A DFT Study of the Photochemical Dimerization of Methyl 3-(2-Furyl)acrylate and Allyl Urocanate

Abstract
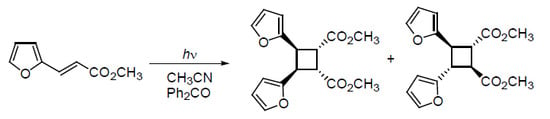
:
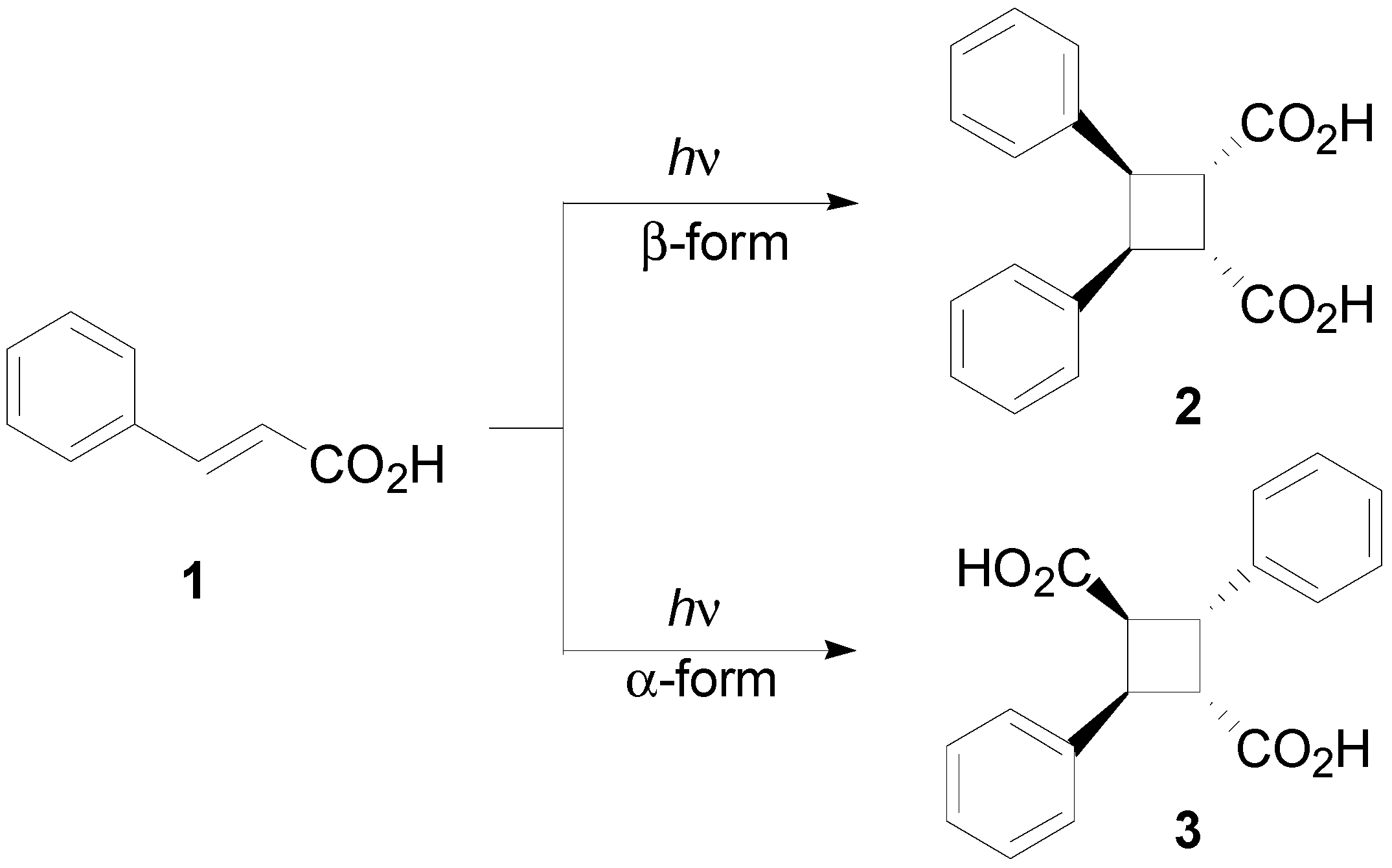
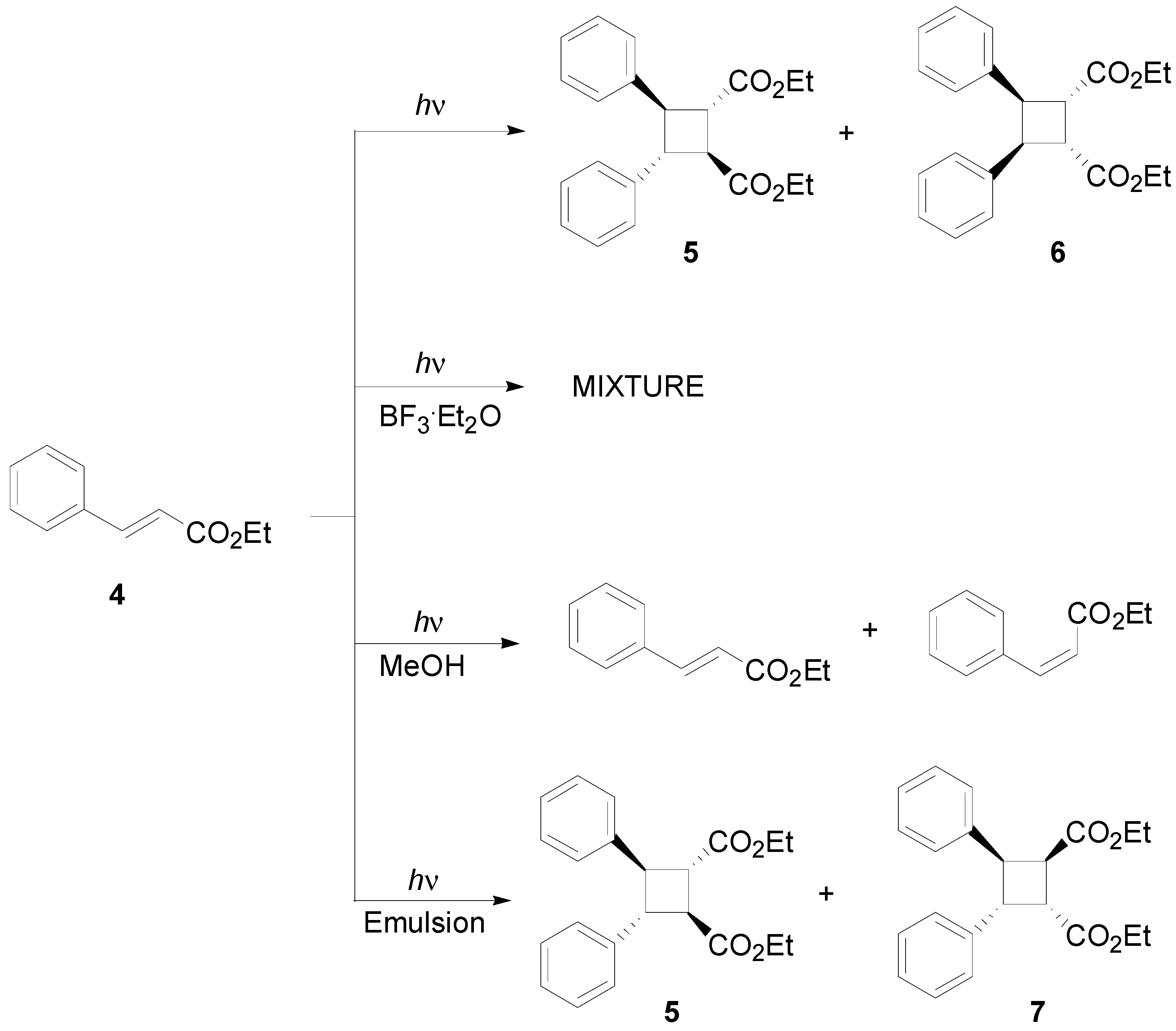
1. Introduction


2. Computational Details
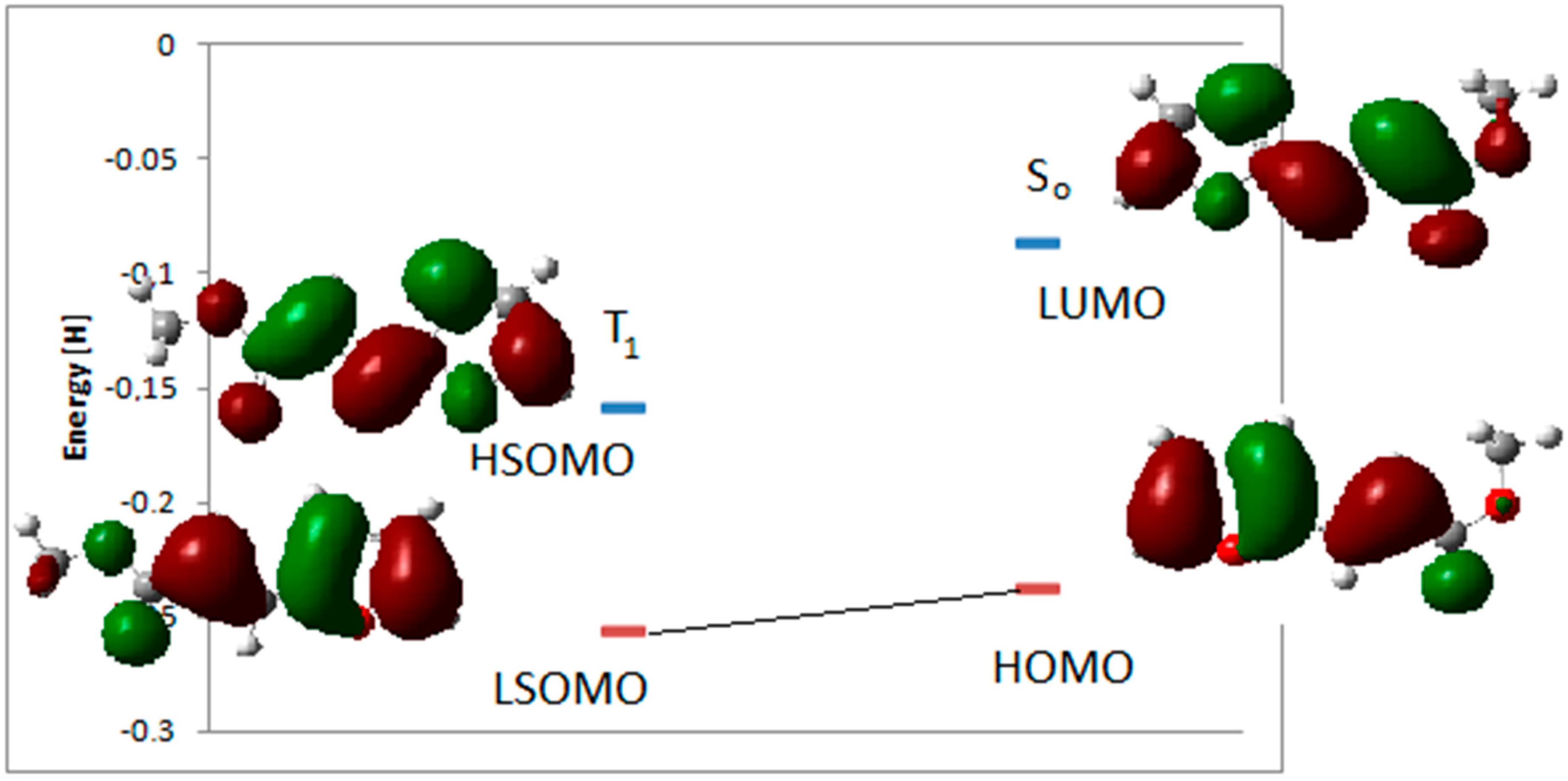
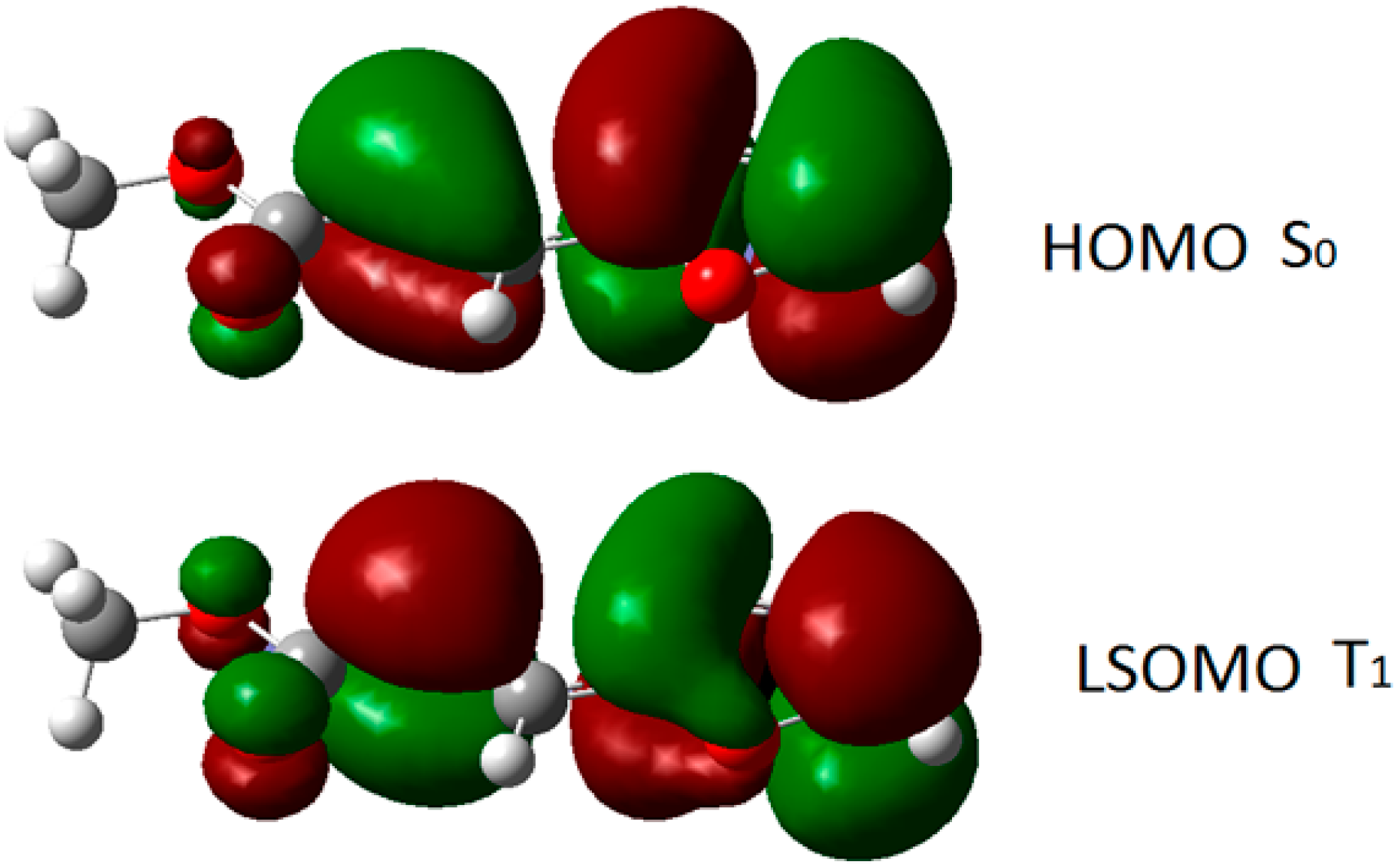
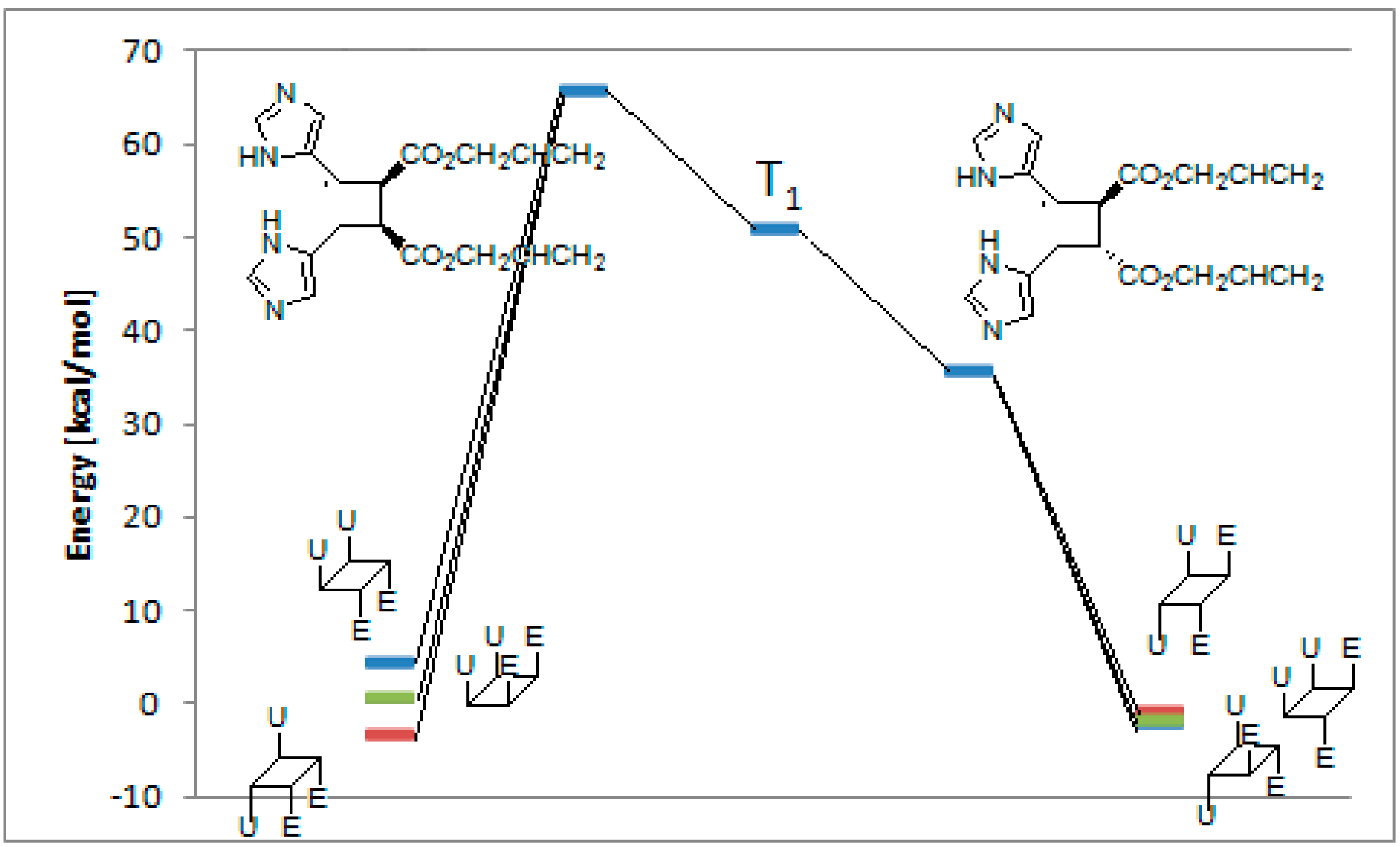
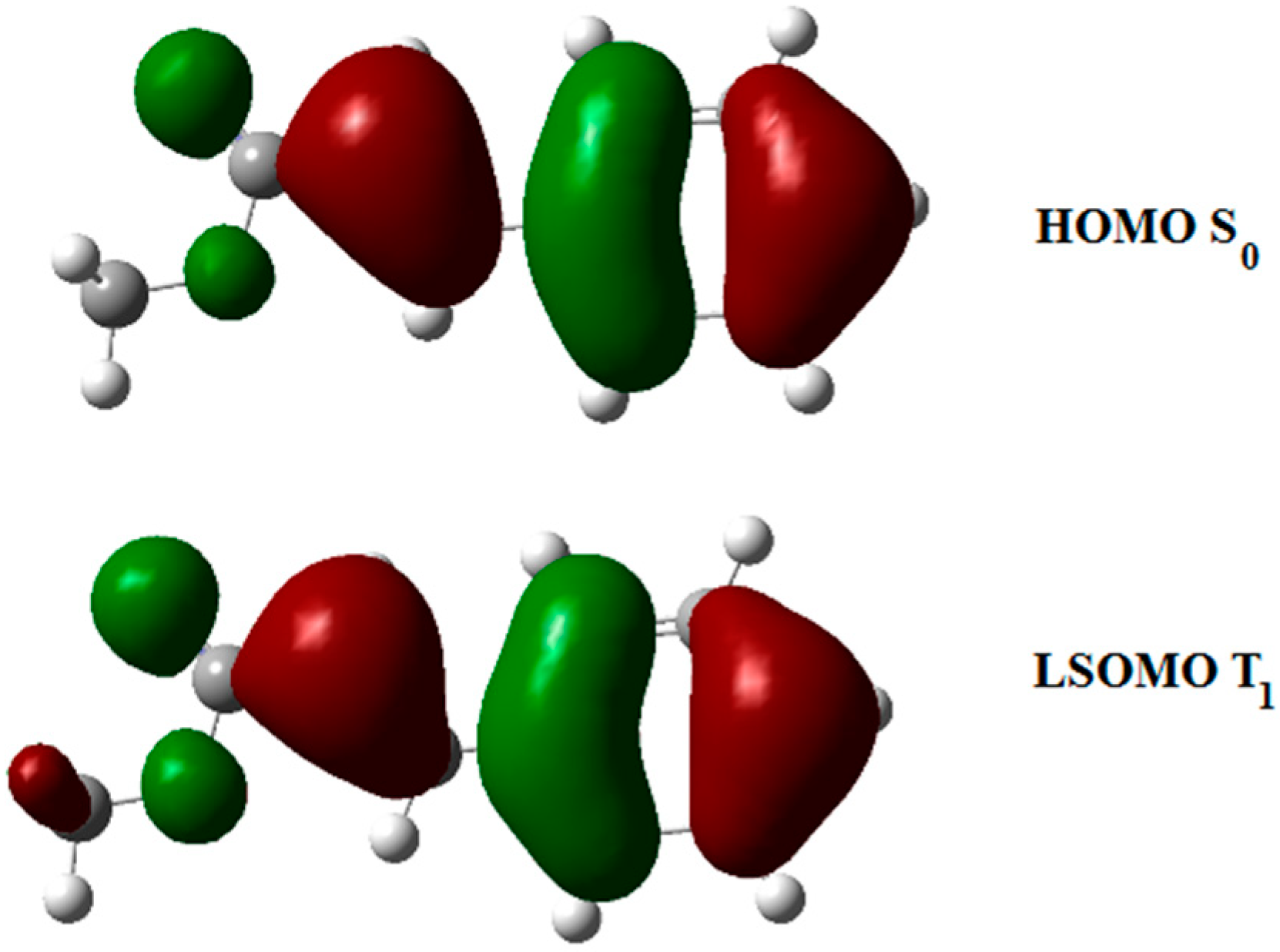
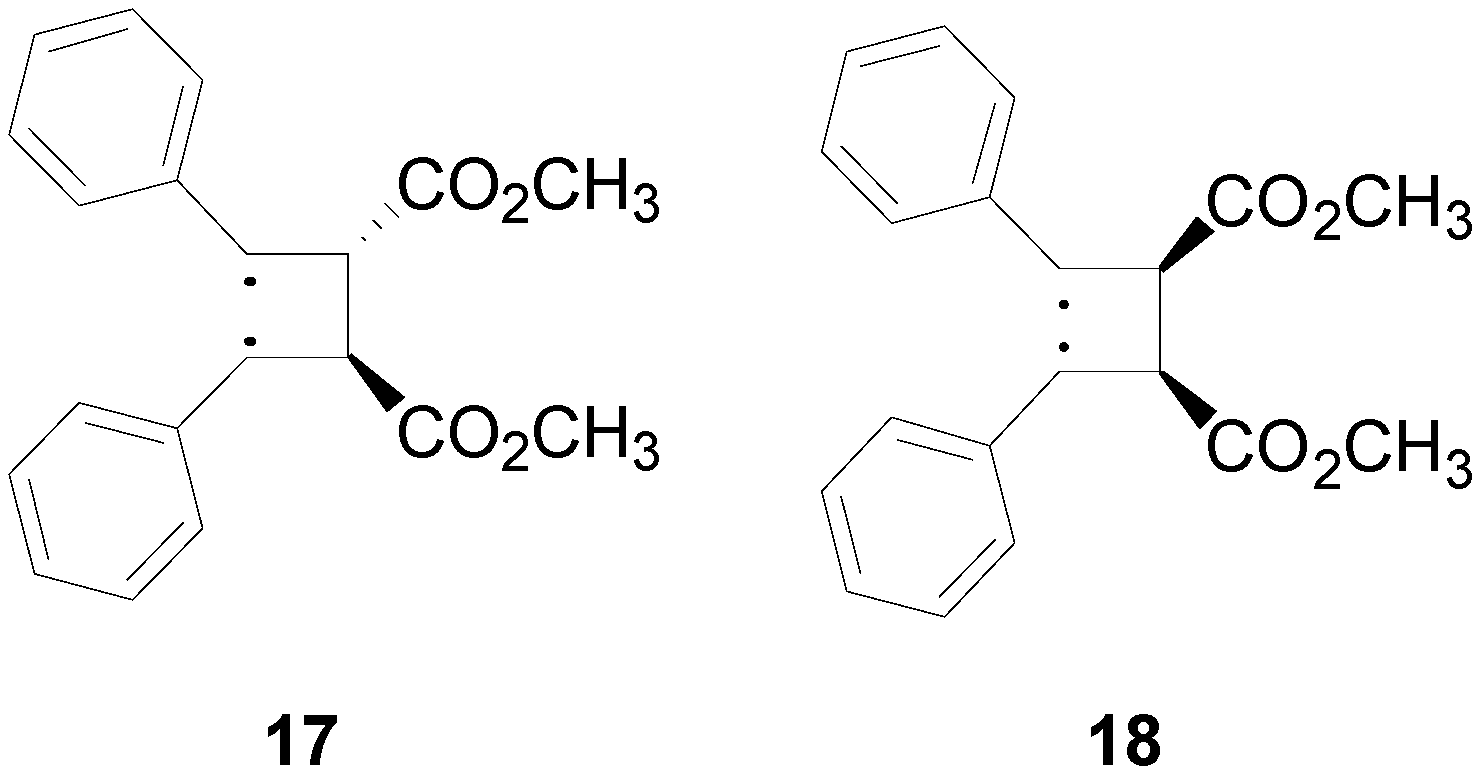
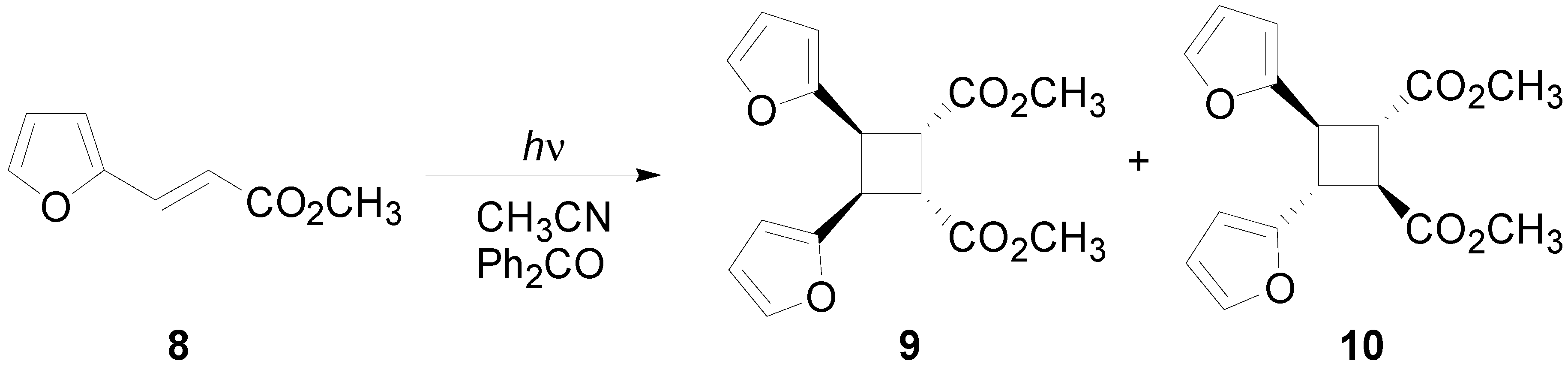
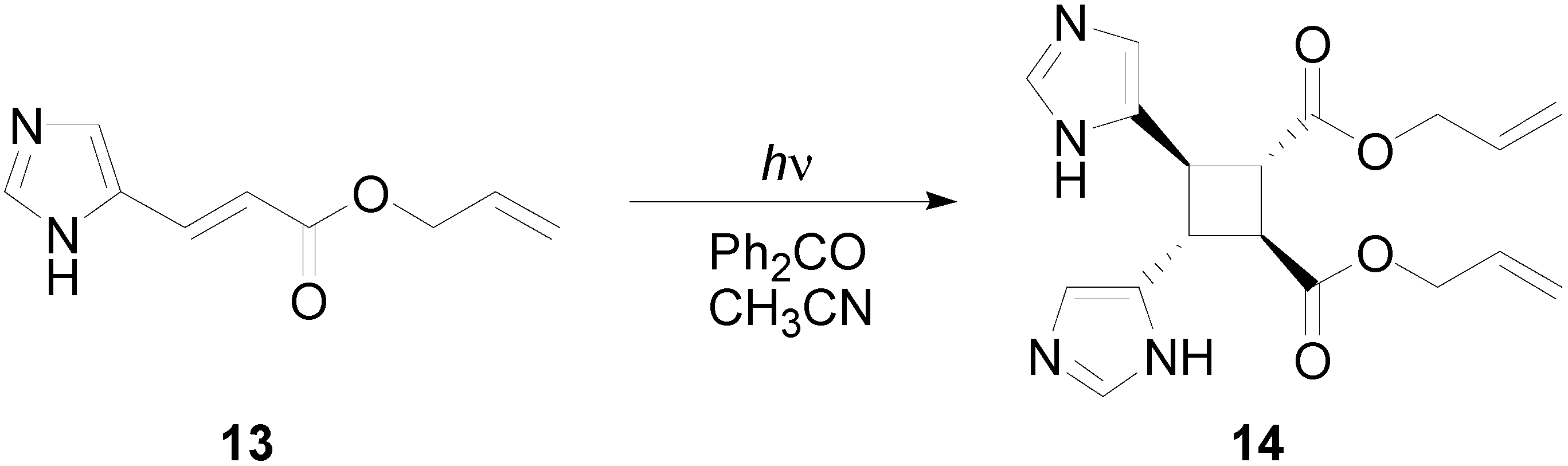
3. Results and Discussion





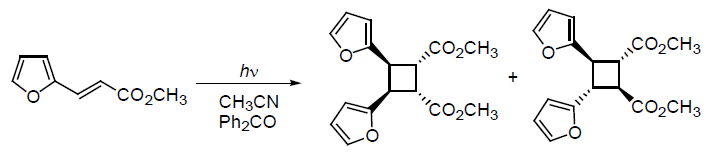{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
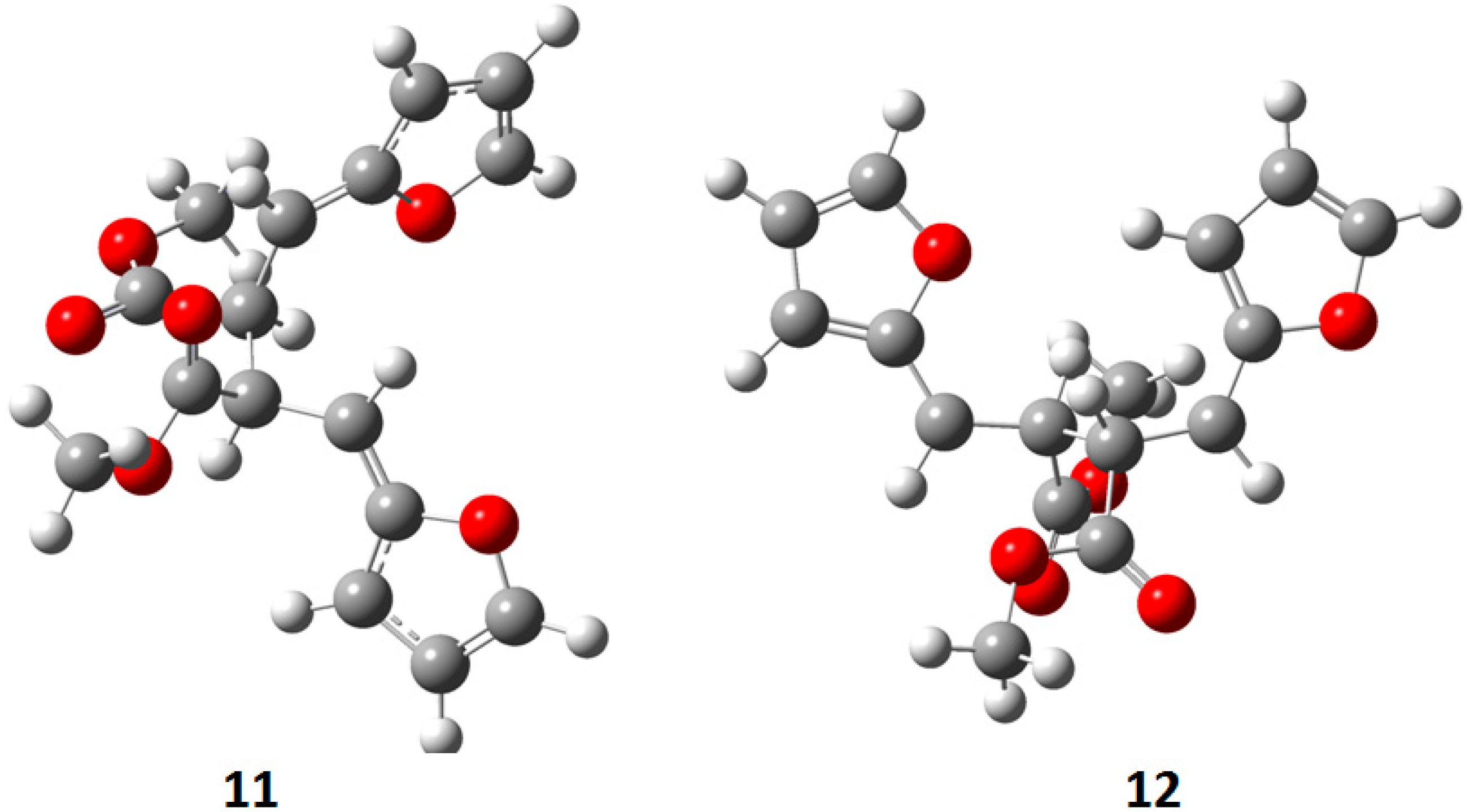{kind=link}
{kind=link}
{kind=link}
{kind=link}
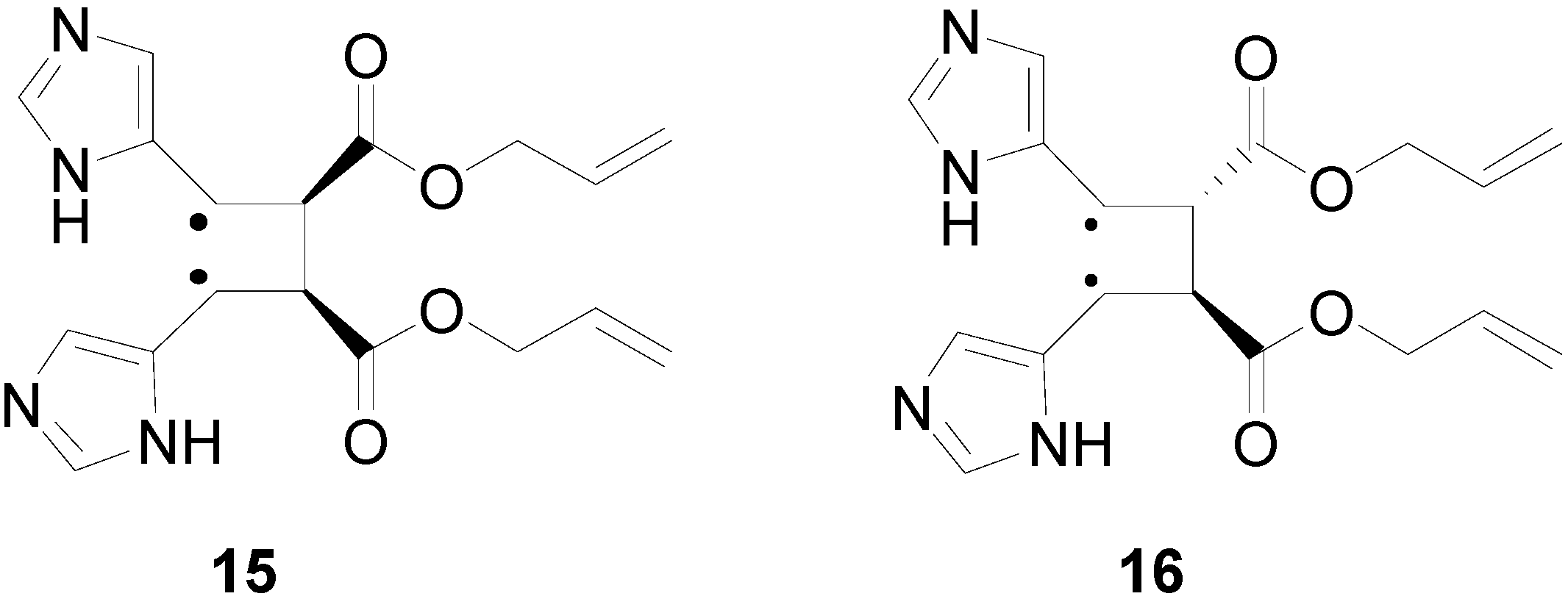
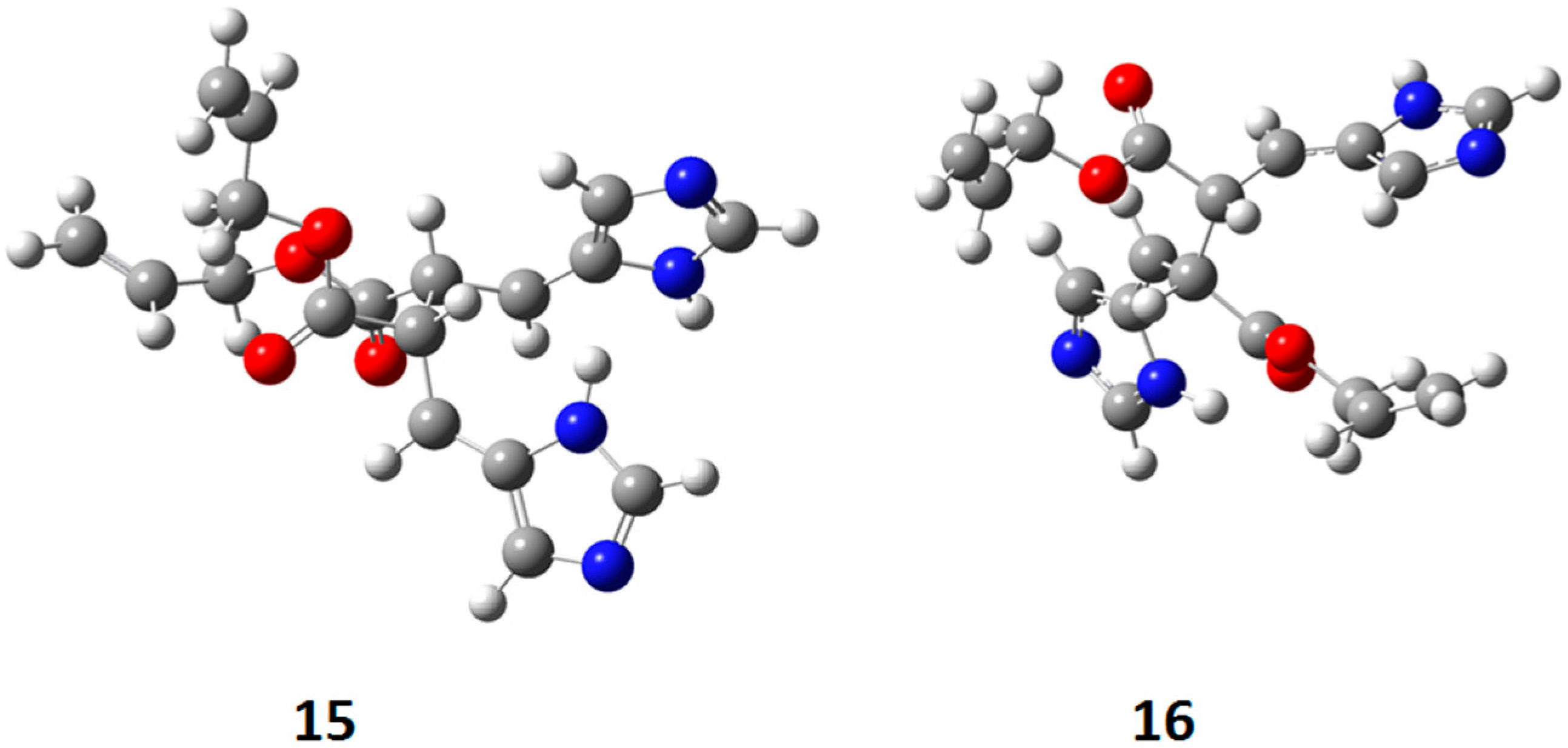
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Atom | S0 | T1 |
|---|---|---|
| 1 | −0.22321 | −0.21131 |
| 2 | 0.16272 | 0.11673 |
| 3 | 0.30938 | 0.31608 |
| 4 | −0.02587 | 0.04881 |
| 5 | −0.29943 | −0.25936 |
| 6 | 0.12484 | 0.07397 |
| 7 | 0.29891 | 0.36801 |
| 8 | 0.02106 | 0.03188 |
| 9 | −0.17997 | −0.18654 |
| 10 | −0.03305 | −0.12168 |
| 11 | −0.01552 | 0.04035 |



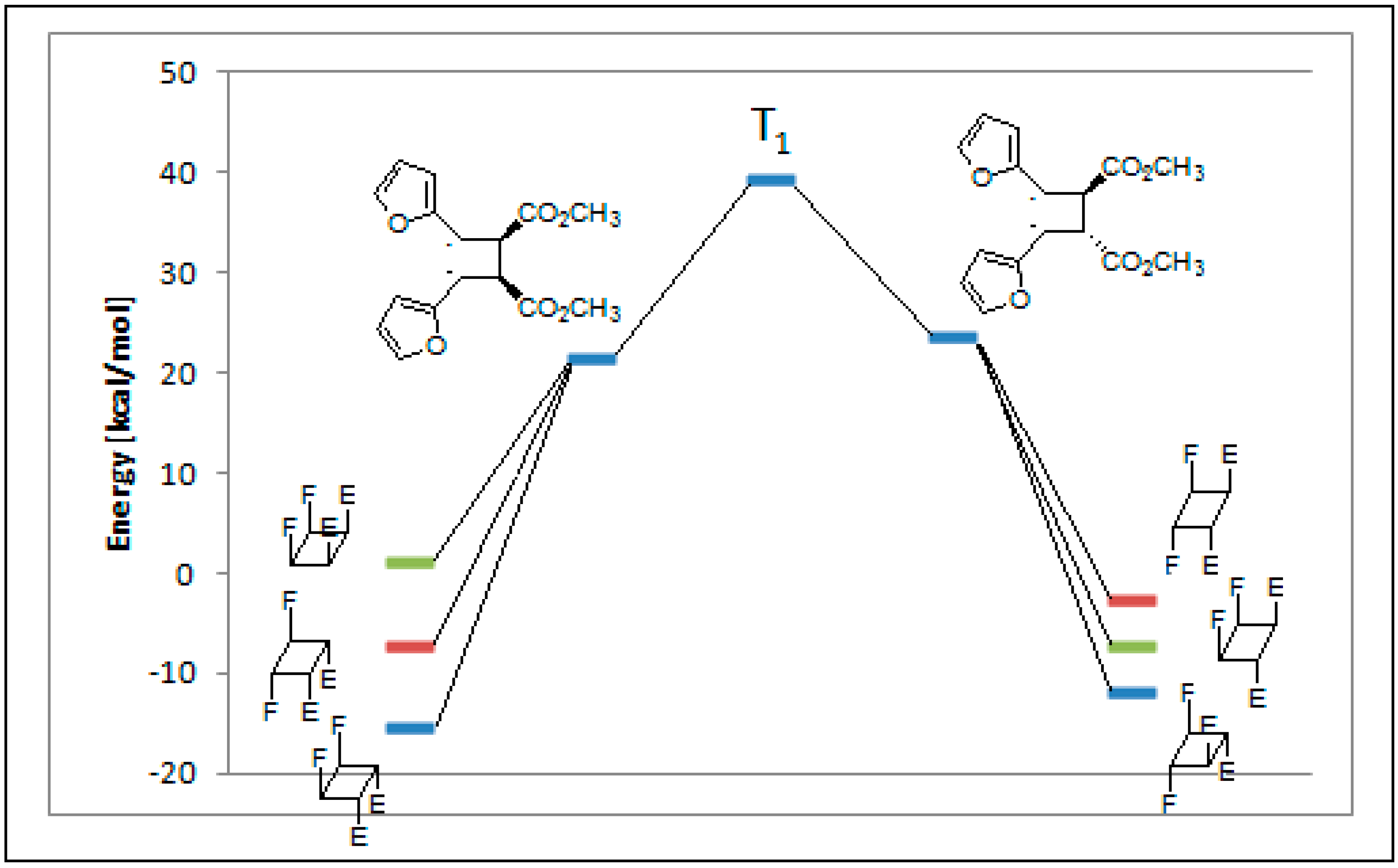
| Entry | Cyclobutane | Relative Energy (kcal·mol−1) | H [kcal·mol−1] | G [kcal·mol−1] | ||
|---|---|---|---|---|---|---|
| a | b | c | aug-cc-pVDZ | |||
| 1 |  | 0 | 0.00 | 5.62 | 5.42 | 5.84 |
| 2 |  | 1010 | 3.73 | 0.00 | 0.00 | 0.00 |
| 3 |  | 34 | 12.87 | 8.94 | 8.62 | 9.42 |
| 4 |  | 214 | 8.34 | 3.63 | 3.90 | 4.69 |
| 5 |  | 21 | 8.26 | 4.51 | 4.37 | 5.09 |
| 6 |  | 247 | 16.69 | 12.55 | 12.30 | 14.05 |


| Atom | S0 | T1 |
|---|---|---|
| 1 | 0.33944 | 0.15839 |
| 2 | −0.06852 | −0.08282 |
| 3 | −0.25121 | −0.17719 |
| 4 | −0.15484 | 0.01599 |
| 5 | 0.28130 | 0.16271 |
| 6 | −0.11141 | −0.08653 |
| 7 | −0.20234 | −0.19414 |
| 8 | −0.00484 | 0.00584 |
| 9 | 0.04775 | 0.11575 |
| 10 | −0.01751 | −0.08307 |
| 11 | 0.00554 | 0.13532 |
| 12 | 0.10280 | 0.08023 |
| 13 | 0.00708 | 0.14128 |



| Entry | Cyclobutane | Relative Energy (kcal·mol−1) | |
|---|---|---|---|
| 6-31G+(d,p) | Solvent | ||
| 1 |  | 4.48 | 3.89 |
| 2 |  | 1.30 | 0.00 |
| 3 |  | 2.58 | 1.63 |
| 4 |  | 0.00 | 0.25 |
| 5 |  | 1.48 | 2.43 |
| 6 |  | 4.21 | 4.59 |



4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ciamician, G.; Silber, P. Chemische Lichtwirkungen. Ber. Dtsch. Chem. Ges. 1902, 35, 1992–2000. [Google Scholar]
- Ciamician, G.; Silber, P. Chemische Lichtwirkungen. Ber. Dtsch. Chem. Ges. 1903, 36, 1575–1583. [Google Scholar]
- Bassani, D.M. The dimerization of cinnamic acid derivatives. In CRC Handbook of Organic Photochemistry and Photobiology; Horspool, W., Lenci, F., Eds.; CRC Press: Boca Raton, FL, USA, 2004; Volume 2, pp. 20.1–20.20. [Google Scholar]
- Stobbe, H. Lichtreaktionen der Allo- und Isozimtsäuren. Ber. Dtsch. Chem. Ges. 1919, 52, 666–672. [Google Scholar]
- Stobbe, H.; Bremer, A. Zur Photochemie der Zimtsäuren, der Chalkone und ihrer Derivate. (II. Mitteilung Über Truxill- und Truxinketone). Mit 4 Figuren. J. Prakt. Chem. 1929, 123, 1–60. [Google Scholar]
- Cohen, M.D.; Schmidt, G.M.J.; Sonntag, F.I. Topochemistry. Part II. The photochemistry of trans-cinnamic acids. J. Chem. Soc. 1964, 2000–2013. [Google Scholar]
- Schmidt, G.M.J. Topochemistry. Part III. The crystal chemistry of some trans-cinnamic acids. J. Chem. Soc. 1964, 2014–2021. [Google Scholar]
- Bregman, J.; Osaki, K.; Schmidt, G.M.J.; Sonntag, F.I. Topochemistry. Part IV. The crystal chemistry of some cis-cinnamic acids. J. Chem. Soc. 1964, 2021–2030. [Google Scholar]
- Schmidt, G.M.J. Photodimerization in the solid state. Pure Appl. Chem. 1971, 27, 647–678. [Google Scholar] [CrossRef]
- Nakanishi, F.; Nakanishi, H.; Tsuchiya, M.; Hasegawa, M. Water-Participation in the Crystalline-State Photodimerization of Cinnamic Acid Derivatives. A New Type of Organic Photoreaction. Bull. Chem. Soc. Jpn. 1976, 49, 3096–3099. [Google Scholar] [CrossRef]
- Khan, M.; Brunklaus, G.; Enkelmann, V.; Spiess, H.-W. Transient states in [2+2] photodimerization of cinnamic acid: Correlation of solid-state NMR and X-ray analysis. J. Am. Chem. Soc. 2008, 130, 1741–1748. [Google Scholar] [CrossRef]
- Lahav, M.; Schmidt, G.M.J. Topochemistry. Part XVIII. The solid-state photochemistry of some heterocyclic analogues of trans-cinnamic acid. J. Chem. Soc. B 1967, 239–243. [Google Scholar]
- Norval, M.; Simpson, T.J.; Bardshiri, E.; Howiè, S.E.M. Urocanic acid analogues and the suppression of the delayed type hypersensitivity response to Herpes simplex virus. Photochem. Photobiol. 1989, 49, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Egerton, P.L.; Hyde, E.M.; Trigg, J.; Payne, A.; Beynon, P.; Mijovic, M.V.; Reiser, A. Photocycloaddition in liquid ethyl cinnamate and in ethyl cinnamate glasses. The photoreaction as a probe into the micromorphology of the solid. J. Am. Chem. Soc. 1981, 103, 3859–3863. [Google Scholar]
- Bolt, J.; Quina, F.H.; Whitten, D.G. Solid state photodimerization of surfactant esters of cinnamic acid. Tetrahedron Lett. 1976, 17, 2595–2598. [Google Scholar] [CrossRef]
- Amarouche, H.; de Bourayne, C.; Riviere, M.; Lattes, A. Réactivité chimique et photochimique dans les milieux micellaires at dan les microémulsions. VIII. Photoréactivité des dérivés cinnamiques dans les microémulsions. C. R. Acad. Sci. Paris 1984, 298, (Serie II). 121–123. [Google Scholar]
- Curme, H.C.; Natale, C.C.; Kelley, D.J. Photosensitized reactions of cinnamate esters. J. Phys. Chem. 1967, 71, 767–770. [Google Scholar] [CrossRef]
- Lewis, F.D.; Oxman, J.D. Photodimerization of Lewis acid complexes of cinnamate esters in solution and the solid state. J. Am. Chem. Soc. 1984, 106, 466–468. [Google Scholar] [CrossRef]
- Lewis, F.D.; Quillen, S.L.; Hale, P.D.; Oxman, J.D. Lewis acid catalysis of photochemical reactions. 7. Photodimerization and cross-cycloaddition of cinnamic esters. J. Am. Chem. Soc. 1988, 110, 1261–1267. [Google Scholar] [CrossRef]
- Botta, B.; Iacomacci, P.; Vinciguerra, V.; Delle Monache, G.; Gacs-Baitz, E.; Botta, M.; Misiti, D. Non-oxidative dimerization of 3,4-dioxygenated cinnamates to aryltetralin lignans. Chem. Pharm. Bull. 1990, 38, 3238–3241. [Google Scholar] [CrossRef]
- Hirayama, F.; Utsuki, T.; Uekama, K. Stoichiometry-dependent photodimerization of tranilast in a γ-cyclodextrin inclusion complex. J. Chem. Soc. Chem. Commun. 1991, 887–888. [Google Scholar]
- D’Auria, M.; Vantaggi, A. Photochemical dimerization of methoxy substituted cinnamic acid methyl esters. Tetrahedron 1992, 48, 2523–2528. [Google Scholar] [CrossRef]
- Pattabimaran, M.; Natarajan, A.; Kaanumalle, L.S.; Ramamurthy, V. Templating photodimerization of trans-cinnamic acids with cucurbit[8]uril and γ-cyclodextrin. Org. Lett. 2005, 7, 529–532. [Google Scholar] [PubMed]
- Pattabiraman, M.; Kaanumalle, L.S.; Natarajan, A.; Ramamurthy, V. Regioselective photodimerization of cinnamic acid in water: templation with cucurbituriis. Langmuir 2006, 22, 7605–7609. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, S.; Ramamurthy, V. Templating photodimerization of trans-cinnamic acid esters with a water-soluble Pd nanocage. J. Org. Chem. 2007, 72, 452–458. [Google Scholar] [PubMed]
- Chowdhury, M.; Kariuki, B.M. Supramolecular assembly in cinnamate structures: The influence of the ammonium ion and halogen interaction. Cryst. Growth Des. 2006, 6, 774–780. [Google Scholar] [CrossRef]
- Novak, M.; Salemink, C.A.; Khan, I. Biological activity of the alkaloids of Erythroxylum coca and Erythroxylum novogranatense. J. Ethnopharmacol. 1984, 10, 261–274. [Google Scholar] [CrossRef]
- Liebermann, C. Ueber ein Nebenalkaloïn des Cocaïns, das Isatropylcocaïn. Chem. Ber. 1888, 21, 2342–2355. [Google Scholar] [CrossRef]
- Ford, C.W.; Hartley, R.D. GC/MS characterisation of cyclodimers from p-coumaric and ferulic acids by photodimerisation—A possible factor influencing cell wall biodegradability. J. Sci. Food Agric. 1989, 46, 301–310. [Google Scholar] [CrossRef]
- Hartley, R.D.; Whatley, E.R.; Harris, P.J. 4,4'-Dihydroxytruxillic acid as a component of cell walls of Lolium multiflorum. Phytochemistry 1988, 27, 349–350. [Google Scholar]
- D’Auria, M.; Piancatelli, G.; Vantaggi, A. Photochemical dimerization of methyl 2-furyl- and 2-thienylacrylate and related compounds in solution. J. Chem. Soc. Perkin Trans. 1 1990, 2999–3002. [Google Scholar]
- D’Auria, M. Regio- and stereochemical control in the photodimerization of methyl 3-(2-furyl)acrylate. Heterocycles 1996, 43, 959–968. [Google Scholar] [CrossRef]
- D’Auria, M.; Racioppi, R. Photochemical dimerization of esters of urocanic acid. J. Photochem. Photobiol. A 1998, 112, 145–148. [Google Scholar] [CrossRef]
- D’Auria, M.; Racioppi, R. Photochemical dimerization in solution of arylacrylonitrile derivatives. Tetrahedron 1997, 53, 17307–17316. [Google Scholar] [CrossRef]
- D’Auria, M.; Emanuele, L.; Esposito, V.; Racioppi, R. The Photochemical dimerization of 3-heteroaryl-acrylates. Arkivoc 2002, 11, 65–78. [Google Scholar] [CrossRef]
- González-Ramírez, I.; Roca-Sanjuán, D.; Climent, T.; Serrano-Pérez, J.J.; Merchán, M.; Serrano-Andrés, L. On the photoproductionof DNA/RNA cyclobutane pyrimidine dimers. Theor. Chem. Acc. 2011, 128, 705–711. [Google Scholar]
- Climent, T.; González-Ramírez, I.; González-Luque, R.; Merchán, M.; Serrano-Andrés, L. Cyclobutane pyrimidine photodimerization of DNA/RNA nucleobases in the triplet state. J. Phys. Chem. Lett. 2010, 1, 2072–2076. [Google Scholar] [CrossRef]
- Roca-Sajuán, D.; Olaso-González-Ramírez, I.; Serrano-Andrés, L.; Merchán, M. Molecular basis of DNA photodimerization: Intrinsic production of cyclobutane cytosine dimers. J. Am. Chem. Soc. 2008, 130, 10768–10779. [Google Scholar] [CrossRef] [PubMed]
- Ko, K.C.; Park, Y.G.; Cho, D.; Lee, J.Y. Simple but useful scheme toward understanding of intramolecular magnetic interactions: Benzene-bridged oxoverdazyl diradicals. J. Phys. Chem. A 2014, 118, 9596–9606. [Google Scholar] [CrossRef] [PubMed]
- Gromov, O.I.; Golubeva, E.N.; Khrustalev, V.N.; Kalai, T.; Hideg, T.; Kokorin, A.I. EPR, X-ray structure and DFT calculations of the nitroxide biradical with one acetylene group in the bridge. Appl. Magn. Reson. 2014, 45, 981–992. [Google Scholar] [CrossRef]
- Sumanovac Ramlja, T.; Sohora, M.; Antol, I.; Kontrec, D.; Basaric, N.; Mlinaric-Majerski, K. Memory of chirality in the phthalilide photocyclization of adamantane dipeptides. Tetrahedron Lett. 2014, 55, 4078–4081. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Casida, M.E. Time-dependent density-functional response theory for molecules. In Recent Advances in Density Functional Methods; Chong, D.P., Ed.; World Scientific: Singapore, Singapore, 1995; Volume 1, pp. 155–192. [Google Scholar]
- Casida, M.E.; Jamorski, C.; Casida, K.C.; Salahub, D.R. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys. 1998, 108, 4439–4449. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- D’Auria, M.; D’Annibale, A.; Ferri, T. Photochemical behaviour of furylidene carbonyl compounds. Tetrahedron 1992, 48, 9323–9336. [Google Scholar] [CrossRef]
- Wiberg, K.B. Basis set effects on calculated geometries: 6-311++G** vs. aug-cc-pVDZ. J. Comput. Chem. 2004, 25, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- D’Auria, M.; Emanuele, L.; Racioppi, R. Regio- and stereoselectivity in the Paternò-Büchi reaction between 2,3-dihydrofuran and furan with benzaldehyde. Lett. Org. Chem. 2006, 3, 244–246. [Google Scholar] [CrossRef]
- D’Auria, M.; Racioppi, R. A DFT study of 1,4-biradical intermediates involved in stereoselective Paternò-Büchi reactions. Eur. J. Org. Chem. 2010, 2010, 3831–3836. [Google Scholar] [CrossRef]
- D’Auria, M. Regio- and stereochemistry of the [2+2]-cycloaddition reaction between enones and alkenes. A DFT study. Tetrahedron 2012, 68, 8699–8703. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 9, 10, and 14 are not available from the authors, others are available form authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D'Auria, M. A DFT Study of the Photochemical Dimerization of Methyl 3-(2-Furyl)acrylate and Allyl Urocanate. Molecules 2014, 19, 20482-20497. https://doi.org/10.3390/molecules191220482
D'Auria M. A DFT Study of the Photochemical Dimerization of Methyl 3-(2-Furyl)acrylate and Allyl Urocanate. Molecules. 2014; 19(12):20482-20497. https://doi.org/10.3390/molecules191220482
Chicago/Turabian StyleD'Auria, Maurizio. 2014. "A DFT Study of the Photochemical Dimerization of Methyl 3-(2-Furyl)acrylate and Allyl Urocanate" Molecules 19, no. 12: 20482-20497. https://doi.org/10.3390/molecules191220482
APA StyleD'Auria, M. (2014). A DFT Study of the Photochemical Dimerization of Methyl 3-(2-Furyl)acrylate and Allyl Urocanate. Molecules, 19(12), 20482-20497. https://doi.org/10.3390/molecules191220482