Pharmacokinetics, Tissue Distribution and Excretion of Verticinone from F. hupehensis in Rats

Abstract
:1. Introduction

2. Results and Discussion
2.1. Pharmacokinetic Study


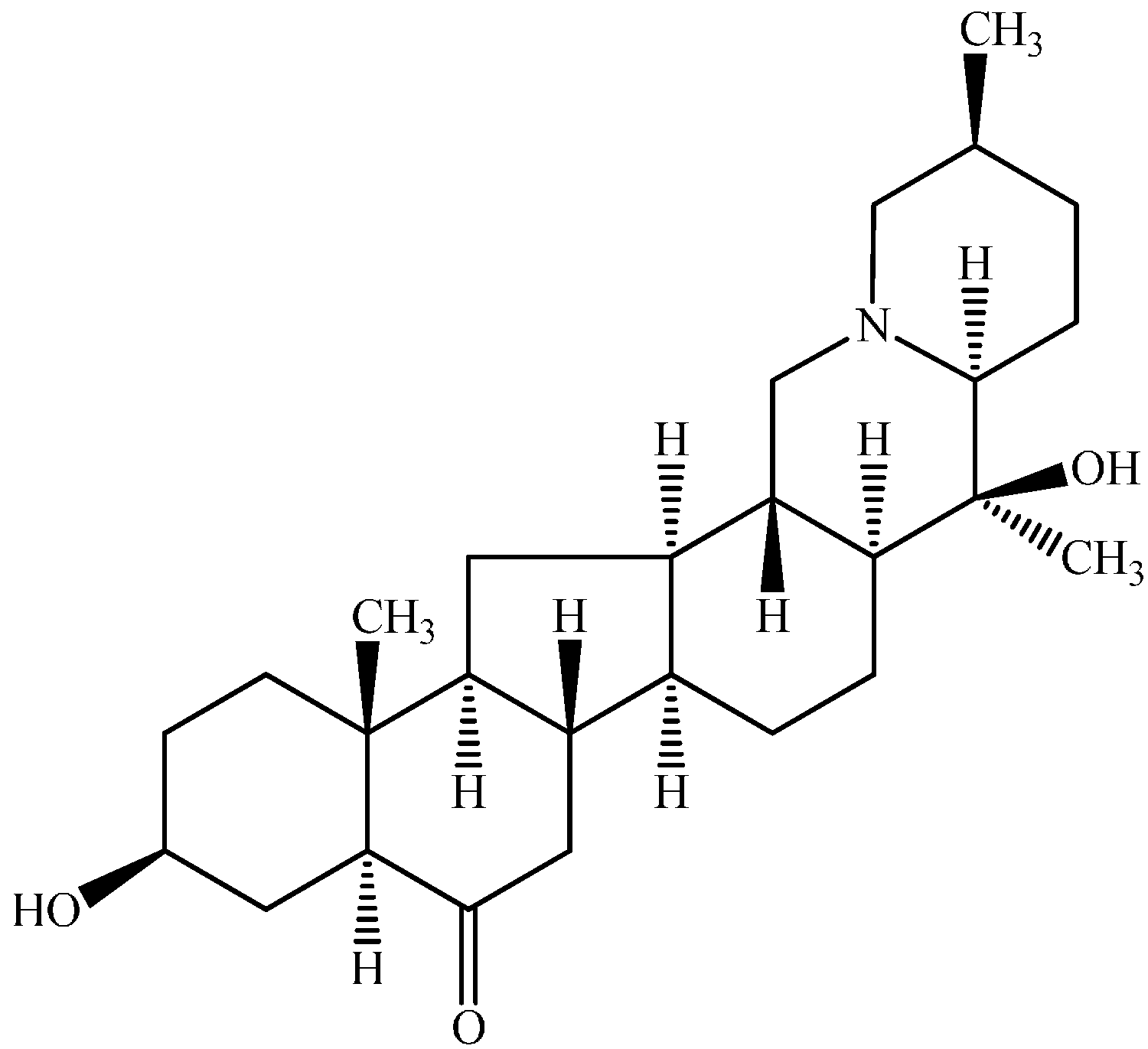{kind=link}
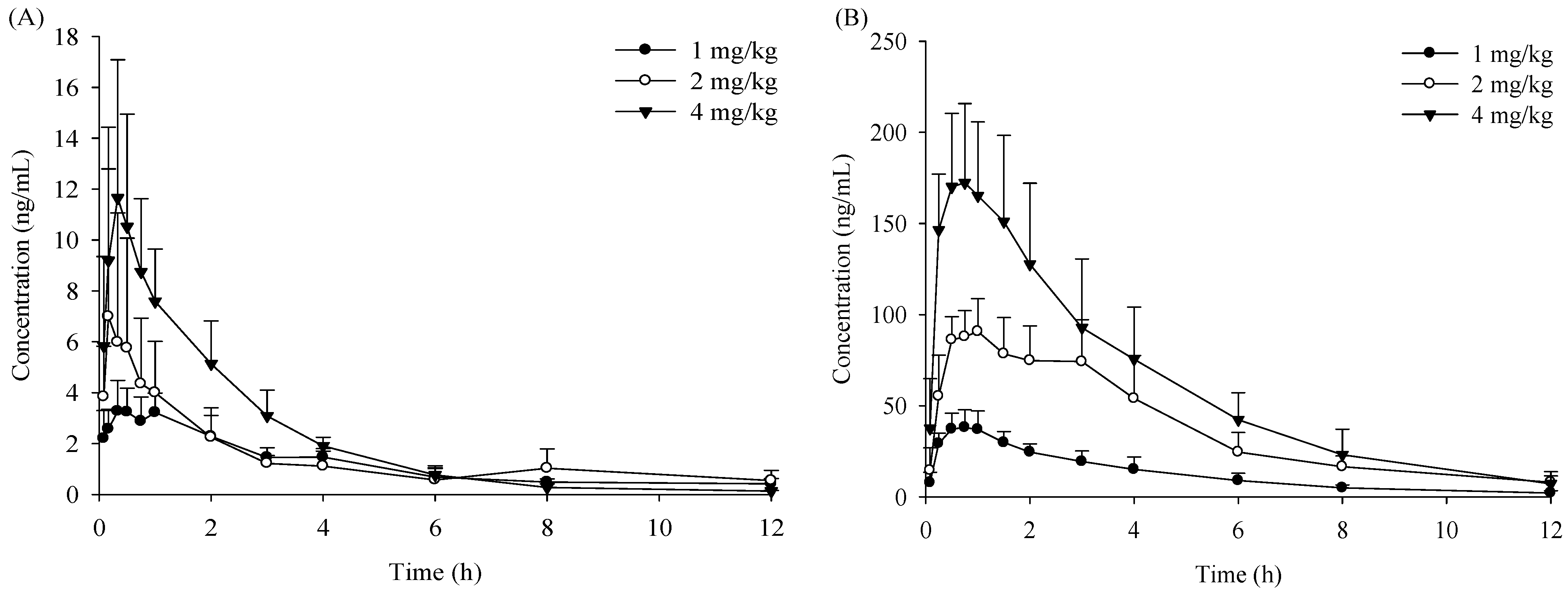{kind=link}
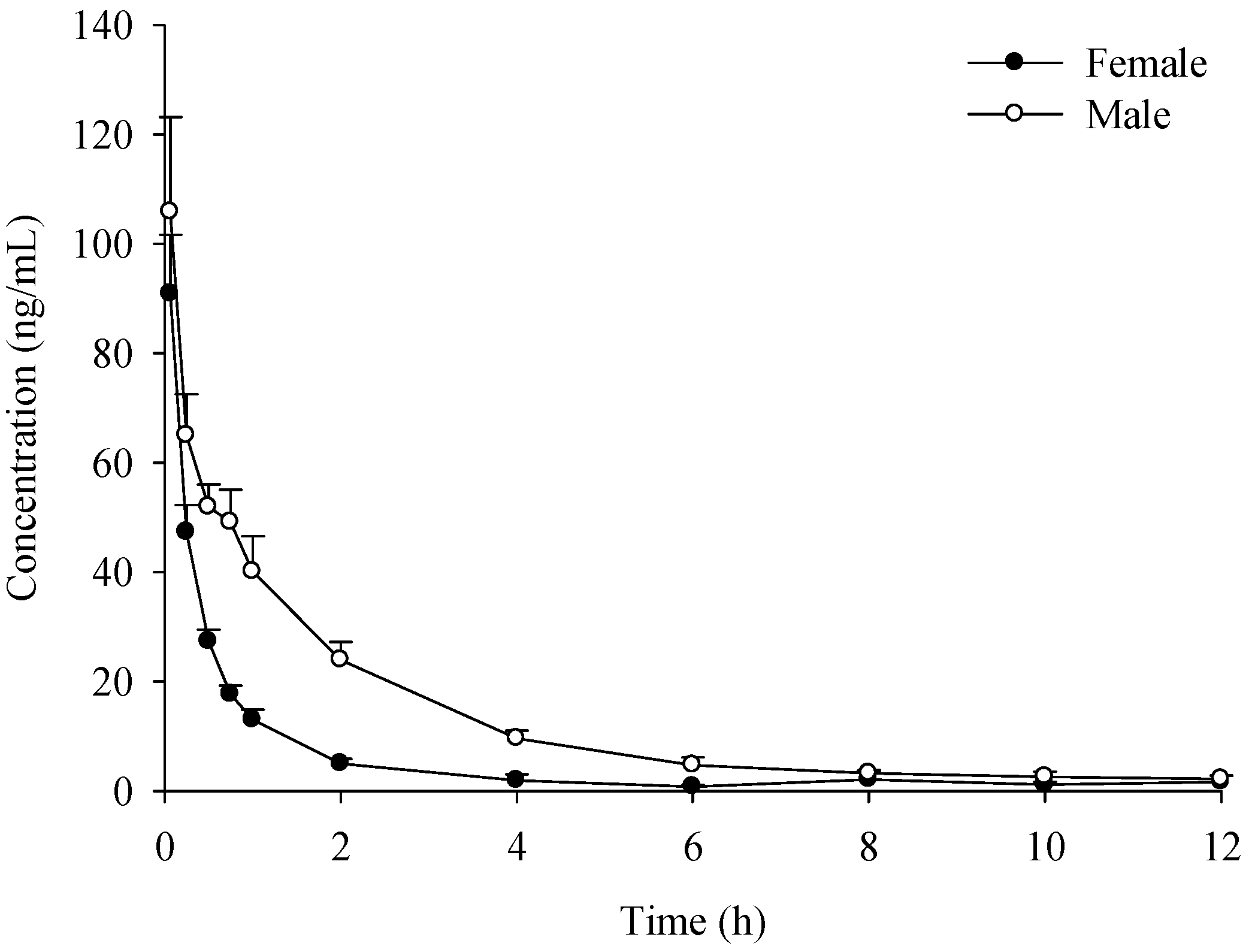{kind=link}
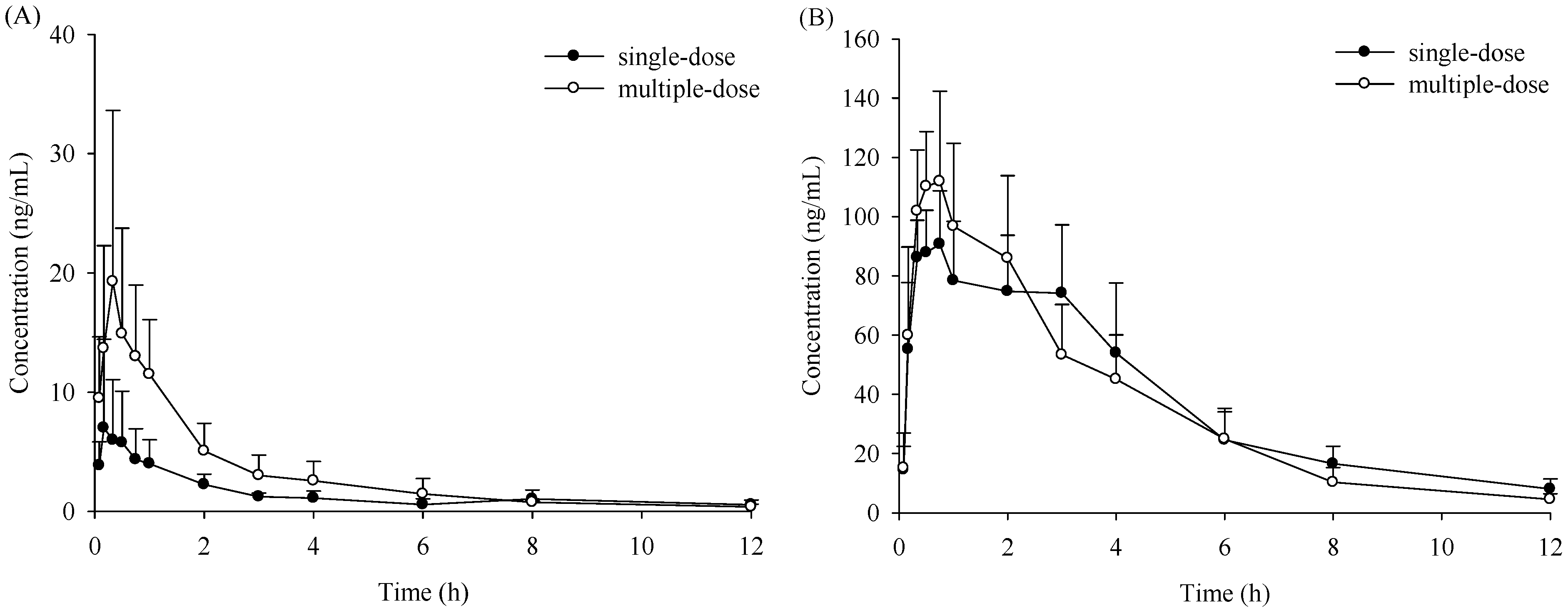{kind=link}
{kind=link}
{kind=link}
| Para-Meters | Unit | i.g. | i.v. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 mg/kg (Mean ± S.D.) | 2 mg/kg (Mean ± S.D.) | 4 mg/kg (Mean ± S.D.) | 0.4 mg/kg (Mean ± S.D.) | ||||||
| ♀ | ♂ | ♀ | ♂ | ♀ | ♂ | ♀ | ♂ | ||
| Cmax | μg/L | 3.67 ± 1.13 | 38.9 ± 9.34 ** | 7.45 ± 7.10 | 94.8 ± 17.1 ** | 12.6 ± 5.14 | 176 ± 45.2 ** | - | - |
| Tmax | h | 0.67 ± 0.31 | 0.70 ± 0.11 | 0.40 ± 0.36 | 1.45 ± 1.42 | 0.40 ± 0.35 | 0.70 ± 0.11 | - | - |
| AUC0–t | μg·h/L | 14.5 ± 3.27 | 159 ± 50.1 ** | 17.3 ± 5.40 | 446 ± 111 ** | 26.6 ± 5.84 | 737 ± 236 ** | 65.6 ± 7.03 | 159.5 ± 20.2 ** |
| AUC0–∞ | μg·h/L | 15.3 ± 3.06 | 167 ± 52.5 ** | 18.8 ± 5.01 | 475 ± 119 ** | 27.1 ± 5.78 | 769 ± 257 ** | 98.9 ± 35.8 | 167.6 ± 23.0 ** |
| MRT0–t | h | 3.59 ± 0.18 | 3.21 ± 0.34 | 3.42 ± 1.22 | 3.62 ± 0.37 | 2.04 ± 0.21 | 3.19 ± 0.48 | 2.04 ± 0.34 | 2.37 ± 0.14 |
| t1/2z | h | 2.86 ± 1.29 | 2.78 ± 0.85 | 4.05 ± 2.48 | 3.05 ± 0.78 | 1.84 ± 0.58 | 2.33 ± 0.86 | - | - |
| CLz/F | L·kg/h | 67.0 ± 11.6 | 6.53 ± 2.35 ** | 113 ± 33.5 | 4.40 ± 0.98 ** | 152 ± 28.2 | 5.59 ± 1.48 ** | 4.47 ± 1.48 | 2.43 ± 0.37 * |
| Vz/F | L/kg | 280 ± 160 | 25.5 ± 9.07 * | 649 ± 419 | 19.4 ± 6.70 ** | 387 ± 74.2 | 18.6 ± 9.14 ** | 82.5 ± 70.3 | 10.9 ± 1.56 |

| Paramters | Unit | ♀ (Mean ± S.D.) | ♂ (Mean ± S.D.) | ||
|---|---|---|---|---|---|
| Single i.g. | Successive i.g. | Single i.g. | Successive i.g. | ||
| Cmax | μg/L | 7.45 ± 7.10 | 19.9 ± 14.0 | 94.8 ± 17.1 | 119 ± 26.8 b |
| Tmax | h | 0.40 ± 0.36 | 0.38 ± 0.22 | 1.45 ± 1.42 | 0.80 ± 0.21 b |
| AUC0–t | μg·h/L | 17.3 ± 5.40 | 36.1 ± 15.2 a | 446 ± 111 | 439 ± 126 b |
| AUC0–∞ | μg·h/L | 18.8 ± 5.01 | 43.4 ± 12.0 a | 475 ± 119 | 455 ± 125 b |
| MRT0–t | h | 3.42 ± 1.22 | 2.24 ± 0.28 | 3.62 ± 0.37 | 2.98 ± 0.30 b |
| t1/2z | h | 4.05 ± 2.48 | 5.94 ± 4.84 | 3.05 ± 0.78 | 2.08 ± 0.51 |
| CLz/F | L·kg/h | 113 ± 33.5 | 49.0 ± 13.1 | 4.40 ± 0.98 | 4.71 ± 1.47 b |
| Vz/F | L/kg | 649 ± 419 | 485 ± 499 | 19.4 ± 6.70 | 13.8 ± 4.07 |

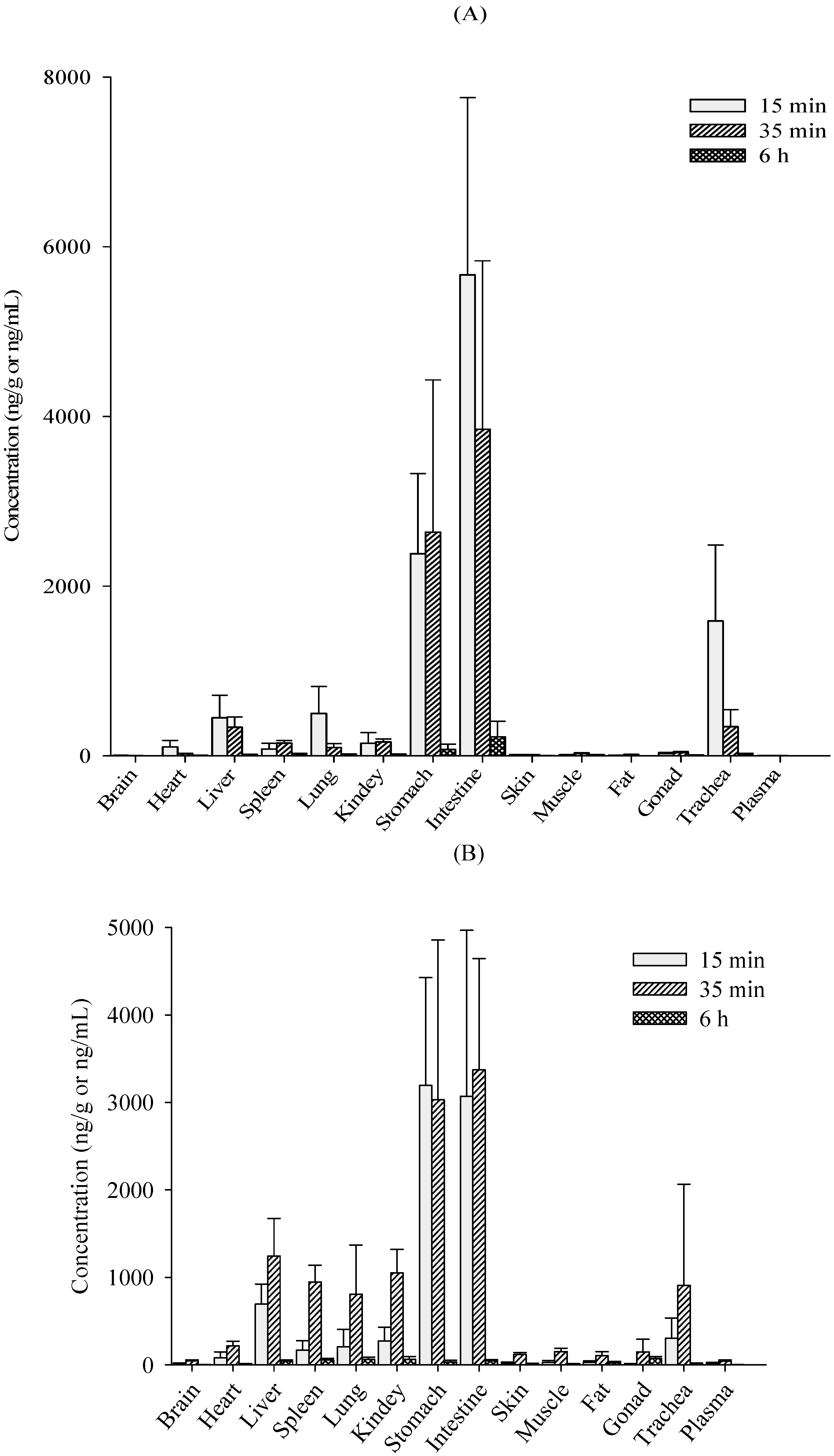
2.2. Tissue Distribution Study
2.3. Plasma Protein Binding Study
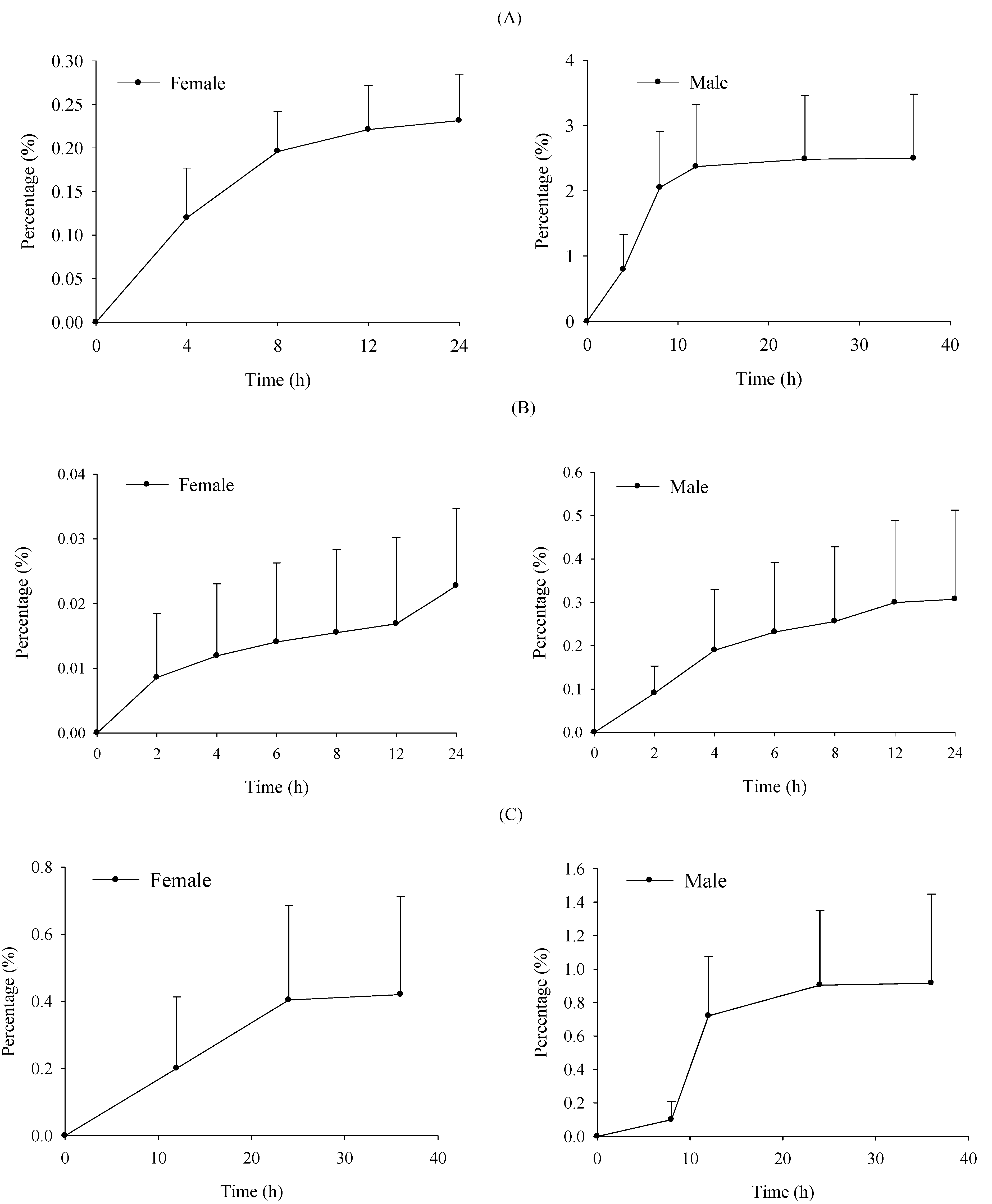
2.4. Excretion Study

| 5 ng/mL | 30 ng/mL | 100 ng/mL | ||
|---|---|---|---|---|
| Protein binding rates (%) | rats | 91.3 ± 2.7 | 91.0 ± 1.3 | 89.8 ± 1.1 |
| humans | 92.6 ± 1.0 | 89.5 ± 0.8 | 94.6 ± 0.5 |

2.5. Discussion
3. Experimental
3.1. Chemicals and Reagents
3.2. Animals
3.3. Single- and Multiple-Dose Plasma Pharmacokinetics
3.4. Tissue Distribution Study
3.5. Plasma Protein Binding Study
3.6. Excretion Study
3.7. Quantitative Analysis
3.8. Pharmacokinetic and Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yao, L.N.; Sun, H.; Jiang, Z.; Qi, Y.D.; Lu, C.B. The effect of F. Ebeiensis, F. Ebeiensis Var. Purpura and F. Hupehensis Hsinoes K.C. Hsia on respiratory function. J. Tongji Med. Univ. 1993, 22, 47–49. [Google Scholar]
- Li, P.; Ji, H.; Xu, G.J.; Xu, L.S. Studies on the antitussive and expectorant effects of Chinese drug beimu. J. China Pharm. Univ. 1993, 24, 360–362. [Google Scholar]
- Xiao, P.G. A Review on the Study of Hubeibeimu. China J. Chin. Mater. Medica 2002, 27, 726–728. [Google Scholar]
- Zhang, Y.H.; Ruan, H.L.; Zeng, F.B.; Pi, H.F.; Zhao, W.; Wu, J.Z. Effective part screening on antitussive, expectorant and antiasthmatic activities of Fritillaria hupehensis. Chin. Tradit. Herb. Drugs 2003, 34, 1016–1018. [Google Scholar]
- Qian, B.C.; Xu, H.J. Studies on the antitussive and sedative activities of peimine and peiminine. Acta Pharm. Sin. 1985, 20, 306–308. [Google Scholar]
- Zhang, J.L.; Wang, H.; Chen, C.; Pi, H.F.; Ruan, H.L.; Zhang, P.; Wu, J.Z. Addictive evaluation of cholic acid-verticinone ester, a potential cough therapeutic agent with agonist action of opioid receptor. Acta Pharm. Sin. 2009, 30, 559–566. [Google Scholar] [CrossRef]
- Xu, F.Z.; Chen, C.; Zhou, X.Q.; Wang, L.J.; He, G.M. Study on the anagelsic effect and the physical dependence of verticinone. Jin Ri Yao Xue 2008, 18, 4–6. [Google Scholar]
- Yun, Y.G.; Jeon, B.H.; Lee, J.H.; Lee, S.K.; Lee, H.J.; Jung, K.H.; Lee, J.; Jun, C.D.; Lee, S.K.; Kim, E.C. Verticinone induces cell cycle arrest and apoptosis in immortalized and malignant human oral keratinocytes. Phytother. Res. 2008, 22, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.Z.; Chen, C.; Zhang, Y.H.; Ruan, H.L.; Pi, H.F.; Zhang, P.; Wu, J.Z. Synthesis and antitussive evaluation of verticinone-cholic acid salt, a novel and potential cough therapeutic agent. Acta Pharmacol. Sin. 2007, 28, 1591–1596. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.L.; Wang, H.; Pi, H.F.; Ruan, H.L.; Zhang, P.; Wu, J.Z. Structural analysis and antitussive evaluation of five novel esters of verticinone and bile acids. Steroids 2009, 74, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, H.; Sun, J.G.; Peng, Y.; Liang, Y.; Wang, G.J.; Wu, J.Z.; Zhang, P. Development and validation of a liquid chromatography-mass spectrometry method for the determination of verticinone in rat plasma and its application to pharmacokinetic study. J. Chromatogr. B 2010, 878, 2067–2071. [Google Scholar] [CrossRef]
- Yeh, Y.H.; Wang, D.Y.; Liau, M.Y.; Wu, M.L.; Deng, J.F.; Noguchi, T. Bile acid composition in snake bile juice and toxicity of snake bile acids to rats. Comp. Biochem. Physiol. C: Toxicol. Pharmacol. 2003, 136, 277–284. [Google Scholar] [CrossRef]
- Chen, L.H.; Zhang, H.M.; Guan, Z.Y.; Zhu, W.F.; Yi, W.J.; Guan, Y.M.; Wang, S.; Liu, H.N. Sex dependent pharmacokinetics, tissue distribution and excretion of peimine and peiminine in rats assessed by liquid chromatography–tandem mass spectrometry. J. Ethnopharmacol. 2013, 145, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Brain, D.; Tim, M. Physiological parameters in laboratory animals and humans. Pharmaceut. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef]
- Kato, R.; Yamazoe, Y. Sex-specific cytochrome P450 as a cause of sex- and species-related differences in drug toxicity. Toxicol. Lett. 1992, 64–65, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Czerniak, R. Gender-based differences in pharmacokinetics in laboratory animal models. Int. J. Toxicol. 2001, 20, 161–163. [Google Scholar] [CrossRef] [PubMed]
- Martignoni, M.; Groothuis, G.M.; de Kanter, R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2006, 2, 875–894. [Google Scholar] [CrossRef] [PubMed]
- Kedderis, G.L.; Mugford, C.A. Sex-Dependent Metabolism of Xenobiotics. Drug Metab. Rev. 1998, 30, 441–198. [Google Scholar] [CrossRef] [PubMed]
- Dunn, R.T., 2nd; Klaassen, C.D. Tissue-specific expression of rat sulfotransferase messenger RNAs. Drug Metab. Dispos. 1998, 26, 598–604. [Google Scholar] [PubMed]
- Singer, S.S.; Giera, D.; Johnson, J.; Sylvester, S. Enzymatic sulfation of steroids: I. the enzymatic basis for the sex difference in cortisol sulfation by rat liver preparations. Endocrinology 1976, 98, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, K.; Tokuma, Y.; Noda, K.; Noguchi, H. Age- and sex related changes of sulfotransferase activities in the rat. Chem. Biol. Interact. 1994, 92, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Meloche, C.A.; Sharma, V.; Swedmark, S.; Andersson, P.; Falany, C.N. Sulfation of budesonide by human cytosolic sulfotransferase, dehydroepiandrosterone-sulfotransferase (DHEA-ST). Drug Metab. Dispos. 2002, 30, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Falany, J.L.; Falany, C.N. Interactions of the human cytosolic sulfotransferases and steroid sulfatase in the metabolism of tibolone and raloxifene. J. Steroid Biochem. Mol. Biol. 2007, 107, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, C.H.J.; Vos, R.M.E.; Delbressine, L.P.C. The in vivo metabolism of tibolone in animal species. Eur. J. Drug Metab. Pharmacokinet. 2002, 27, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.Z.; Zhan, J.; Kang, P.; Yamazaki, S. Gender specific drug metabolism of PF-02341066 in rats- role of sulfoconjugation. Curr. Drug Metab. 2010, 11, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Sample of the compound verticinone is available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Sun, J.-G.; Peng, Y.; Liang, Y.; Wang, G.-J.; Chen, H.; Wu, J.-Z.; Zhang, P. Pharmacokinetics, Tissue Distribution and Excretion of Verticinone from F. hupehensis in Rats. Molecules 2014, 19, 20613-20626. https://doi.org/10.3390/molecules191220613
Wu X, Sun J-G, Peng Y, Liang Y, Wang G-J, Chen H, Wu J-Z, Zhang P. Pharmacokinetics, Tissue Distribution and Excretion of Verticinone from F. hupehensis in Rats. Molecules. 2014; 19(12):20613-20626. https://doi.org/10.3390/molecules191220613
Chicago/Turabian StyleWu, Xiao, Jian-Guo Sun, Ying Peng, Yan Liang, Guang-Ji Wang, Hui Chen, Ji-Zhou Wu, and Peng Zhang. 2014. "Pharmacokinetics, Tissue Distribution and Excretion of Verticinone from F. hupehensis in Rats" Molecules 19, no. 12: 20613-20626. https://doi.org/10.3390/molecules191220613
APA StyleWu, X., Sun, J. -G., Peng, Y., Liang, Y., Wang, G. -J., Chen, H., Wu, J. -Z., & Zhang, P. (2014). Pharmacokinetics, Tissue Distribution and Excretion of Verticinone from F. hupehensis in Rats. Molecules, 19(12), 20613-20626. https://doi.org/10.3390/molecules191220613