, and efflux of tea polyphenol (−)-epigallocatechin-3-gallate in HT-29 human colon adenocarcinoma cells. Cancer Res. 2002, 62, 7241–7246. [Google Scholar]
- Sanchez-del-Campo, L.; Oton, F.; Tarraga, A.; Cabezas-Herrera, J.; Chazarra, S.; Rodriguez-Lopez, J.N. Synthesis and biological activity of a 3,4,5-trimethoxybenzoyl ester analogue of epicatechin-3-gallate. J. Med. Chem. 2008, 51, 2018–2026. [Google Scholar] [CrossRef]
- Babich, H.; Zuckerbraun, H.L.; Weinerman, S.M. In vitro cytotoxicity of (-)-catechin gallate, a minor polyphenol in green tea. Toxicol Lett 2007, 171, 171–180. [Google Scholar] [CrossRef]
- Saez-Ayala, M.; Sanchez-del-Campo, L.; Montenegro, M.F.; Chazarra, S.; Tarraga, A.; Cabezas-Herrera, J.; Rodriguez-Lopez, J.N. Comparison of a pair of synthetic tea-catechin-derived epimers: Synthesis, antifolate activity, and tyrosinase-mediated activation in melanoma. ChemMedChem 2011, 6, 440–449. [Google Scholar] [CrossRef]
- Tückmantel, W.; Kozikowski, A.P.; Romanczyk, L.J. Studies in Polyphenol Chemistry and Bioactivity. 1. Preparation of building blocks from (+)-Catechin. Procyanidin formation. synthesis of the cancer cell growth inhibitor, 3-O-Galloyl-(2R,3R)-epicatechin-4β,8-[3-O-galloyl-(2R,3R)-epicatechin]. J. Am. Chem. Soc. 1999, 121, 12073–12081. [Google Scholar] [CrossRef]
- Park, K.D.; Lee, S.G.; Kim, S.U.; Kim, S.H.; Sun, W.S.; Cho, S.J.; Jeong, D.H. Anticancer activity of 3-O-acyl and alkyl-(-)-epicatechin derivatives. Bioorg. Med. Chem. Lett. 2004, 14, 5189–5192. [Google Scholar] [CrossRef]
- Hirasawa, M.; Takada, K. Multiple effects of green tea catechin on the antifungal activity of antimycotics against Candida albicans. J. Antimicrob. Chemother. 2004, 53, 225–229. [Google Scholar] [CrossRef]
- Cocco, L.; Roth, B.; Temple, C., Jr.; Montgomery, J.A.; London, R.E.; Blakley, R.L. Protonated state of methotrexate, trimethoprim, and pyrimethamine bound to dihydrofolate reductase. Arch. Biochem. Biophys. 1983, 226, 567–577. [Google Scholar] [CrossRef]
- Vyas, S.; Manon, B.; Vir Singh, T.; Dev Sharma, P.; Sharma, M. Potential O-acyl-substituted (−)-Epicatechin gallate prodrugs as inhibitors of DMBA/TPA-induced squamous cell carcinoma of skin in Swiss albino mice. Chem. Biodivers. 2011, 8, 599–613. [Google Scholar] [CrossRef]
- Ishii, T.; Minoda, K.; Bae, M.J.; Mori, T.; Uekusa, Y.; Ichikawa, T.; Aihara, Y.; Furuta, T.; Wakimoto, T.; Kan, T.; Nakayama, T. Binding affinity of tea catechins for HSA: characterization by high-performance affinity chromatography with immobilized albumin column. Mol. Nutr. Food Res. 2010, 54, 816–822. [Google Scholar]
- Pal, S.; Saha, C.; Hossain, M.; Dey, S.K.; Kumar, G.S. Influence of galloyl moiety in interaction of epicatechin with bovine serum albumin: A spectroscopic and thermodynamic characterization. PLoS One 2012, 7, e43321. [Google Scholar]
- Boulton, D.W.; Walle, U.K.; Walle, T. Extensive binding of the bioflavonoid quercetin to human plasma proteins. J. Pharm. Pharmacol. 1998, 50, 243–249. [Google Scholar] [CrossRef]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Controlled Release 2008, 132, 171–183. [Google Scholar] [CrossRef]
- Wunder, A.; Muller-Ladner, U.; Stelzer, E.H.; Funk, J.; Neumann, E.; Stehle, G.; Pap, T.; Sinn, H.; Gay, S.; Fiehn, C. Albumin-based drug delivery as novel therapeutic approach for rheumatoid arthritis. J. Immunol. 2003, 170, 4793–4801. [Google Scholar]
- Gao, S.; Feng, N.; Yu, S.; Yu, D.; Wang, X. Vasodilator constituents from the roots of Lysidice rhodostega. Planta Med. 2004, 70, 1128–1134. [Google Scholar] [CrossRef]
- Chazarra, S.; Aznar-Cervantes, S.; Sanchez-del-Campo, L.; Cabezas-Herrera, J.; Xiaofeng, W.; Cenis, J.L.; Rodriguez-Lopez, J.N. Purification and kinetic properties of human recombinant dihydrofolate reductase produced in Bombyx mori chrysalides. Appl. Biochem. Biotechnol. 2010, 162, 1834–1846. [Google Scholar] [CrossRef]
- Smith, S.L.; Patrick, P.; Stone, D.; Phillips, A.W.; Burchall, J.J. Porcine liver dihydrofolate reductase. Purification, properties, and amino acid sequence. J. Biol. Chem. 1979, 254, 11475–11484. [Google Scholar]
- Montenegro, M.F.; Saez-Ayala, M.; Pinero-Madrona, A.; Cabezas-Herrera, J.; Rodriguez-Lopez, J.N. Reactivation of the tumour suppressor RASSF1A in breast cancer by simultaneous targeting of DNA and E2F1 methylation. PLoS One 2012, 7, e52231. [Google Scholar]
- Sáez-Ayala, M.; Montenegro, M.F.; Sánchez-del-Campo, L.; Fernández-Pérez, M.P.; Chazarra, S.; Freter, R.; Middleton, M.; Piñero-Madrona, A.; Cabezas-Herrera, J.; Goding, C.R.; Rodríguez-López, J.N. Directed phenotype-switching as an effective anti-melanoma strategy. Cancer Cell. 2013, 24, 105–119. [Google Scholar] [CrossRef]
- Sánchez-del-Campo, L.; Rodríguez-López, J.N. Targeting the methionine cycle for melanoma therapy with 3-O-(3,4,5-trimethoxybenzoyl)-(-)-epicatechin. Int. J. Cancer 2008, 123, 2446–2455. [Google Scholar] [CrossRef]
- Fujita, T. Hydrophobic bonding of sulfonamide drugs with serum albumin. J. Med. Chem. 1972, 15, 1049–1056. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 12b and 13b are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sáez-Ayala, M.; Fernández-Pérez, M.P.; Chazarra, S.; Mchedlishvili, N.; Tárraga-Tomás, A.; Rodríguez-López, J.N. Factors Influencing the Antifolate Activity of Synthetic Tea-Derived Catechins. Molecules 2013, 18, 8319-8341. https://doi.org/10.3390/molecules18078319
Sáez-Ayala M, Fernández-Pérez MP, Chazarra S, Mchedlishvili N, Tárraga-Tomás A, Rodríguez-López JN. Factors Influencing the Antifolate Activity of Synthetic Tea-Derived Catechins. Molecules. 2013; 18(7):8319-8341. https://doi.org/10.3390/molecules18078319
Chicago/Turabian StyleSáez-Ayala, Magalí, María Piedad Fernández-Pérez, Soledad Chazarra, Nani Mchedlishvili, Alberto Tárraga-Tomás, and José Neptuno Rodríguez-López. 2013. "Factors Influencing the Antifolate Activity of Synthetic Tea-Derived Catechins" Molecules 18, no. 7: 8319-8341. https://doi.org/10.3390/molecules18078319
APA StyleSáez-Ayala, M., Fernández-Pérez, M. P., Chazarra, S., Mchedlishvili, N., Tárraga-Tomás, A., & Rodríguez-López, J. N. (2013). Factors Influencing the Antifolate Activity of Synthetic Tea-Derived Catechins. Molecules, 18(7), 8319-8341. https://doi.org/10.3390/molecules18078319