Development of an Advanced Synthetic Route to Macrosphelides and Its Application to the Discovery of a More Potent Macrosphelide Derivative



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
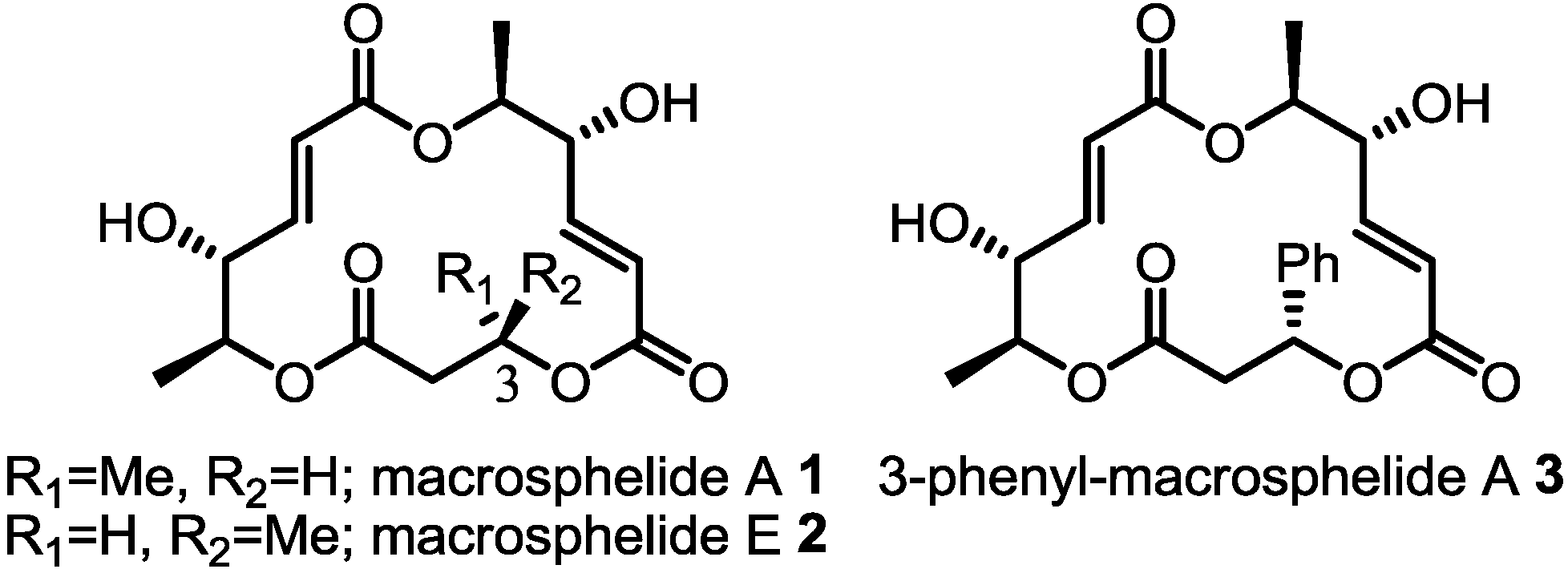
:1. Introduction

2. Results and Discussion
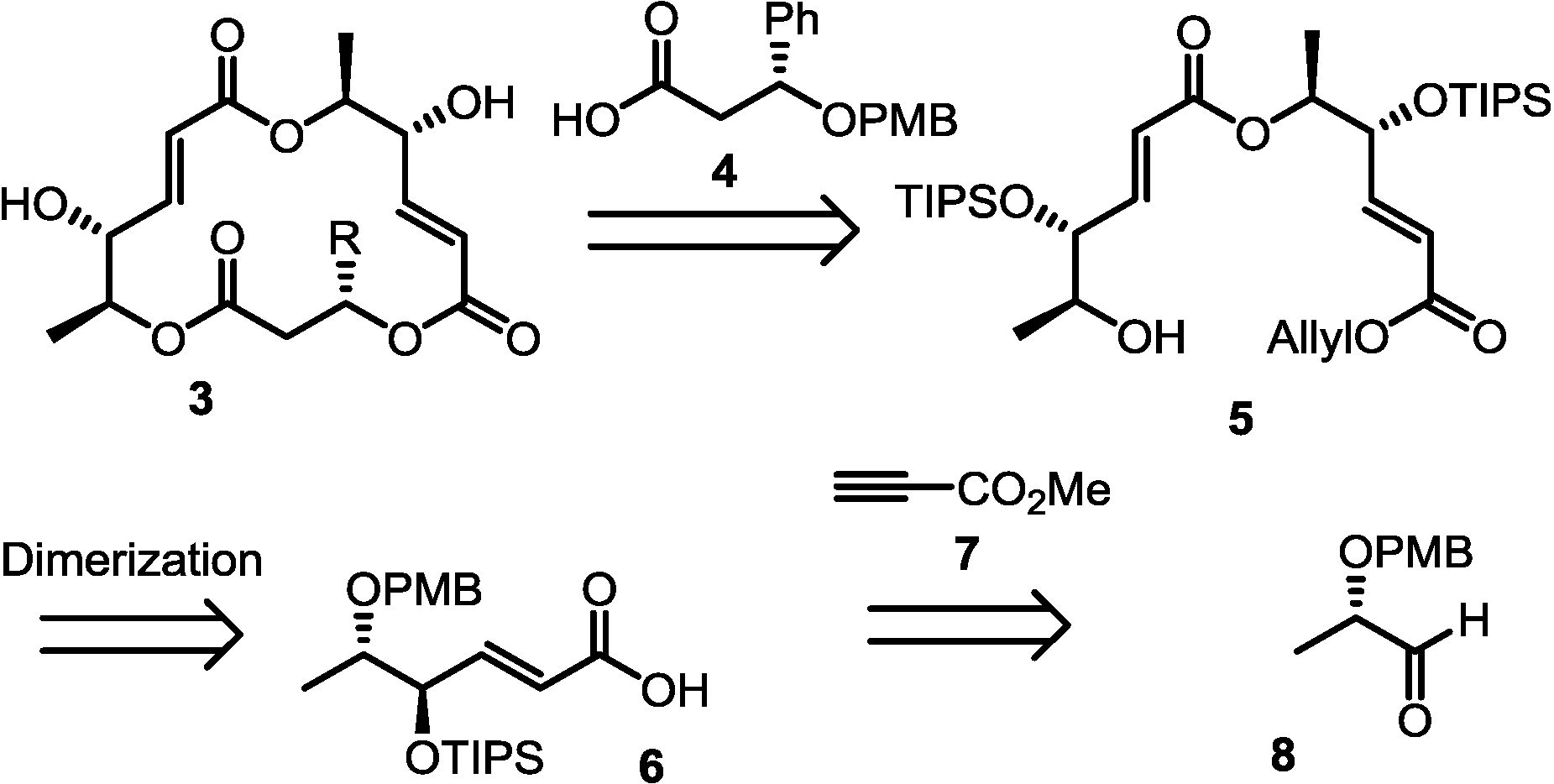
2.1. Retrosynthesis

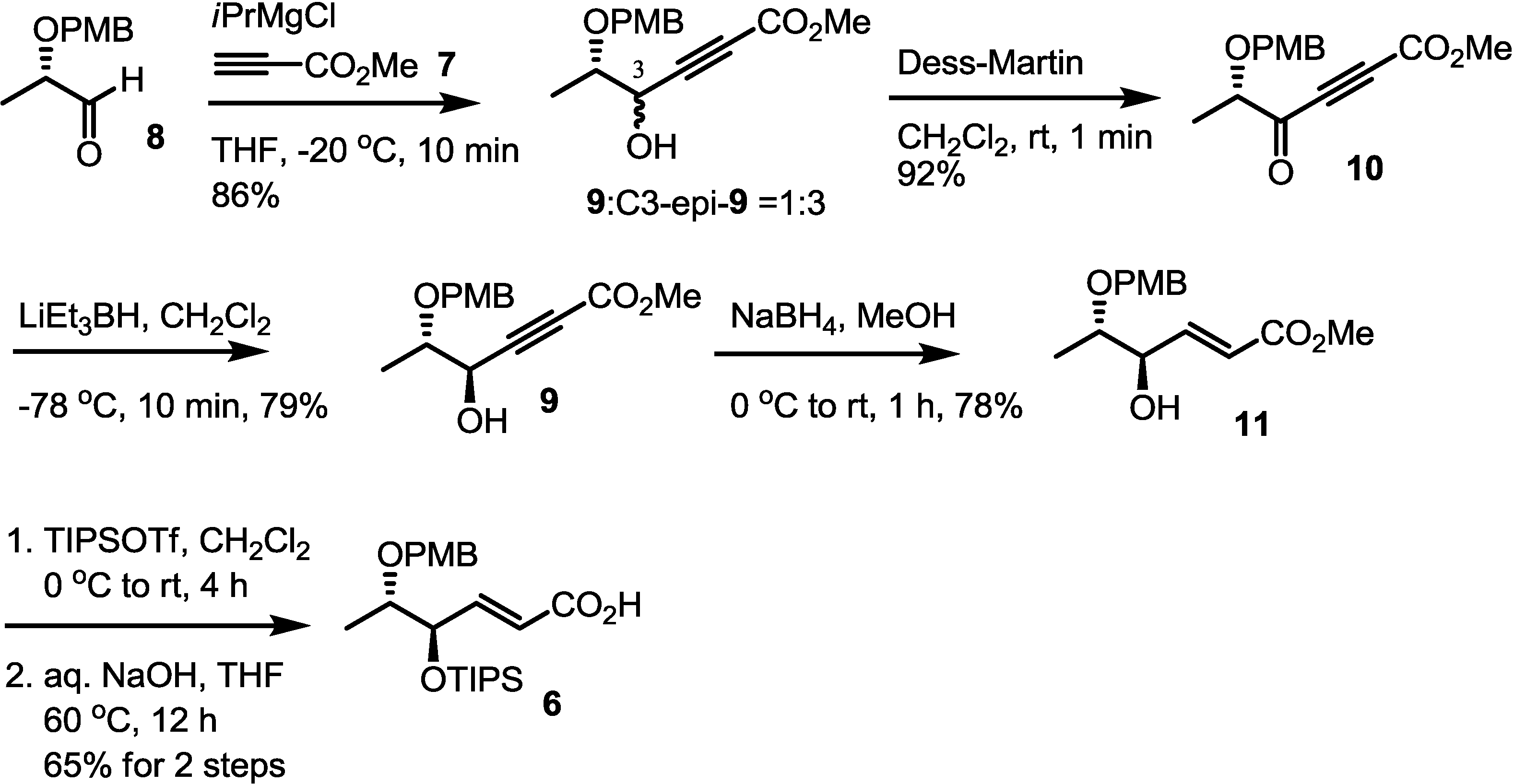
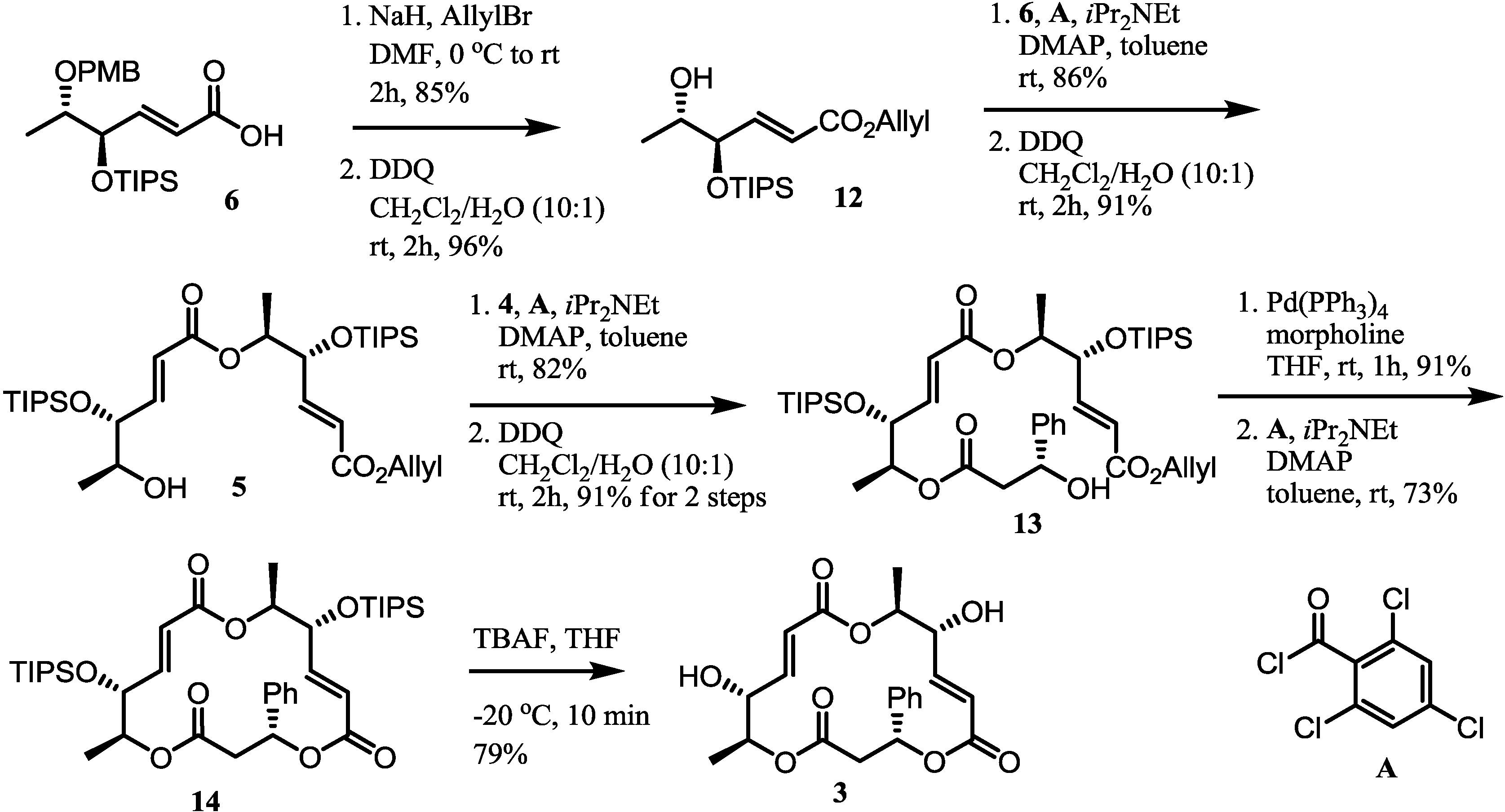
2.2. Synthesis of Monomer 6 of Macrosphelide A

2.3. Completion of the Synthesis

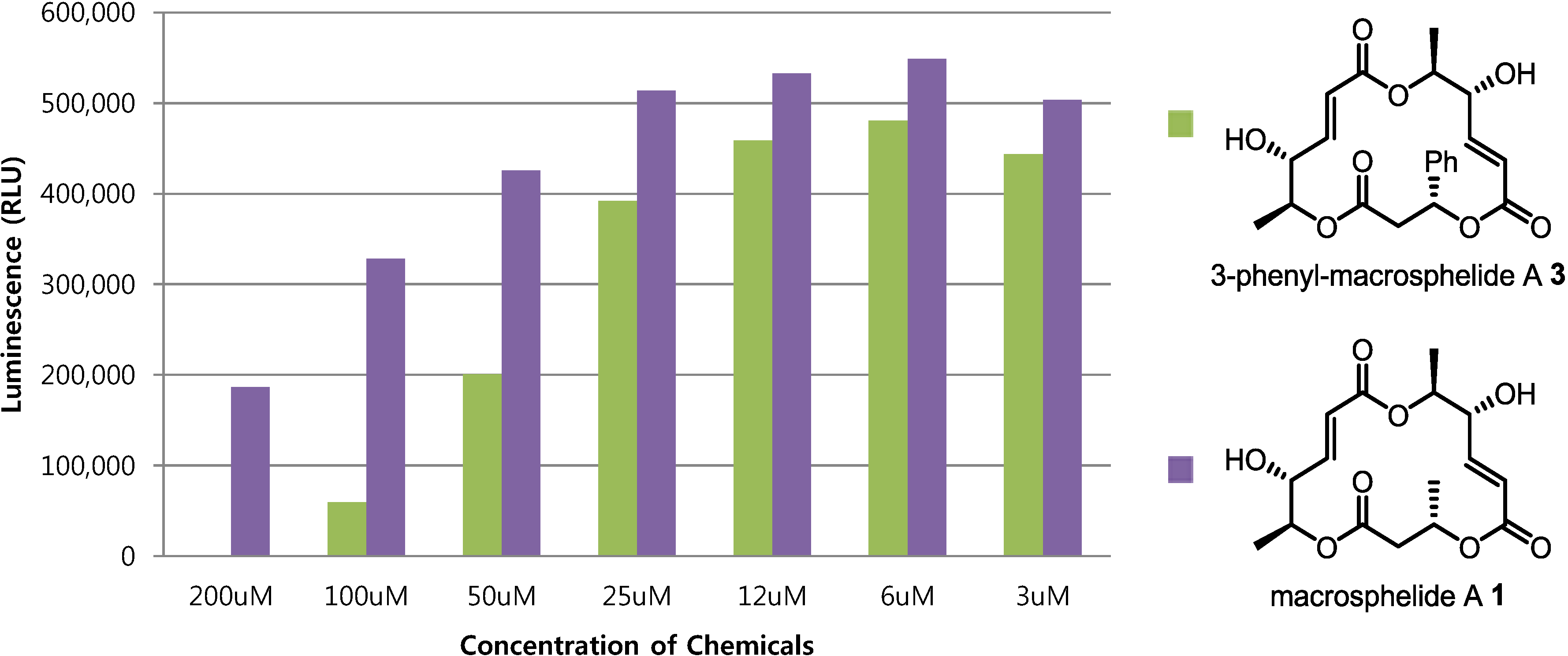
2.4. Cytotoxic Activity

3. Experimental Section
3.1. General Information
 −18.9 (c 0.76, CHCl3); FT-IR (KBr) νmax 2949, 1771, 1720, 1610 cm−1; 1H-NMR (CDCl3, 300 MHz) δ 7.26 (d, 2H, J = 6.3 Hz), 6.93–6.88 (m, 3H), 6.15 (dd, 1H, J = 15.6, 1.8 Hz), 4.65–4.44 (m, 3H), 3.83 (s, 3H), 3.76 (s, 3H), 3.70 (m, 1H), 2.37 (d, 1H, J = 4.5 Hz), 1.15 (d, 3H, J = 6.5 Hz); 13C-NMR (CDCl3, 75 MHz) δ 166.7, 159.3, 145.9, 129.9, 129.3, 121.4, 113.9, 76.2, 72.6, 70.5, 55.2, 51.5, 14.0. −9.0 (c 1.0, CHCl3); FT-IR (KBr) νmax 2942, 2865, 2678, 1698, 1655 cm−1; 1H-NMR (CDCl3, 500 MHz) δ 7.26 (d, 2H, J = 6.5 Hz), 7.11 (dd, 1H, J = 15.5, 5.5 Hz), 6.88 (d, 2H, J = 6.5 Hz), 6.08 (dd, 1H, J = 16.0, 1.0 Hz), 4.55 (d, 2H, J = 4.0 Hz), 4.47 (m, 1H), 3.82 (s, 3H), 3.60 (m, 1H), 1.28 (d, 3H, J = 6.6 Hz), 1.17 (m, 21H); 13C-NMR (CDCl3, 125 MHz) δ 159.5, 151.5, 130.9, 129.6, 114.2, 114.1, 78.4, 76.3, 71.7, 55.6, 18.4 (2C), 16.1, 13.1; LR-MS (ESI) m/z 461 (M+K+). −12.3 (c 0.8, CHCl3); FT-IR (KBr) νmax 2942, 2866, 1360, 1819, 1721 cm−1; 1H-NMR (CDCl3, 300 MHz) δ 6.96 (dd, 1H, J = 15.6, 6.3 Hz), 6.04 (dd, 1H, J = 15.6, 1.2 Hz), 5.92 (m, 1H), 5.30 (m, 2H), 4.66 (d, 2H, J = 5.7 Hz), 4.35 (m, 1H), 3.92 (m, 1H), 1.13 (d, 3H, J = 6.6 Hz); 13C-NMR (CDCl3, 75 MHz) δ 165.5, 146.4, 132.1, 122.6, 118.1, 76.1, 70.7, 65.1, 17.9, 17.4, 12.3; LR-MS (FAB+) m/z 343 (M+H+). −14.2 (c 0.76, CHCl3); FT-IR (KBr) νmax 2942, 2866, 1359, 1723, 1655 cm−1; 1H-NMR (CDCl3, 500 MHz) δ 6.89 (m, 2H), 6.00 (dd, 1H, J = 15.3, 1.2 Hz), 5.96 (dd, 1H, J = 15.3, 1.2 Hz), 5.88 (m, 1H), 5.24 (m, 1H), 5.00 (dd, 1H, J = 8.5, 6.5 Hz), 4.58 (d, 2H, J = 3.3 Hz), 4.52 (m, 1H), 4.28 (m, 1H), 3.84 (dd, 1H, J = 6.5, 3.5 Hz), 1.16 (d, 3H, J = 6.6 Hz), 1.04 (d, 3H, J = 6.4 Hz), 0.98 (m, 21H); 13C-NMR (CDCl3, 125 MHz) δ 166.0, 165.8, 147.5, 146.9, 132.5, 123.1, 122.6, 118.4, 76.4, 74.6, 73.6, 71.1, 65.5, 60.7, 18.3, 17.8, 14.5, 12.8, 12.7; LR-MS (ESI) m/z 649 (M+Na+). −0.5 (c 0.86, CHCl3); 1H-NMR (CDCl3, 500 MHz) δ 7.36 (m, 5H), 6.97 (dd, 1H, J = 15.5, 5.5 Hz), 6.92 (dd, 1H, J = 15.5, 5.5 Hz), 6.11 (m, 2H), 5.95 (m, 1H), 5.23 (m, 2H), 5.15 (dd, 1H, J = 8.0, 4.5 Hz), 5.00 (m, 2H), 4.68 (m, 2H), 4.63 (m, 1H), 4.58 (m, 1H), 2.75 (m, 2H), 1.45 (d, 3H, J = 6.6 Hz), 1.35 (d, 3H, J = 6.5 Hz), 1.07 (m, 42H); LR-MS (FAB+) m/z 775 (M+H+). −6.7 (c 0.76, CHCl3); FT-IR (KBr) νmax 2942, 2866, 2359, 1721 cm−1; 1H-NMR (CDCl3, 500 MHz) δ 7.36 (m, 5H), 6.88 (m, 2H), 6.22 (dd, 1H, J = 8.5, 2.5 Hz), 5.96 (d, 1H, J = 15.5 Hz), 5.93 (d, 1H, J = 15.5 Hz), 5.02 (m, 1H), 4.91 (m, 1H), 4.28 (t, 1H, J = 7.5 Hz), 4.24 (t, 1H, J = 7.5 Hz), 2.89 (dd, 1H, J = 15.5, 10.0 Hz), 2.75 (dd, 1H, J = 15.5, 2.5 Hz), 1.45 (d, 3H, J = 6.6 Hz), 1.35 (d, 3H, J = 6.5 Hz), 1.07 (m, 42H); 13C-NMR (CDCl3, 125 MHz) δ 169.5, 164.9, 164.3, 148.5, 147.9, 139.5, 129.0, 128.6, 126.4, 123.0, 122.6, 76.4, 74.9, 74.0, 73.0, 72.4, 42.3, 31.9, 18.3, 18.2, 18.1, 12.9, 12.8; LR-MS (FAB+) m/z 717 (M+H+). +34.2 (c 0.21, CHCl3); 1H-NMR (CDCl3, 500 MHz) δ 7.39–7.31 (m, 5H), 7.02 (dd, 1H, J = 16.0, 4.0 Hz), 6.95 (dd, 1H, J = 16.0, 4.0 Hz), 6.31 (dd, 1H, J = 11.5, 2.0 Hz), 6.12 (dd, 1H, J = 16.0, 1.5 Hz ), 6.10 (dd, 1H, J = 16.0, 1.5 Hz ), 5.04–4.96 (m, 1H), 4.94–4.89 (m, 1H), 4.28 (m, 1H), 4.18 (m, 1H), 3.03 (dd, 1H, J = 16.0, 11.5 Hz ), 2.94 (br, 1H), 2.75 (dd, 1H, J = 16.0, 2.0 Hz ), 2.50 (br, 1H), 1.50 (d, 3H, J = 6.5 Hz), 1.41 (d, 3H, J = 6.5 Hz); 13C-NMR (CDCl3, 125 MHz) δ170.1, 166.3, 164.3, 146.6, 145.5, 139.3, 129.1, 128.8, 126.5, 122.7 (2C), 75.9, 75.1, 74.5, 73.6, 72.5, 42.0, 18.5, 18.4; FT-IR (KBr) νmax 3429, 2923, 2852, 1710, 1627; LR-MS (FAB) m/z 405 (M+H+); HR-MS (FAB) calcd for C21H25O8 405.1549 (M+H+) found 405.1569.
−18.9 (c 0.76, CHCl3); FT-IR (KBr) νmax 2949, 1771, 1720, 1610 cm−1; 1H-NMR (CDCl3, 300 MHz) δ 7.26 (d, 2H, J = 6.3 Hz), 6.93–6.88 (m, 3H), 6.15 (dd, 1H, J = 15.6, 1.8 Hz), 4.65–4.44 (m, 3H), 3.83 (s, 3H), 3.76 (s, 3H), 3.70 (m, 1H), 2.37 (d, 1H, J = 4.5 Hz), 1.15 (d, 3H, J = 6.5 Hz); 13C-NMR (CDCl3, 75 MHz) δ 166.7, 159.3, 145.9, 129.9, 129.3, 121.4, 113.9, 76.2, 72.6, 70.5, 55.2, 51.5, 14.0. −9.0 (c 1.0, CHCl3); FT-IR (KBr) νmax 2942, 2865, 2678, 1698, 1655 cm−1; 1H-NMR (CDCl3, 500 MHz) δ 7.26 (d, 2H, J = 6.5 Hz), 7.11 (dd, 1H, J = 15.5, 5.5 Hz), 6.88 (d, 2H, J = 6.5 Hz), 6.08 (dd, 1H, J = 16.0, 1.0 Hz), 4.55 (d, 2H, J = 4.0 Hz), 4.47 (m, 1H), 3.82 (s, 3H), 3.60 (m, 1H), 1.28 (d, 3H, J = 6.6 Hz), 1.17 (m, 21H); 13C-NMR (CDCl3, 125 MHz) δ 159.5, 151.5, 130.9, 129.6, 114.2, 114.1, 78.4, 76.3, 71.7, 55.6, 18.4 (2C), 16.1, 13.1; LR-MS (ESI) m/z 461 (M+K+). −12.3 (c 0.8, CHCl3); FT-IR (KBr) νmax 2942, 2866, 1360, 1819, 1721 cm−1; 1H-NMR (CDCl3, 300 MHz) δ 6.96 (dd, 1H, J = 15.6, 6.3 Hz), 6.04 (dd, 1H, J = 15.6, 1.2 Hz), 5.92 (m, 1H), 5.30 (m, 2H), 4.66 (d, 2H, J = 5.7 Hz), 4.35 (m, 1H), 3.92 (m, 1H), 1.13 (d, 3H, J = 6.6 Hz); 13C-NMR (CDCl3, 75 MHz) δ 165.5, 146.4, 132.1, 122.6, 118.1, 76.1, 70.7, 65.1, 17.9, 17.4, 12.3; LR-MS (FAB+) m/z 343 (M+H+). −14.2 (c 0.76, CHCl3); FT-IR (KBr) νmax 2942, 2866, 1359, 1723, 1655 cm−1; 1H-NMR (CDCl3, 500 MHz) δ 6.89 (m, 2H), 6.00 (dd, 1H, J = 15.3, 1.2 Hz), 5.96 (dd, 1H, J = 15.3, 1.2 Hz), 5.88 (m, 1H), 5.24 (m, 1H), 5.00 (dd, 1H, J = 8.5, 6.5 Hz), 4.58 (d, 2H, J = 3.3 Hz), 4.52 (m, 1H), 4.28 (m, 1H), 3.84 (dd, 1H, J = 6.5, 3.5 Hz), 1.16 (d, 3H, J = 6.6 Hz), 1.04 (d, 3H, J = 6.4 Hz), 0.98 (m, 21H); 13C-NMR (CDCl3, 125 MHz) δ 166.0, 165.8, 147.5, 146.9, 132.5, 123.1, 122.6, 118.4, 76.4, 74.6, 73.6, 71.1, 65.5, 60.7, 18.3, 17.8, 14.5, 12.8, 12.7; LR-MS (ESI) m/z 649 (M+Na+). −0.5 (c 0.86, CHCl3); 1H-NMR (CDCl3, 500 MHz) δ 7.36 (m, 5H), 6.97 (dd, 1H, J = 15.5, 5.5 Hz), 6.92 (dd, 1H, J = 15.5, 5.5 Hz), 6.11 (m, 2H), 5.95 (m, 1H), 5.23 (m, 2H), 5.15 (dd, 1H, J = 8.0, 4.5 Hz), 5.00 (m, 2H), 4.68 (m, 2H), 4.63 (m, 1H), 4.58 (m, 1H), 2.75 (m, 2H), 1.45 (d, 3H, J = 6.6 Hz), 1.35 (d, 3H, J = 6.5 Hz), 1.07 (m, 42H); LR-MS (FAB+) m/z 775 (M+H+). −6.7 (c 0.76, CHCl3); FT-IR (KBr) νmax 2942, 2866, 2359, 1721 cm−1; 1H-NMR (CDCl3, 500 MHz) δ 7.36 (m, 5H), 6.88 (m, 2H), 6.22 (dd, 1H, J = 8.5, 2.5 Hz), 5.96 (d, 1H, J = 15.5 Hz), 5.93 (d, 1H, J = 15.5 Hz), 5.02 (m, 1H), 4.91 (m, 1H), 4.28 (t, 1H, J = 7.5 Hz), 4.24 (t, 1H, J = 7.5 Hz), 2.89 (dd, 1H, J = 15.5, 10.0 Hz), 2.75 (dd, 1H, J = 15.5, 2.5 Hz), 1.45 (d, 3H, J = 6.6 Hz), 1.35 (d, 3H, J = 6.5 Hz), 1.07 (m, 42H); 13C-NMR (CDCl3, 125 MHz) δ 169.5, 164.9, 164.3, 148.5, 147.9, 139.5, 129.0, 128.6, 126.4, 123.0, 122.6, 76.4, 74.9, 74.0, 73.0, 72.4, 42.3, 31.9, 18.3, 18.2, 18.1, 12.9, 12.8; LR-MS (FAB+) m/z 717 (M+H+). +34.2 (c 0.21, CHCl3); 1H-NMR (CDCl3, 500 MHz) δ 7.39–7.31 (m, 5H), 7.02 (dd, 1H, J = 16.0, 4.0 Hz), 6.95 (dd, 1H, J = 16.0, 4.0 Hz), 6.31 (dd, 1H, J = 11.5, 2.0 Hz), 6.12 (dd, 1H, J = 16.0, 1.5 Hz ), 6.10 (dd, 1H, J = 16.0, 1.5 Hz ), 5.04–4.96 (m, 1H), 4.94–4.89 (m, 1H), 4.28 (m, 1H), 4.18 (m, 1H), 3.03 (dd, 1H, J = 16.0, 11.5 Hz ), 2.94 (br, 1H), 2.75 (dd, 1H, J = 16.0, 2.0 Hz ), 2.50 (br, 1H), 1.50 (d, 3H, J = 6.5 Hz), 1.41 (d, 3H, J = 6.5 Hz); 13C-NMR (CDCl3, 125 MHz) δ170.1, 166.3, 164.3, 146.6, 145.5, 139.3, 129.1, 128.8, 126.5, 122.7 (2C), 75.9, 75.1, 74.5, 73.6, 72.5, 42.0, 18.5, 18.4; FT-IR (KBr) νmax 3429, 2923, 2852, 1710, 1627; LR-MS (FAB) m/z 405 (M+H+); HR-MS (FAB) calcd for C21H25O8 405.1549 (M+H+) found 405.1569.3.2. Cell Culture
3.3. Cell Viability
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Koehn, F.E.; Carter, G.T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 2005, 3, 206–220. [Google Scholar]
- Kong, D.-X.; Jiang, Y.-Y.; Zhang, H.-Y. Marine natural products as sources of novel scaffolds: Achievement and concern. Drug Discov. Today 2010, 15, 884–886. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar]
- Li, J.W.-H.; Vederas, J.C. Drug discovery and natural products: End of an era or an endless frontier? Science 2009, 325, 161–165. [Google Scholar]
- John, J.E. Natural products as lead-structures: A role for biotechnology. Drug Discov. Today 2010, 15, 409–410. [Google Scholar]
- Hayashi, M.; Kim, Y.-P.; Hiraoka, H.; Natori, M.; Takamatsu, S.; Kawakubo, T.; Masuma, R.; Komiyama, K.; Ōmura, S. Macrosphelide, a novel inhibitor of cell-cell adhesion molecule I. Taxonomy, fermentation, isolation and biological activities. J. Antibiot. 1995, 48, 1435–1439. [Google Scholar]
- Ahmed, K.; Zhao, Q.-L.; Matsuya, Y.; Yu, D.-Y.; Feril, L.B.; Nemoto, H.; Kondo, T. Rapid and transient intracellular oxidative stress due to novel macrosphelides trigger apoptosis via Fas/caspase-8-dependent pathway in human lymphoma U937 cells. Chem.-Biol. Interact. 2007, 170, 86–99. [Google Scholar]
- Ahmed, K.; Matsuya, Y.; Nemoto, H.; Zaidi, S.F.H.; Sugiyama, T.; Yoshihisa, Y.; Shimizu, T.; Kondo, T. Mechanism of apoptosis induced by a newly synthesized derivative of macrosphelides with a thiazole side chain. Chem.-Biol. Interact 2009, 177, 218–226. [Google Scholar]
- Tomprefa, N.; McQuilken, M.P.; Hill, R.A.; Whipps, J.M. Antimicrobial activity of Coniothyrium minitans and its macrolide antibiotic macrosphelide A. J. Appl. Microbiol. 2009, 106, 2048–2056. [Google Scholar]
- Sunazuka, T.; Hirose, T.; Harigaya, Y.; Takamatsu, S.; Hayashi, M.; Komiyama, K.; Ōmura, S.; Sprengeler, P.A.; Smith, A.B. Relative and absolute stereochemistries and total synthesis of (+)-macrosphelides A and B, potent, orally bioavailable inhibitors of cell-cell adhesion. J. Am. Chem. Soc. 1997, 119, 10247–10248. [Google Scholar]
- Nakamura, H.; Ono, M.; Shida, Y.; Akita, H. New total syntheses of (+)-macrosphelides C, F and G. Tetrahedron: Asymmetry 2002, 13, 705–713. [Google Scholar]
- Kobayashi, Y.; Wang, Y.-G. Synthesis of macrosphelides H and G. Tetrahedron Lett. 2002, 43, 4381–4384. [Google Scholar]
- Sharma, G.V.M.; Mouli, C.C. The total synthesis of macrosphelides A and E fromcarbohydrate precursors. Tetrahedron Lett. 2002, 43, 9159–9161. [Google Scholar]
- Matsuya, Y.; Kawaguchi, T.; Nemoto, H. New strategy for the total synthesis ofmacrosphelides A and B based onring-closing metathesis. Org. Lett. 2003, 5, 2939–2941. [Google Scholar]
- Sharma, G.V.M.; Mouli, C.C. A total synthesis of macrosphelides C and F from l-(+)-arabinose. Tetrahedron Lett. 2003, 44, 8161–8163. [Google Scholar]
- Kusaka, S.-i.; Dohi, S.; Doi, T.; Takahashi, T. Total synthesis of macrosphelide A by way ofpalladium-catalyzed carbonylative esterification. Tetrahedron Lett. 2003, 44, 8857–8859. [Google Scholar]
- Paek, S.-M.; Seo, S.-Y.; Kim, S.-H.; Jung, J.-W.; Lee, Y.-S.; Jung, J.-K.; Suh, Y.-G. Concise Syntheses of(+)-Macrosphelides A and B. Org. Lett. 2005, 7, 3159–3162. [Google Scholar]
- Yun, H.; Paek, S.-M.; Jung, J.-W.; Kim, N.-J.; Kim, S.-H.; Suh, Y.-G. First total syntheses of (−)-macrosphelides J and K and elucidation of their absolute configuration. Chem. Commun. 2009, 2463–2465. [Google Scholar]
- Prasad, K.R.; Gutala, P. Enantioselective total synthesis of macrosphelides A and E. Tetrahedron 2011, 67, 4514–4520. [Google Scholar]
- Sharma, G.V.M.; Reddy, P.S. Total synthesis of macrosphelide M from diacetone glucose. Eur. J. Org. Chem. 2012, 12, 2414–2421. [Google Scholar]
- Matsuya, Y.; Kawaguchi, T.; Ishihara, K.; Ahmed, K.; Zhao, Q.-L.; Kondo, T.; Nemoto, H. Synthesis of macrosphelides with athiazole side chain: New antitumorcandidates having apoptosis-inducing property. Org. Lett. 2006, 8, 4609–4612. [Google Scholar]
- Wang, B.-L.; Jiang, Z.-X.; You, Z.-W.; Qing, F.-L. Total synthesis of trifluoromethylated analogs of macrosphelide A. Tetrahedron 2007, 63, 12671–12680. [Google Scholar]
- Matsuya, Y.; Kobayashi, Y.; Kawaguchi, T.; Hori, A.; Watanabe, Y.; Ishihara, K.; Ahmed, K.; Wei, Z.-L.; Yu, D.-Y.; Zhao, Q.-L.; et al. Design, synthesis, and biological evaluation of artificial macrosphelides in the search for new apoptosis-inducing agents. Chem. Eur. J. 2009, 15, 5799–5813. [Google Scholar]
- Takahashi, T.; Kusaka, S.; Doi, T.; Sunazuka, T.; Ōmura, S. A combinatorial synthesis of a macrosphelide library utilizing a palladium-catalyzed carbonylation on a polymer support. Angew. Chem. Int. Ed. 2003, 42, 5230–5234. [Google Scholar]
- Curran, D.P.; Sinha, M.K.; Zhang, K.; Sabatini, J.J.; Cho, D.-H. Binary fluorous tagging enables the synthesis and separation of a 16-stereoisomer library of macrosphelides. Nat. Chem. 2012, 4, 124–129. [Google Scholar]
- Instead of methyl group in C3 position, introduction of a thiazole moiety was published. Matsuya, Y.; Nemoto, H. Artificial macrosphelides as a novel apoptosis-inducing compound. Heterocycles 2010, 81, 57–66. [Google Scholar]
- Numata, A.; Iritani, M.; Yamada, T.; Minoura, K.; Matsumura, E.; Yamori, T.; Tsuruo, T. Novel antitumour metabolites produced by a fungal strain from a sea hare. Tetrahedron Lett. 1997, 38, 8215–8218. [Google Scholar]
- Yamada, T.; Iritani, M.; Doi, M.; Minoura, K.; Ito, T.; Numata, A. Absolute stereostructures of cell-adhesion inhibitors, macrosphelides C, E–G and I, produced by a Periconia species separated from an Aplysiasea hare. J. Chem. Soc. Perkin Trans. 1 2001, 3046–3053. [Google Scholar]
- Meta, C.T.; Koide, K. Trans-selective conversions of γ-hydroxy-α,β-alkynoic esters to γ-hydroxy-α,β-alkenoic esters. Org. Lett. 2004, 6, 1785–1787. [Google Scholar]
- Carr, K.; Greener, N.A.; Mullah, K.; Somerville, F.M.; Sutherland, J.K. A synthesis of (±)-demethoxydaunomycinone. J. Chem. Soc. Perkin Trans. 1 1992, 15, 1975–1980. [Google Scholar]
- Faucher, A.-M.; Brochu, C.; Landry, S.R.; Duchesne, I.; Hantos, S.; Roy, A.; Myles, A.; Legault, C. Chelation-controlled reduction of α- and β-oxygenated ketones with lithium tri-n-butylborohydride. Tetrahedron Lett. 1998, 39, 8425–8428. [Google Scholar]
- Formal synthesis of 1 using LiAlH4 was reported. Srinivasa Rao, K.; Mukkanti, K.; Srinivasa Reddy, D.; Pal, M.; Iqbal, J. A simple procedure for the synthesis of γ-hydroxy-α,β-(E)-alkenoic esters: Formal synthesis of (+)-macrosphelides A and B. Tetrahedron Lett. 2005, 46, 2287–2290. [Google Scholar]
- Oikawa, Y.; Yoshioka, T.; Yonemitsu, O. Specific removal of o-methoxybenzyl protection by DDQ oxidation. Tetrahedron Lett. 1982, 23, 885–888. [Google Scholar]
- Eh, M.; Schomburg, D.; Schicht, K.; Kalesse, M. An efficient synthesis of radicinin analogues. Tetrahedron 1995, 51, 8983–8992. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heo, Y.M.; Lee, H.; Shin, Y.K.; Paek, S.-M. Development of an Advanced Synthetic Route to Macrosphelides and Its Application to the Discovery of a More Potent Macrosphelide Derivative. Molecules 2014, 19, 15572-15583. https://doi.org/10.3390/molecules191015572
Heo YM, Lee H, Shin YK, Paek S-M. Development of an Advanced Synthetic Route to Macrosphelides and Its Application to the Discovery of a More Potent Macrosphelide Derivative. Molecules. 2014; 19(10):15572-15583. https://doi.org/10.3390/molecules191015572
Chicago/Turabian StyleHeo, Yu Mi, Hunseok Lee, Young Kee Shin, and Seung-Mann Paek. 2014. "Development of an Advanced Synthetic Route to Macrosphelides and Its Application to the Discovery of a More Potent Macrosphelide Derivative" Molecules 19, no. 10: 15572-15583. https://doi.org/10.3390/molecules191015572
APA StyleHeo, Y. M., Lee, H., Shin, Y. K., & Paek, S. -M. (2014). Development of an Advanced Synthetic Route to Macrosphelides and Its Application to the Discovery of a More Potent Macrosphelide Derivative. Molecules, 19(10), 15572-15583. https://doi.org/10.3390/molecules191015572