A Straightforward Diphenylmethyl Protection Method and Deprotection of Some Pyrimidine Nucleosides

Abstract
:1. Introduction
2. Results and Discussion


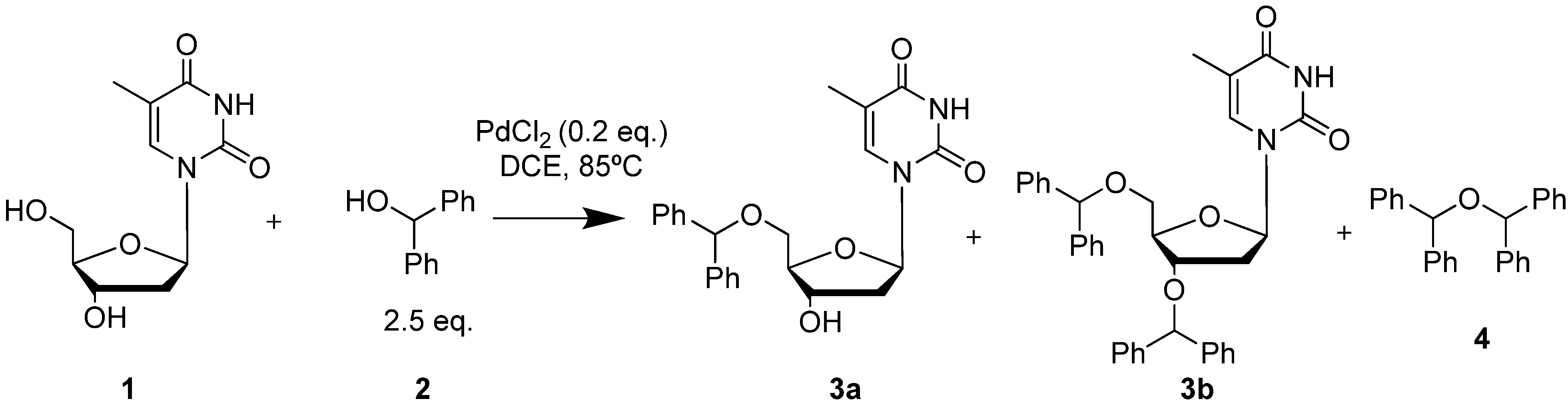{kind=link}
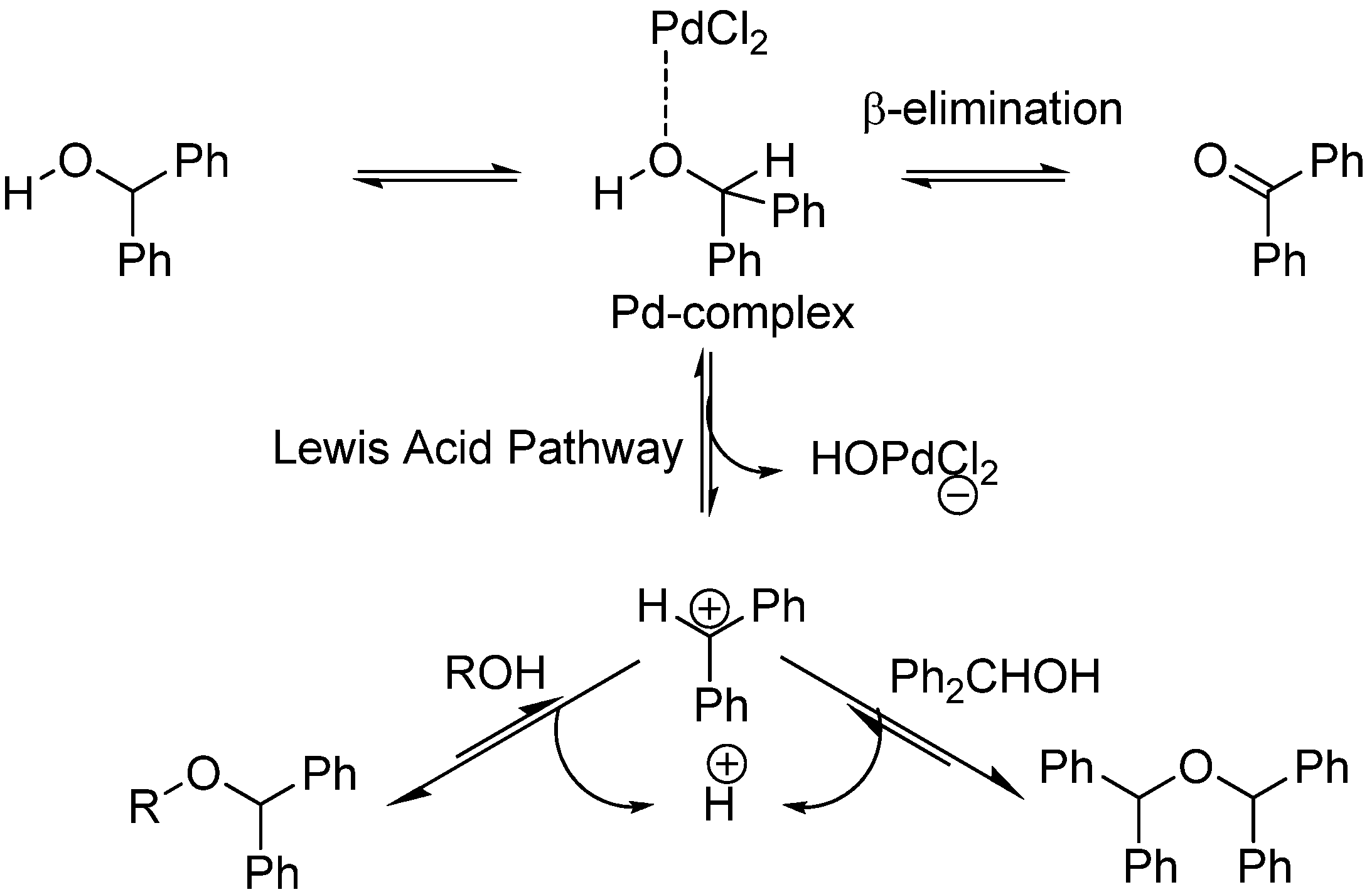{kind=link}
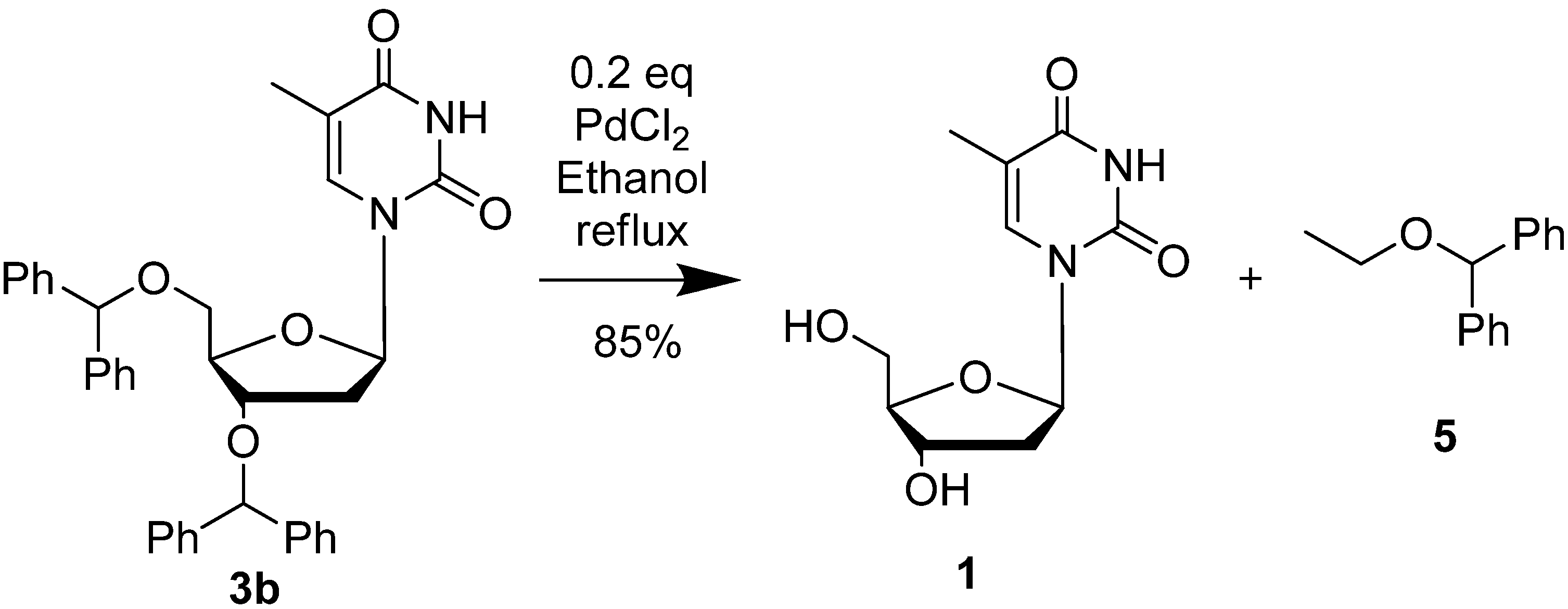{kind=link}
| Exp. | Catalyst | Recovered 1 (%) | Recovered 2 (%) | Yield 3b (%) a | Yield 4 (%) |
|---|---|---|---|---|---|
| 1 | PdCl2 | - | 6 | 87 | Trace |
| 2 | NiCl2 | 95 | 94 | 2 | - |
| 3 | CuCl2·2H2O | 43 | 0 | 55 (24 h) | Trace |
| 4 | Pd(OAc)2 | 10 | 0 | 65 | - |
| 5 | Ni(OAc)2 | 90 | 0 | 0 | Trace |
| 6 | Cu(OAc)2 | 32 | - | 45 | Trace |

| Entry | Solvent | Time (h) | Yield 3b (%) |
|---|---|---|---|
| 1 | DCE | 16 | 87 |
| 2 | Acetonitrile | 48 | 47 |
| 3 | THF | 48 | 42 |
| 4 | Dioxane | 16 | Degradation |
| 5 | DMF | 16 | Degradation |
| Entry | Substrate | Eq DPM-OH | Product | Yield (%) | |
|---|---|---|---|---|---|
| 1 | thymidine | 1.2 | 5′-O-benzhydryl-thymidine 3a | 67 | |
| 2 | 2.5 | 3′,5′-di-O-benzhydryl-thymidine 3b | 87 | ||
| 3 | 2′-deoxyuridine | 1.2 | 5′-O-benzhydryl-2′-deoxyuridine 6a | 65 | |
| 4 | 2.5 | 3′,5′-di-O-benzhydryl-2′-deoxyuridine 6b | 88 | ||
| 5 | 5-fluoro-2′-deoxyuridine | 1.2 | 5′-O-benzhydryl-5-fluoro-2′-deoxyuridine 7a | 64 | |
| 6 | 2.5 | 3′,5′-di-O-benzhydryl-5-fluoro-2′-deoxyuridine 7b | 85 | ||
| 7 | uridine | 1.2 | 5′-O-benzhydryl-uridine 8a | Trace | |
| 8 | 2.5 | 2′,5′-di-O-benzhydryl-uridine 8b | 54 |  | |
| 3′,5′-di-O-benzhydryl-uridine 8c | 32 | ||||
| 9 | 3.7 | 2′,3′,5′-tris-O-benzhydryl-uridine 8d | 81 | ||
| 10 | 5-fluorouridine | 1.2 | 5′-O-benzhydryl-5-fluorouridine 9a | Trace | |
| 11 | 2.5 | 2′,5′-di-O-benzhydryl-5-fluorouridine 9b | 59 |  | |
| 3′,5′-di-O-benzhydryl-5-fluorouridine 9c | 23 | ||||
| 12 | 3.7 | 2′,3′,5′-tris-O-benzhydryl-5-fluorouridine 9d | 79 | ||

| Entry | Catalyst | Time (h) | Yield of 1 (%) a |
|---|---|---|---|
| 1 | PdCl2 | 16 | 85 |
| 2 | CuBr2 | 16 | 43 |
3. Experimental
3.1. General
3.2. General Procedure for Protection
3.3. General Procedure for Deprotection
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Lalonde, M.; Chan, T.H. Use of organosilicon reagents as protective groups in organic synthesis. Synthesis 1985, 1985, 817–845. [Google Scholar] [CrossRef]
- Corey, E.J.; Venkateswarlu, A. Protection of hydroxyl groups as tert-butyldimethylsilyl derivatives. J. Am. Chem. Soc. 1972, 94, 6190–6191. [Google Scholar]
- Somoza, A. Protecting groups for RNA synthesis: an increasing need for selective preparative methods. Chem. Soc. Rev. 2008, 37, 2668–2675. [Google Scholar]
- Pitsch, S. Protecting groups for the synthesis of ribonucleic acids. Chimia 2001, 55, 320–324. [Google Scholar]
- Greene, T.W.; Wuts, P.G.M.; Wiley, J. Protective Groups in Organic Synthesis; Wiley: New York, NY, USA, 1999; Volume 168. [Google Scholar]
- Weissman, S.A.; Zewge, D. Recent advances in ether dealkylation. Tetrahedron 2005, 61, 7833–7863. [Google Scholar] [CrossRef]
- Marco, J.L.; Hueso-Rodríguez, J.A. Synthesis of optically pure 1-(3-furyl)-1,2-dihydroxyethane derivatives. Tetrahedron Lett. 1988, 29, 2459–2462. [Google Scholar] [CrossRef]
- Tilve, M.J.; Gallo-Rodriguez, C. Glycosylation studies on conformationally restricted 3,5-O-(di-tert-butylsilylene)-d-galactofuranosyl trichloroacetimidate donors for 1,2-cis α-d-galactofuranosylation. Carbohydr. Res. 2011, 346, 2838–2848. [Google Scholar]
- Dobson, D.; Todd, A.; Gilmore, J. The Synthesis of 7-Alkoxyindoles. Synthetic Commun 1991, 21, 611–617. [Google Scholar] [CrossRef]
- Paredes, R.; Pérez, R.L. Borderline mechanisms involving ion-molecule pairs for the nucleophilic substitution reactions of benzhydrol and its derivatives. Facile formation and cleavage of diphenylmethyl ethers for the protection of hydroxyl groups. Tetrahedron Lett. 1998, 39, 2037–2038. [Google Scholar] [CrossRef]
- Kolovos, M.; Froussios, C. O-diphenylmethylation of alcohols and carboxylic acids using diphenylmethyl diphenyl phosphate as alkylating agent. Tetrahedron Lett. 1984, 25, 3909–3912. [Google Scholar] [CrossRef]
- Roberts, J.D.; Watanabe, W. The Kinetics and Mechanism of the Acid-Catalyzed Reaction of Diphenyldiazomethane with Ethyl Alcohol. J. Am. Chem. Soc. 1950, 72, 4869–4879. [Google Scholar] [CrossRef]
- Di Gioia, M.L.; Leggio, A.; Liguori, A.; Perri, F.; Siciliano, C.; Viscomi, M.C. A preparation of N-Fmoc-N-methyl-α-amino acids and N-nosyl-N-methyl-a-amino acids. Amino Acids 2010, 38, 133–143. [Google Scholar]
- Gazizov, M.B.; Ibragimov, S.N.; Khamidullina, O.D.; Karimova, R.F.; Pudovik, M.A.; Sinyashin, O.G. New Reaction of Organic Monohalides with Orthoformates. Russ. J. Gen. Chem. 2005, 75, 1325–1326. [Google Scholar]
- Noji, M.; Ohno, T.; Fuji, K.; Futaba, N.; Tajima, H.; Ishii, K. Secondary Benzylation Using Benzyl Alcohols Catalyzed by Lanthanoid, Scandium, and Hafnium Triflate. J. Org. Chem. 2003, 68, 9340–9347. [Google Scholar] [CrossRef]
- Bikard, Y.; Mezaache, R.; Weibel, J.-M.; Benkouider, A.; Sirlin, C.; Pale, P. Diarylmethyl ethers and Pd salts or complexes: a perfect combination for the protection and deprotection of alcohols. Tetrahedron 2008, 64, 10224–10232. [Google Scholar] [CrossRef]
- Bikard, Y.; Weibel, J.-M.; Sirlin, C.; Dupuis, L.; Loeffler, J.-P.; Pale, P. PdCl2, a useful catalyst for protection of alcohols as diphenylmethyl (DPM) ethers. Tetrahedron Lett. 2007, 48, 8895–8899. [Google Scholar]
- Mezaache, R.; Dembelé, Y.A.; Bikard, Y.; Weibel, J.-M.; Blanc, A.L.; Pale, P. Copper(II) bromide as an efficient catalyst for the selective protection and deprotection of alcohols as bis(4-methoxyphenyl)methyl ethers. Tetrahedron Lett. 2009, 50, 7322–7326. [Google Scholar]
- Yuan, F.-Q.; Sun, F.-Y.; Han, F.-S. A simple and efficient method for the allylation of heteroarenes catalyzed by PdCl2. Tetrahedron 2012, 68, 6837–6842. [Google Scholar]
- Yasuda, M.; Somyo, T.; Baba, A. Direct Carbon-Carbon Bond Formation from Alcohols and Active Methylenes, Alkoxyketones, or Indoles Catalyzed by Indium Trichloride. Angew. Chem. Int. Ed. 2006, 45, 793–796. [Google Scholar]
- Liu, Y.-L.; Liu, L.; Wang, D.; Chen, Y.-J. A highly α-regioselective In(OTf)3-catalyzed N-nucleophilic substitution of cyclic Baylis-Hillman adducts with aromatic amines. Tetrahedron 2009, 65, 3473–3479. [Google Scholar] [CrossRef]
- Olah, G.A.; Surya Prakash, G.K.; Narang, S.C. Synthetic Methods and Reactions; 53. Convenient Reductive Cleavage of Benzylic (Benzhydrilic) Ethers and Acetals Using AlCl3/Pd-Catalyzed Hydrogen Transfer from Cyclohexene. Synthesis 1978, 1978, 825–825. [Google Scholar] [CrossRef]
- Froussios, C.; Kolovos, M. Preparation of Diphenylmethyl Esters and Ethers of Unprotected Amino Acids and β-Hydroxy-α-amino Acids. Synthesis 1987, 1987, 1106–1108. [Google Scholar] [CrossRef]
- Mairanovsky, V.G. Electro-Deprotection—Electrochemical Removal of Protecting Groups. Ang. Chem. Int. Ed. Engl. 1976, 15, 281–292. [Google Scholar] [CrossRef]
- Codington, J.F.; Fox, J.J. Nucleosides. XXXIV. 1-(2,3,5-Tri-O-trityl-β-D-ribosyl)uracil (2′,3′,5′-tri-O-trityluridine). Carbohydr. Res. 1966, 3, 124–127. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Saudi, M.; Van Aerschot, A. A Straightforward Diphenylmethyl Protection Method and Deprotection of Some Pyrimidine Nucleosides. Molecules 2013, 18, 8524-8534. https://doi.org/10.3390/molecules18078524
Saudi M, Van Aerschot A. A Straightforward Diphenylmethyl Protection Method and Deprotection of Some Pyrimidine Nucleosides. Molecules. 2013; 18(7):8524-8534. https://doi.org/10.3390/molecules18078524
Chicago/Turabian StyleSaudi, Milind, and Arthur Van Aerschot. 2013. "A Straightforward Diphenylmethyl Protection Method and Deprotection of Some Pyrimidine Nucleosides" Molecules 18, no. 7: 8524-8534. https://doi.org/10.3390/molecules18078524
APA StyleSaudi, M., & Van Aerschot, A. (2013). A Straightforward Diphenylmethyl Protection Method and Deprotection of Some Pyrimidine Nucleosides. Molecules, 18(7), 8524-8534. https://doi.org/10.3390/molecules18078524