Inhibitory Activity of Synthesized Acetylated Procyanidin B1 Analogs against HeLa S3 Cells Proliferation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
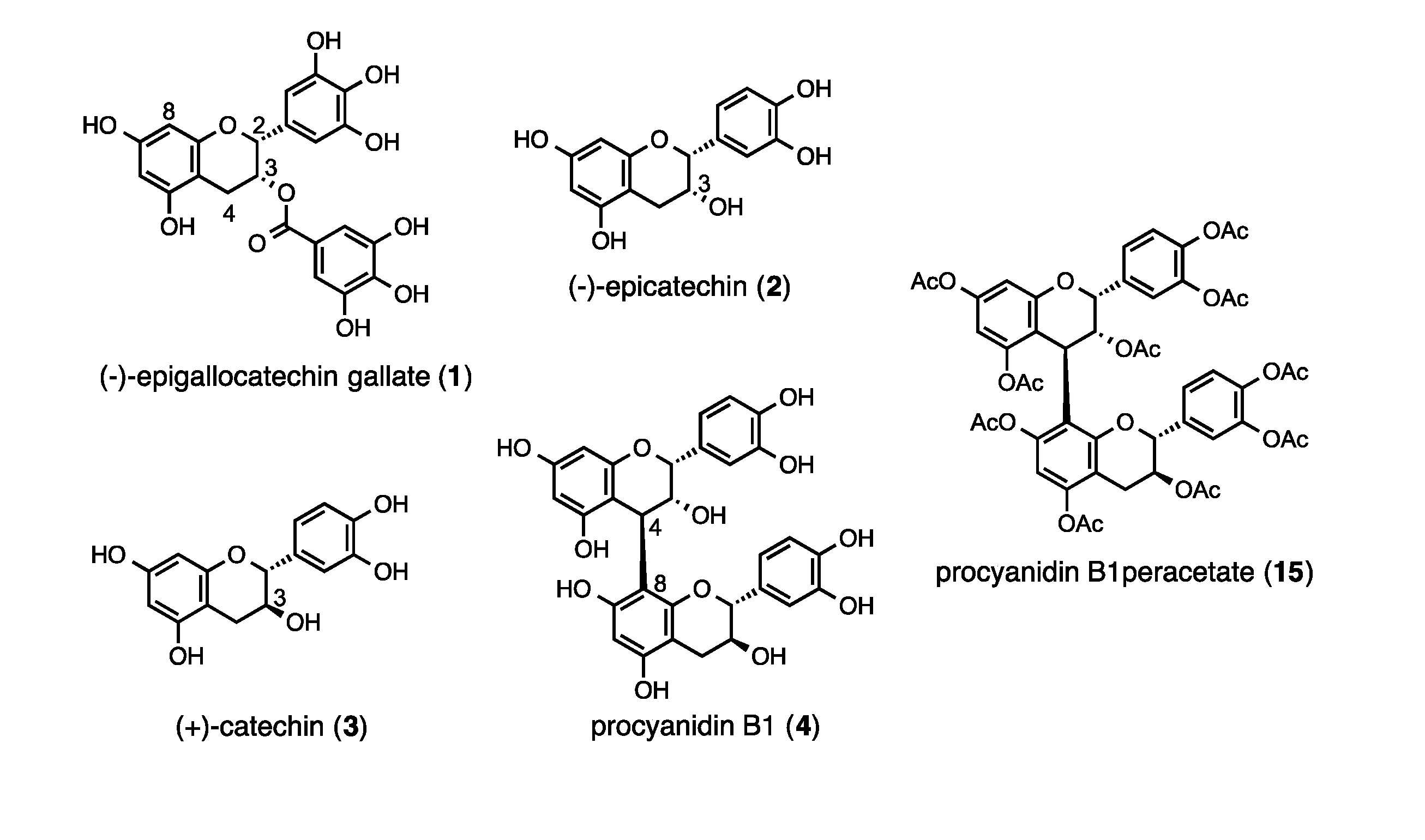
:1. Introduction

2. Results and Discussion




3. Experimental
3.1. General
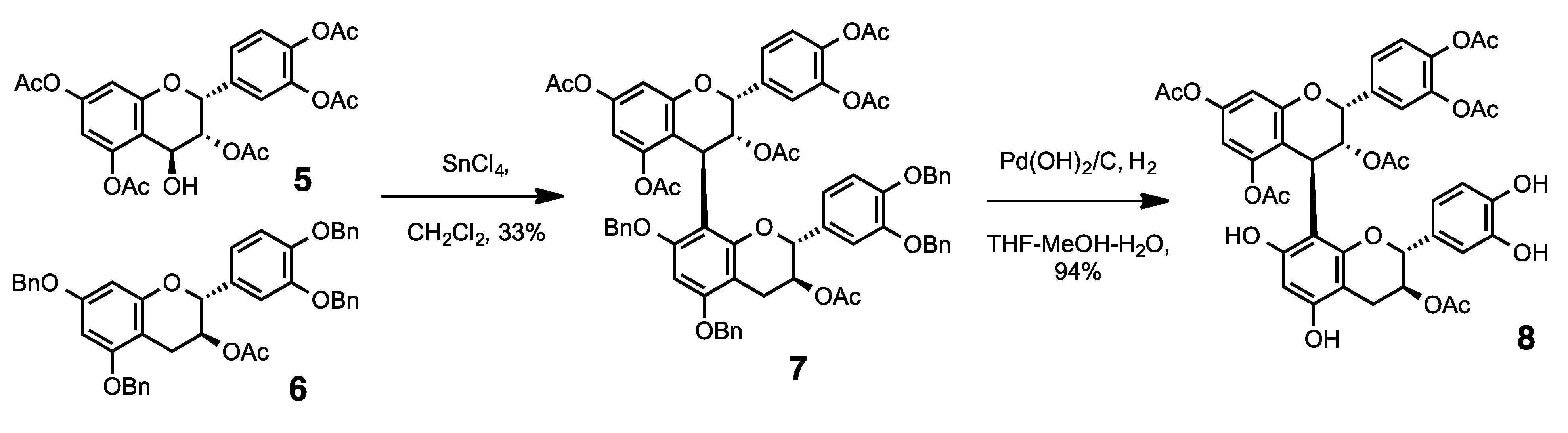
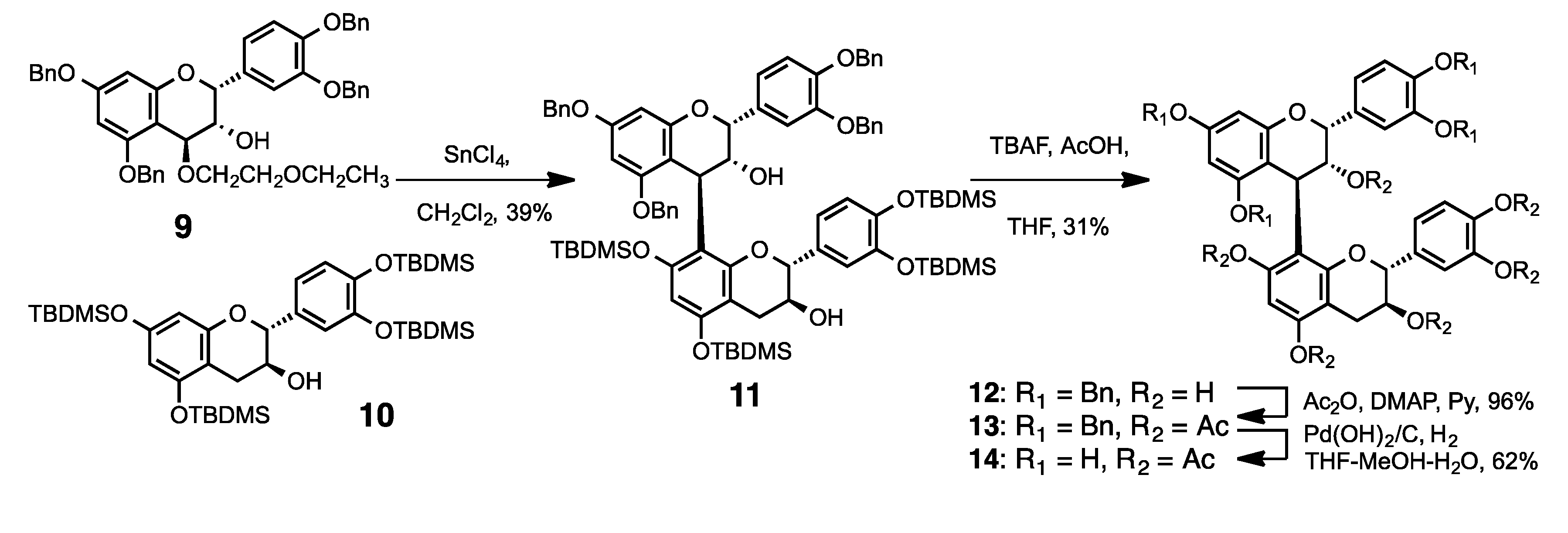
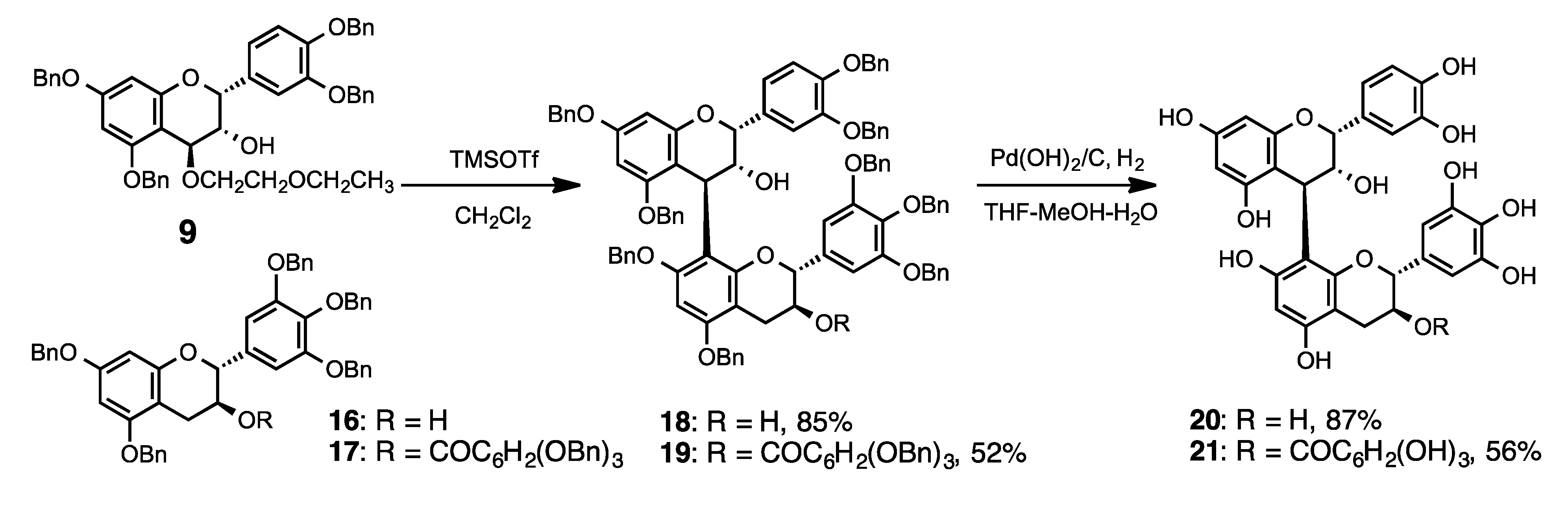
3.2. Synthesis
 +56.4 (c 1.93, CHCl3); 1H-NMR (500 MHz, CDCl3, 0.8 : 0.2 mixture of rotational isomers) major isomer: 7.46-6.94 (18.4H, m), 6,71 (0.8H, d, J = 8.5 Hz), 6.49 (0.8H, d, J = 2.2 Hz), 6.39 (0.8H, dd, J = 2.2, 8.5 Hz), 6.12 (0.8H, s), 6.01 (0.8H, d, J = 2.2 Hz), 5.64 (0.8H, d, J = 2.2 Hz), 5.26–4.56 (0.8H, m), 3.96–3.95 (0.8H, m), 3.83-3.80 (0.8H, m), 3.65 (0.8H, d, J = 8.5 Hz), 3.67 (0.8H, dd, J = 6.0, 16.5 Hz), 2.52 (0.8H, dd, J = 8.9, 16.5 Hz), 1.70 (0.8H, d, J = 6.5 Hz), 1.33 (0.8H, d, J = 3.0 Hz), 1.04 (7.2H, s), 0.97 (7.2H, s), 0.95 (7.2H, s), 0.86 (7.2H, s), 0.30 (3H, s), 0.29 (3H, s), 0.26 (3H, s), 0.19 (3H, s), 0.16 (3H, s), 0.15 (3H, s), 0.04 (3H, s), 0.02 (3H, s); minor isomer: 7.46–6.94 (4.8H, m), 6.78 (0.2H, d, J = 2.2 Hz), 6.65 (0.2H, d, J = 2.2 Hz), 6.29 (0.2H, d, J = 2.2 Hz), 6.20 (0.2H, d, J = 2.2 Hz), 5.96 (0.2H, s), 5.33–4.54 (2H, m), 3.95–3.98 (0.2H, m), 3.89–3.88 (0.2H, m), 3.63–3.61 (0.2H, m), 3.57–3.50 (0.2H, m), 3.09–3.04 (0.2H, m), 2.62 (0.2H, dd, J = 9.4, 16.5 Hz), 1.65–1.50 (0.2H, m), 1.33–1.25 (0.2H, m), 1.07-0.75 (7.2H, m), 0.30–0.01 (4.8H, m); 13C-NMR (125 MHz, CDCl3, 0.8: 0.2 mixture of rotational isomers) major isomer: 158.0, 157.0, 155.4, 154.8, 152.9, 152.7, 149.0, 148.5, 147.0, 146.6, 137.54, 137.52, 137.48, 137.37, 132.9, 130.0, 128.7-126.8 (C×7), 120.8, 119.8, 119.3, 115.1, 113.8, 113.4, 106.2, 104.4, 102.2, 94.1, 93.0, 81.2, 75.3, 72.1, 71.50, 71.49, 69.7, 69.0, 68.4, 36.1, 28.9, 25.95, 25.91, 25.89, 25.81, 18.42, 18.40, 18.31, 18.29, −3.7–4.6 (C×4); minor isomer: 158.53, 158.46, 155.2, 154.7, 153.6, 152.4, 148.7, 148.1, 147.10, 147.07, 137.9, 137.7, 137.6, 137.3, 132.6, 130.8, 128.7–126.8 (C×7), 120.1, 119.1, 118.9, 115.0, 114.2, 112.0, 104.8, 104.5, 102.2, 95.0, 93.6, 81.3, 76.0, 72.5, 71.0, 70.6, 70.2, 69.6, 68.8, 36.7, 28.5, 26.4, 25.1–25.8 (Cx3), 19.1-18.0 (C×4), −3.7–4.2 (C×4); FABMS (m/z) 1398 (31), 1397 (43), 1396 ([M+H]+, 53), 838 (28), 837 (38), 836 (57), 725 (26), 722 (19), 721 (28), 654 (55), 653 (100); FABHRMS calcd for C82H107O12Si4 [M+H]+, 1395.6840; found: 1395.6913. + 61.9 (c 0.63, CHCl3); 1H-NMR (500 MHz, CDCl3) 7.47–6.83 (22H, m), 7.08 (1H, d, J = 2.2 Hz), 7.01 (1H, d, J = 8.5 Hz), 6.96 (1H, d, H = 2.2 Hz), 6.81 (1H, dd, J = 2.2, 8.5 Hz), 6.46 (1H, s), 6.33 (1H, d, J = 2.2 Hz), 6.25 (1H, d, J = 2.2 Hz), 5.27 (2H, s), 5.26 (2H, s), 5.11–4.79 (9H, m), 4.95 (1H, d, J = 8.0 Hz), 4.77 (1H, d, J = 10.0 Hz), 4.71 (1H, d, J = 10.0 Hz), 4.47 (1H, br s), 2.99 (1H, dd, J = 5.5, 16.5 Hz), 2.60 (1H, dd, J = 8.5, 16.5 Hz), 2.33 (3H, s), 2.24 (3H, s), 2.22 (3H, s), 1.93 (3H, s), 1.57 (3H, 2), 1.52 (3H, s); 13C-NMR (125 MHz, CDCl3) 170, 168.9, 168.8, 168.7, 168.0, 167.9, 159.0, 158.0, 155.2, 152.2, 149.0, 148.7, 147.8, 147.5, 141.9, 141.6, 137.30, 137.28, 137.0, 136.6, 136.1, 131.1, 128.6-127.4 (Cx13), 124.6, 123.3, 122.3, 119.8, 119.1, 114.8, 113.5, 110.5, 102.4, 94.7, 93.6, 77.8, 77.2, 74.9, 71.5, 71.3, 70.2, 70.0, 68.3, 34.0, 25.1, 20.89, 20.87, 20.7, 20.6, 20.3, 19.9. FABMS (m/z) 1192 (32), 1191 ([M+H]+, 44), 1133 (17), 1132 (41), 1131 (66), 1130 (53), 1042 (37), 1041 (62), 1040 (82), 950 (18), 949 (28), 605 (23), 604 (45), 603 (100); FABHRMS calcd for C70H63O18 [M+H]+, 1191.4014; found: 1191.3949. 0 (c 0.012, CHCl3); 1H-NMR (500 MHz, CDCl3, 0.7:0.3 mixture of rotational isomers) major isomer: 7.25–6.41 (4.2H, m), 6.12–6.10 (1.4H, m), 6.12 (0.7H, s), 5.19–5.13 (1.4H, m), 5.02 (0.7H, d, H = 10.0 Hz), 4.70 (0.7H, t, J = 10.0 Hz), 4.43 (0.7H, br s), 2.92 (0.7H, dd, J = 5.0, 16.0 Hz), 2.67 (0.7H, dd, J = 10.0, 16.0 Hz), 2.33 (2.1H, s), 2.23 (2.1H, s), 2.21 (2.1H. s), 1.92 (2.1H. s), 1.71 (2.1H, s), 1.62 (2.1H, s); minor isomer: 7.25–6.08 (2.7H, m), 5.19–4.68 (1.5H, m), 4.47 (0.3H, br s), 2.85–2.81 (0.3H, m), 2.75–2.61 (0.3H, m), 2.32 (1.8H, s), 2.01 (0.9H, s), 1.90 (0.9H, s), 1.69 (0.9H, s), 1.65(0.9H, s); 13C-NMR (125 MHz, CDCl3) major isomer: 176.1, 174.3, 173.4, 173.2, 173.0, 173.4, 162.2, 161.4, 159.4, 159.3, 151.9, 151.7, 148.7 (×2), 140.8, 133.4, 132.9, 131.9-131.4 (×7), 123.0, 122.9, 121.6, 118.4, 117.3 (×2), 114.6, 109.0, 104.7, 99.4, 98.6, 78.5, 76.3, 73.7, 72.4, 38.0, 27.8, 23.4, 23.12, 23.05 (×2), 22.8, 22.6. Minor isomer was not identified. FABMS (m/z) 832 (3.6), 831 ([M+H]+, 9.1), 605 (27), 604 (45), 603 (100); FABHRMS calcd for C42H39O18 [M+H]+, 831.2136; found: 831.2116. 0 (c 0.030, CHCl3); 1H NMR (500 MHz, CDCl3, 0.7:0.3 mixture of rotational isomers) major isomer: 7.51-6.80 (32.9H, m), 6.45 (1.4H, s), 6.40 (0.7H, s), 6.07 (0.7H, d, J = 2.0 Hz), 5.75 (0.7H, d, J = 2.0 Hz), 5.61 (0.7H, s), 5.19–4.56 (12.6H, m), 4.71 (0.7H, d, J = 11.0 Hz), 4.56 (0.7H, d, J = 11.0 Hz), 4.12 (0.7H, br s), 4.04 (0.7H, s), 3.92 (0.7H, d, J = 4.5 Hz), 3.04 (0.7H, d, J = 17.5 Hz), 2.95 (0.7H, dd, J = 5.0, 17.5 Hz), 1.85 (0.7H, br s), 1.60 (0.7H, br s); minor isomer: 7.51–6.80 (13.8H, m), 6.34 (0.3H, s), 6.29 (0.3H, d, J = 2.5 Hz), 6.24 (0.3H, s), 6.12 (0.3H, d, J = 2.2 Hz), 5.19–3.95 (6.0H, m), 4.68 (0.3H, d, J = 12.0 Hz), 4.42 (0.3H, d, J = 12.0 Hz), 3.20–3.12 (0.3H, m), 3.05–3.00 (0.3H, m), 1.80–1.40 (0.6H, m); 13C-NMR (125 MHz, CDCl3, 0.7:0.3 mixture of rotational isomers) major isomer: 158.3, 157.0, 156.6, 156.9, 155.2, 154.4, 152.9, 152.5, 149.3, 148.8, 138.4–136.9 (C×11), 133.3, 132.7, 128.7–126.6 (C×21), 119.8, 115.2, 113.5, 111.3, 105.4, 104.4, 102.4, 93.8, 93.3, 91.7, 79.2, 75.7, 75.2, 72.5, 71.4, 71.3, 71.1, 70.5, 70.1, 70.0, 69.1, 66.4, 35.8, 28.6; minor isomer: 158.4, 156.9, 156.7, 155.6, 155.2, 153.1, 152.8, 149.1, 148.7, 138.4–136.9 (C×12), 133.6, 132.5, 128.7–126.6 (C×21), 120.1, 115.1, 114.2, 111.7, 105.6, 104.5, 101.8, 94.5, 93.5, 93.1, 78.3, 75.6, 75.2, 72.2, 71.6, 71.4, 71.3, 70.6, 70.1, 69.9, 69.6, 65.2, 36.1, 28.9. −4.6 (c 1.64, CHCl3); 1H-NMR (500 MHz, CDCl3) major isomer: 7.45–6.05 (67H, m), 5.63–5.62 (3H, m), 5.51 (1H, d, J = 6.0 Hz), 5.30–4.51 (26H, m), 4.14–4.10 (2H, m), 3.20 (1H, dd, J = 6.0, 18.5 Hz), 3.04 (1H, d, J = 18.5 Hz), 1.78 (1H, d, J = 6.0 Hz); 13C-NMR (125 MHz, CDCl3) 165.3, 158.4, 156.9, 156.2, 155.4, 154.8, 152.8, 152.5, 149.3, 148.9, 143.4, 138.1–136.5 (C×7), 132.8, 132.3, 128.7–125.7 (C×26), 119.7, 114.5, 113.6, 111.9, 109.9, 106.0, 105.8, 104.2, 102.6, 93.8, 93.4, 91.8, 78.4, 75.9, 75.3, 75.1, 75.1, 71.8, 71.4, 71.2, 70.8, 70.7, 70.2, 69.2, 68.9, 35.9, 26.9. −12.5 (c 0.016, CH3OH); 1H NMR (400 MHz, CD3OD, −40 °C) 6.82 (1H, br s), 6.87 (1H, d, J = 8.5 Hz), 6.59 (1H, br d, J = 8.5 Hz), 6.59 (2H, s), 5.86 (1H, s), 6.10–5.15 (2H, m), 5.03 (1H, br s), 4.86 (1H, br s), 4.60 (1H, br s), 4.25 (1H, br s), 3.79 (1H, br s), 2.92 (1H, br d, J = 16.0 Hz), 2.79 (1H, d, J = 16.0 Hz); FABMS (m/z) 596 (29), 595 ([M+H]+, 27), 581 (33), 561 (18), 537 (26), 493 (26), 329 (100); FABHRMS calcd for C30H27O13 [M+H]+, 595.1452; found: 595.1402. 0 (c 0.025, CH3OH); 1H NMR (400 MHz, CD3OD, −40°C) 7.01 (2H, s), 6.89 (1H, br s), 6.87 (1H, br s), 6.80 (6.60 (2H, m), 6.58 (2H, s), 6.13 (1H, br s), 5.89 (1H, br s), 5.55 (1H, br s), 5.14 (1H, br s), 5.12 (1H, br s), 4.78 (1H, br s), 4.54 (1H, br s), 3.84 (1H, br s), 3.14–2.72 (2H, m); FABMS (m/z) 749 (18), 748 (25), 747 (27), 746 ([M+H]+, 17), 719 (17), 603 (21), 545 (23), 503 (20), 487 (23), 329 (100); FABHRMS calcd for C37H31O17 [M+H]+, 747.1561; found: 747.1519.
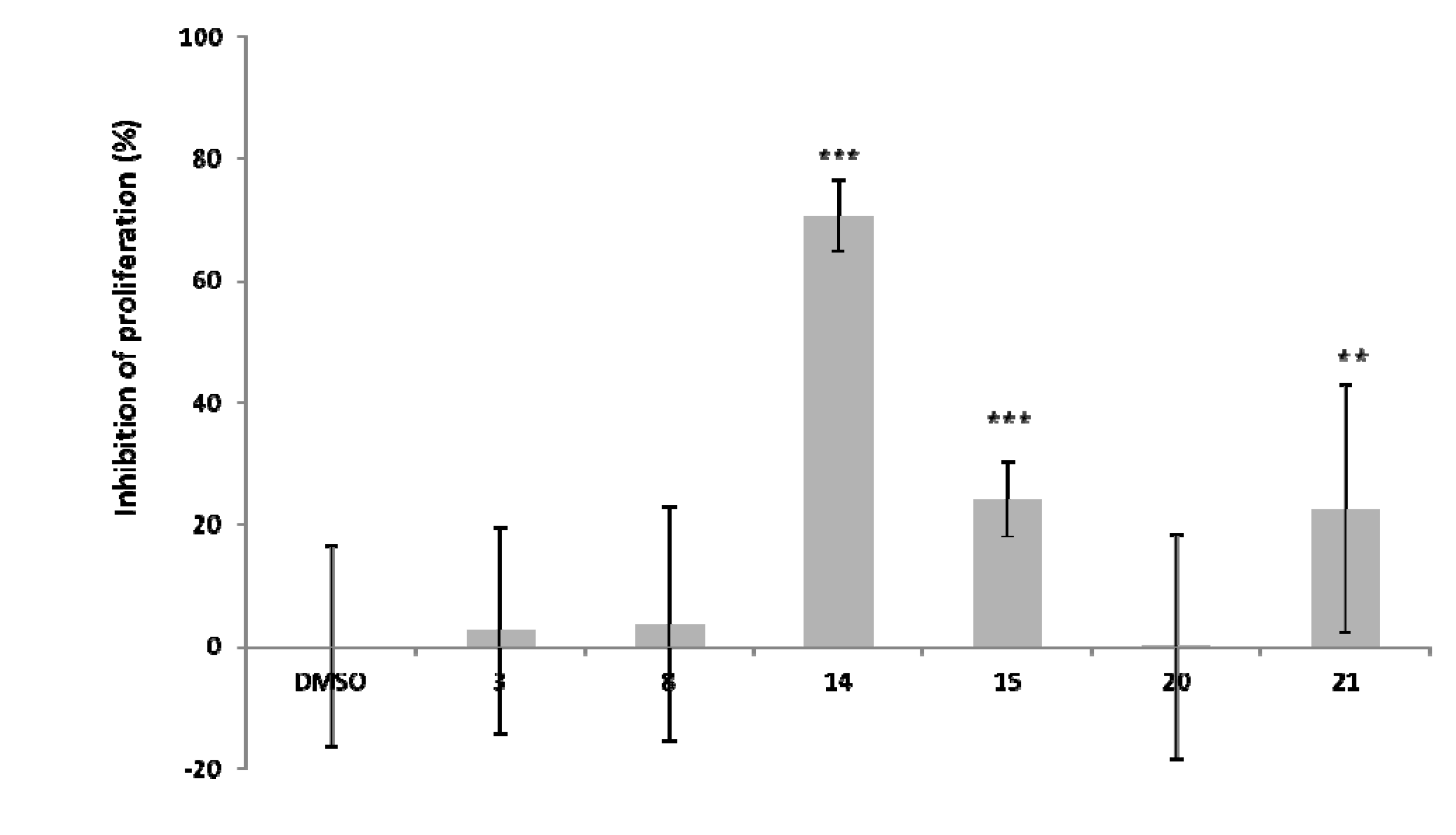
+56.4 (c 1.93, CHCl3); 1H-NMR (500 MHz, CDCl3, 0.8 : 0.2 mixture of rotational isomers) major isomer: 7.46-6.94 (18.4H, m), 6,71 (0.8H, d, J = 8.5 Hz), 6.49 (0.8H, d, J = 2.2 Hz), 6.39 (0.8H, dd, J = 2.2, 8.5 Hz), 6.12 (0.8H, s), 6.01 (0.8H, d, J = 2.2 Hz), 5.64 (0.8H, d, J = 2.2 Hz), 5.26–4.56 (0.8H, m), 3.96–3.95 (0.8H, m), 3.83-3.80 (0.8H, m), 3.65 (0.8H, d, J = 8.5 Hz), 3.67 (0.8H, dd, J = 6.0, 16.5 Hz), 2.52 (0.8H, dd, J = 8.9, 16.5 Hz), 1.70 (0.8H, d, J = 6.5 Hz), 1.33 (0.8H, d, J = 3.0 Hz), 1.04 (7.2H, s), 0.97 (7.2H, s), 0.95 (7.2H, s), 0.86 (7.2H, s), 0.30 (3H, s), 0.29 (3H, s), 0.26 (3H, s), 0.19 (3H, s), 0.16 (3H, s), 0.15 (3H, s), 0.04 (3H, s), 0.02 (3H, s); minor isomer: 7.46–6.94 (4.8H, m), 6.78 (0.2H, d, J = 2.2 Hz), 6.65 (0.2H, d, J = 2.2 Hz), 6.29 (0.2H, d, J = 2.2 Hz), 6.20 (0.2H, d, J = 2.2 Hz), 5.96 (0.2H, s), 5.33–4.54 (2H, m), 3.95–3.98 (0.2H, m), 3.89–3.88 (0.2H, m), 3.63–3.61 (0.2H, m), 3.57–3.50 (0.2H, m), 3.09–3.04 (0.2H, m), 2.62 (0.2H, dd, J = 9.4, 16.5 Hz), 1.65–1.50 (0.2H, m), 1.33–1.25 (0.2H, m), 1.07-0.75 (7.2H, m), 0.30–0.01 (4.8H, m); 13C-NMR (125 MHz, CDCl3, 0.8: 0.2 mixture of rotational isomers) major isomer: 158.0, 157.0, 155.4, 154.8, 152.9, 152.7, 149.0, 148.5, 147.0, 146.6, 137.54, 137.52, 137.48, 137.37, 132.9, 130.0, 128.7-126.8 (C×7), 120.8, 119.8, 119.3, 115.1, 113.8, 113.4, 106.2, 104.4, 102.2, 94.1, 93.0, 81.2, 75.3, 72.1, 71.50, 71.49, 69.7, 69.0, 68.4, 36.1, 28.9, 25.95, 25.91, 25.89, 25.81, 18.42, 18.40, 18.31, 18.29, −3.7–4.6 (C×4); minor isomer: 158.53, 158.46, 155.2, 154.7, 153.6, 152.4, 148.7, 148.1, 147.10, 147.07, 137.9, 137.7, 137.6, 137.3, 132.6, 130.8, 128.7–126.8 (C×7), 120.1, 119.1, 118.9, 115.0, 114.2, 112.0, 104.8, 104.5, 102.2, 95.0, 93.6, 81.3, 76.0, 72.5, 71.0, 70.6, 70.2, 69.6, 68.8, 36.7, 28.5, 26.4, 25.1–25.8 (Cx3), 19.1-18.0 (C×4), −3.7–4.2 (C×4); FABMS (m/z) 1398 (31), 1397 (43), 1396 ([M+H]+, 53), 838 (28), 837 (38), 836 (57), 725 (26), 722 (19), 721 (28), 654 (55), 653 (100); FABHRMS calcd for C82H107O12Si4 [M+H]+, 1395.6840; found: 1395.6913. + 61.9 (c 0.63, CHCl3); 1H-NMR (500 MHz, CDCl3) 7.47–6.83 (22H, m), 7.08 (1H, d, J = 2.2 Hz), 7.01 (1H, d, J = 8.5 Hz), 6.96 (1H, d, H = 2.2 Hz), 6.81 (1H, dd, J = 2.2, 8.5 Hz), 6.46 (1H, s), 6.33 (1H, d, J = 2.2 Hz), 6.25 (1H, d, J = 2.2 Hz), 5.27 (2H, s), 5.26 (2H, s), 5.11–4.79 (9H, m), 4.95 (1H, d, J = 8.0 Hz), 4.77 (1H, d, J = 10.0 Hz), 4.71 (1H, d, J = 10.0 Hz), 4.47 (1H, br s), 2.99 (1H, dd, J = 5.5, 16.5 Hz), 2.60 (1H, dd, J = 8.5, 16.5 Hz), 2.33 (3H, s), 2.24 (3H, s), 2.22 (3H, s), 1.93 (3H, s), 1.57 (3H, 2), 1.52 (3H, s); 13C-NMR (125 MHz, CDCl3) 170, 168.9, 168.8, 168.7, 168.0, 167.9, 159.0, 158.0, 155.2, 152.2, 149.0, 148.7, 147.8, 147.5, 141.9, 141.6, 137.30, 137.28, 137.0, 136.6, 136.1, 131.1, 128.6-127.4 (Cx13), 124.6, 123.3, 122.3, 119.8, 119.1, 114.8, 113.5, 110.5, 102.4, 94.7, 93.6, 77.8, 77.2, 74.9, 71.5, 71.3, 70.2, 70.0, 68.3, 34.0, 25.1, 20.89, 20.87, 20.7, 20.6, 20.3, 19.9. FABMS (m/z) 1192 (32), 1191 ([M+H]+, 44), 1133 (17), 1132 (41), 1131 (66), 1130 (53), 1042 (37), 1041 (62), 1040 (82), 950 (18), 949 (28), 605 (23), 604 (45), 603 (100); FABHRMS calcd for C70H63O18 [M+H]+, 1191.4014; found: 1191.3949. 0 (c 0.012, CHCl3); 1H-NMR (500 MHz, CDCl3, 0.7:0.3 mixture of rotational isomers) major isomer: 7.25–6.41 (4.2H, m), 6.12–6.10 (1.4H, m), 6.12 (0.7H, s), 5.19–5.13 (1.4H, m), 5.02 (0.7H, d, H = 10.0 Hz), 4.70 (0.7H, t, J = 10.0 Hz), 4.43 (0.7H, br s), 2.92 (0.7H, dd, J = 5.0, 16.0 Hz), 2.67 (0.7H, dd, J = 10.0, 16.0 Hz), 2.33 (2.1H, s), 2.23 (2.1H, s), 2.21 (2.1H. s), 1.92 (2.1H. s), 1.71 (2.1H, s), 1.62 (2.1H, s); minor isomer: 7.25–6.08 (2.7H, m), 5.19–4.68 (1.5H, m), 4.47 (0.3H, br s), 2.85–2.81 (0.3H, m), 2.75–2.61 (0.3H, m), 2.32 (1.8H, s), 2.01 (0.9H, s), 1.90 (0.9H, s), 1.69 (0.9H, s), 1.65(0.9H, s); 13C-NMR (125 MHz, CDCl3) major isomer: 176.1, 174.3, 173.4, 173.2, 173.0, 173.4, 162.2, 161.4, 159.4, 159.3, 151.9, 151.7, 148.7 (×2), 140.8, 133.4, 132.9, 131.9-131.4 (×7), 123.0, 122.9, 121.6, 118.4, 117.3 (×2), 114.6, 109.0, 104.7, 99.4, 98.6, 78.5, 76.3, 73.7, 72.4, 38.0, 27.8, 23.4, 23.12, 23.05 (×2), 22.8, 22.6. Minor isomer was not identified. FABMS (m/z) 832 (3.6), 831 ([M+H]+, 9.1), 605 (27), 604 (45), 603 (100); FABHRMS calcd for C42H39O18 [M+H]+, 831.2136; found: 831.2116. 0 (c 0.030, CHCl3); 1H NMR (500 MHz, CDCl3, 0.7:0.3 mixture of rotational isomers) major isomer: 7.51-6.80 (32.9H, m), 6.45 (1.4H, s), 6.40 (0.7H, s), 6.07 (0.7H, d, J = 2.0 Hz), 5.75 (0.7H, d, J = 2.0 Hz), 5.61 (0.7H, s), 5.19–4.56 (12.6H, m), 4.71 (0.7H, d, J = 11.0 Hz), 4.56 (0.7H, d, J = 11.0 Hz), 4.12 (0.7H, br s), 4.04 (0.7H, s), 3.92 (0.7H, d, J = 4.5 Hz), 3.04 (0.7H, d, J = 17.5 Hz), 2.95 (0.7H, dd, J = 5.0, 17.5 Hz), 1.85 (0.7H, br s), 1.60 (0.7H, br s); minor isomer: 7.51–6.80 (13.8H, m), 6.34 (0.3H, s), 6.29 (0.3H, d, J = 2.5 Hz), 6.24 (0.3H, s), 6.12 (0.3H, d, J = 2.2 Hz), 5.19–3.95 (6.0H, m), 4.68 (0.3H, d, J = 12.0 Hz), 4.42 (0.3H, d, J = 12.0 Hz), 3.20–3.12 (0.3H, m), 3.05–3.00 (0.3H, m), 1.80–1.40 (0.6H, m); 13C-NMR (125 MHz, CDCl3, 0.7:0.3 mixture of rotational isomers) major isomer: 158.3, 157.0, 156.6, 156.9, 155.2, 154.4, 152.9, 152.5, 149.3, 148.8, 138.4–136.9 (C×11), 133.3, 132.7, 128.7–126.6 (C×21), 119.8, 115.2, 113.5, 111.3, 105.4, 104.4, 102.4, 93.8, 93.3, 91.7, 79.2, 75.7, 75.2, 72.5, 71.4, 71.3, 71.1, 70.5, 70.1, 70.0, 69.1, 66.4, 35.8, 28.6; minor isomer: 158.4, 156.9, 156.7, 155.6, 155.2, 153.1, 152.8, 149.1, 148.7, 138.4–136.9 (C×12), 133.6, 132.5, 128.7–126.6 (C×21), 120.1, 115.1, 114.2, 111.7, 105.6, 104.5, 101.8, 94.5, 93.5, 93.1, 78.3, 75.6, 75.2, 72.2, 71.6, 71.4, 71.3, 70.6, 70.1, 69.9, 69.6, 65.2, 36.1, 28.9. −4.6 (c 1.64, CHCl3); 1H-NMR (500 MHz, CDCl3) major isomer: 7.45–6.05 (67H, m), 5.63–5.62 (3H, m), 5.51 (1H, d, J = 6.0 Hz), 5.30–4.51 (26H, m), 4.14–4.10 (2H, m), 3.20 (1H, dd, J = 6.0, 18.5 Hz), 3.04 (1H, d, J = 18.5 Hz), 1.78 (1H, d, J = 6.0 Hz); 13C-NMR (125 MHz, CDCl3) 165.3, 158.4, 156.9, 156.2, 155.4, 154.8, 152.8, 152.5, 149.3, 148.9, 143.4, 138.1–136.5 (C×7), 132.8, 132.3, 128.7–125.7 (C×26), 119.7, 114.5, 113.6, 111.9, 109.9, 106.0, 105.8, 104.2, 102.6, 93.8, 93.4, 91.8, 78.4, 75.9, 75.3, 75.1, 75.1, 71.8, 71.4, 71.2, 70.8, 70.7, 70.2, 69.2, 68.9, 35.9, 26.9. −12.5 (c 0.016, CH3OH); 1H NMR (400 MHz, CD3OD, −40 °C) 6.82 (1H, br s), 6.87 (1H, d, J = 8.5 Hz), 6.59 (1H, br d, J = 8.5 Hz), 6.59 (2H, s), 5.86 (1H, s), 6.10–5.15 (2H, m), 5.03 (1H, br s), 4.86 (1H, br s), 4.60 (1H, br s), 4.25 (1H, br s), 3.79 (1H, br s), 2.92 (1H, br d, J = 16.0 Hz), 2.79 (1H, d, J = 16.0 Hz); FABMS (m/z) 596 (29), 595 ([M+H]+, 27), 581 (33), 561 (18), 537 (26), 493 (26), 329 (100); FABHRMS calcd for C30H27O13 [M+H]+, 595.1452; found: 595.1402. 0 (c 0.025, CH3OH); 1H NMR (400 MHz, CD3OD, −40°C) 7.01 (2H, s), 6.89 (1H, br s), 6.87 (1H, br s), 6.80 (6.60 (2H, m), 6.58 (2H, s), 6.13 (1H, br s), 5.89 (1H, br s), 5.55 (1H, br s), 5.14 (1H, br s), 5.12 (1H, br s), 4.78 (1H, br s), 4.54 (1H, br s), 3.84 (1H, br s), 3.14–2.72 (2H, m); FABMS (m/z) 749 (18), 748 (25), 747 (27), 746 ([M+H]+, 17), 719 (17), 603 (21), 545 (23), 503 (20), 487 (23), 329 (100); FABHRMS calcd for C37H31O17 [M+H]+, 747.1561; found: 747.1519.3.3. Inhibitory Activity of Cell Proliferation
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Yang, C.S.; Maliakai, P.; Meng, X. Inhibition of carcinogenesis by tea. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 25–54. [Google Scholar] [CrossRef]
- Tachibana, H.; Koga, K.; Fujimura, Y.; Yamada, K. A receptor for green tea polyphenol EGCG. Nat. Struct. Mol. Biol. 2004, 11, 380–381. [Google Scholar] [CrossRef]
- Harborne, J.B. The Flavonoids: Advances in research from 1986; Chapman and Hall: London, UK, 1993. [Google Scholar]
- Harborne, J.B.; Baxter, H. The Handbook of Natural Flavonoids; John Wiley & Sons: New York, NY, USA, 1999. [Google Scholar]
- Tuckmantel, W.; Kozikowski, A.P.; Romanczyk, L.J., Jr. Studies in polyphenol chemistry and bioactivity. 1. Preparation of building blocks from (+)-catechin. Procyanidin formation. Synthesis of the cancer cell growth inhibitor, 3-O-galloyl-(2R,3R)-epicatechin-4β,8-[3-O-galloyl-(2R,3R)-epicatechin]. J. Am. Chem. Soc. 1999, 121, 12073–12081. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Tuckmantel, W.; George, C. Studies in polyphenol chemistry and bioactivity. 2. Establishment of interflavan linkage regio- and stereochemistry by oxidative degradation of an O-alkylated derivative of procyanidin B2 to (R)-(−)-2,4-diphenylbutyric acid. J. Org. Chem. 2000, 65, 5371–5381. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Tuckmantel, W.; Boettcher, G.; Romanczyk, L.J., Jr. Studies in polyphenol chemistry and bioactivity. 4.1 synthesis of trimeric, tetrameric, pentameric, and higher oligomeric epicatechin-derived procyanidins having all-4β,8-interflavan connectivity and their inhibition of cancer cell growth through cell cycle arrest. J. Org. Chem. 2003, 68, 1641–1658. [Google Scholar] [CrossRef]
- Ohmori, K.; Ushimaru, N.; Suzuki, K. Oligomeric catechins: An enabling synthetic strategy by orthogonal activation and C(8) protection. Proc. Natl. Acad. Sci. USA 2004, 101, 12002–12007. [Google Scholar] [CrossRef]
- Tarascou, I.; Barathieu, K.; Andre, Y.; Pianet, I.; Dufourc, E.; Fouquet, E. An improved synthesis of procyanidin dimers: regio- and stereocontrol of the interflavan bond. Eur. J. Org. Chem. 2006, 5367–5377. [Google Scholar]
- Sharma, P.K.; Kolchinski, A.; Shea, H.A.; Nair, J.J.; Gou, Y.; Romanczyk, L.J., Jr.; Schmitz, H.H. Scale-up syntheses of two naturally occurring procyanidins: (−)-epicatechin-(4β,8)-(+)-catechin and (−)-epicatechin-3-O-galloyl-(4β,8)-(−)-epicatechin-3-O-gallate. Org. Process. Res. Dev. 2007, 11, 422–430. [Google Scholar] [CrossRef]
- Mohri, Y.; Sagehashi, M.; Yamada, T.; Hattori, Y.; Morimura, K.; Kamo, T.; Hirota, M.; Makabe, H. An efficient synthesis of procyanidins. Rare earth metal Lewis acid catalyzed equimolar condensation of catechin and epicatechin. Tetrahedron Lett. 2007, 48, 5891–5894. [Google Scholar]
- Oyama, K.; Kuwano, M.; Ito, M.; Yoshida, K.; Kondo, T. Synthesis of procyanidins by stepwise- and self-condensation using 3,4-cis-4-acetoxy-3-O-acetyl-4-dehydro-5,7,3′,4′-tetra-O-benzyl-(+)-catechin and (−)-epicatechin as a key building monomer. Tetrahedron Lett. 2008, 49, 3176–3180. [Google Scholar] [CrossRef]
- Matthew, A.C.; Bonnet, S.L.; van der Westhuizen, J.H. Synthesis of proanthocyanidins. Part 1. The first oxidative formation of the interflavanyl bond in procyanidins. Org. Lett. 2008, 10, 3865–3868. [Google Scholar]
- Mohri, Y.; Sagehashi, M.; Yamada, T.; Hattori, Y.; Morimura, Y.; Hamauzu, K.; Kamo, T.; Hirota, M.; Makabe, H. An efficient synthesisof procyanidins using equimolar condensation of catechin and/or epicatechin catalyzed by ytterbium triflate. Heterocycles 2009, 79, 549–563. [Google Scholar] [CrossRef]
- Watanabe, G.; Ohmori, K.; Suzuki, K. First regiocontrolled synthesis of procyanidin B6, a catechin dimer with rare connectivity: A halo-capping strategy for formation of 4,6-interflavan bonds. Chem. Commun. 2013, 49, 5210–5216. [Google Scholar] [CrossRef]
- Saito, A.; Nakajima, N.; Tanaka, A.; Ubukata, M. First regiocontrolled synthesis of procyanidin B6, a catechin dimer with rare connectivity: a halo-capping strategy for formation of 4,6-interflavan bonds. Biosci. Biotechnol. Biochem. 2002, 66, 1764–1767. [Google Scholar] [CrossRef]
- Saito, A.; Nakajima, N.; Tanaka, A.; Ubukata, M. Synthetic studies of proanthocyanidins. Part 2: Stereoselective gram-scale synthesis of procyanidin-B3. Tetrahedron 2002, 58, 7829–7837. [Google Scholar] [CrossRef]
- Saito, A.; Nakajima, N.; Tanaka, A.; Ubukata, M. Synthetic studies of proanthocyanidins. Part 3: Stereoselective 3,4-cis catechin and catechin condensation by TMSOTf-catalyzed intramolecular coupling method. Tetrahedron Lett. 2003, 44, 5449–5452. [Google Scholar] [CrossRef]
- Saito, A.; Nakajima, N.; Tanaka, A.; Ubukata, M. Synthetic studies of proanthocyanidins. Part 4. The synthesis of procyanidin B1 and B4: TMSOTf-catalyzed cyclization of catechin and epicatechin condensation. Heterocycles 2003, 61, 287–298. [Google Scholar] [CrossRef]
- Saito, A.; Nakajima, N.; Matsuura, M.; Tanaka, A.; Ubukata, M. Synthetic studies of proanthocyanidins. Part 5. Highly stereoselective synthesis and inhibitory activity of Maillard reaction of 3,4-trans catechin and epicatechin dimers, procyanidin B1, B2, B3, B4 and their acetates. Heterocycles 2004, 62, 479–489. [Google Scholar] [CrossRef]
- Saito, A.; Tanaka, A.; Ubukata, M.; Nakajima, N. Efficient stereoselective synthesis of proanthocyanidin trimers with TMSOTf-catalyzed intermolecular condensation. Synlett 2004, 1069–1073. [Google Scholar]
- Saito, A.; Tanaka, A.; Ubukata, M.; Nakajima, N. Stereoselection of 3,4-cis and 3,4-trans catechin and catechin condensation under intramolecular coupling method. Synlett 2004, 2040–2042. [Google Scholar]
- Saito, A.; Doi, Y.; Matsuura, N.; Tanaka, A.; Ubukata, M.; Nakajima, N. Systematic synthesis of four epicatechin series procyanidin trimers and their inhibitory activity on the Maillard reaction and antioxidant activity. Bioorg. Med. Chem. 2004, 12, 4783–4790. [Google Scholar] [CrossRef]
- Saito, A.; Enomoto, M.; Tanaka, A.; Doi, Y.; Shoji, K.; Mizushina, Y.; Ikawa, H.; Yoshida, H.; Matsuura, N.; Nakajima, N. Stereoselective synthesis of procyanidin B3-3-O-gallate and procyanidin B3-3,3''-di-O-gallate, and their abilities as antioxidant and DNA polymerase inhibitor. Tetrahedron 2004, 60, 12043–12049. [Google Scholar] [CrossRef]
- Saito, A.; Mizushina, Y.; Ikawa, H.; Yoshida, H.; Doi, Y.; Tanaka, A.; Nakajima, N. Systematic synthesis of galloyl-substituted procyanidin B1 and B2, and their ability of DPPH radical scavenging activity and inhibitory activity of DNA polymerases. Bioorg. Med. Chem. 2005, 13, 2759–2771. [Google Scholar] [CrossRef]
- Sakuda, H.; Saito, A.; Mizushina, Y.; Ikawa, H.; Yoshida, H.; Tanaka, A.; Nakajima, N. Synthesis of galloyl-substituted procyanidin B4 series, and their DPPH radical scavenging activity and DNA polymerase inhibitory activity. Heterocycls 2006, 67, 175–188. [Google Scholar] [CrossRef]
- Saito, A.; Mizushina, Y.; Tanaka, A.; Nakajima, N. Versatile synthesis of epicatechin series procyanidin oligomers, and their antioxidant and DNA polymerase inhibitory activity. Tetrahedron 2009, 65, 7422–7428. [Google Scholar] [CrossRef]
- Ishihara, S.; Doi, S.; Harui, K.; Okamoto, T.; Okamoto, S.; Uenishi, J.; Kawasaki, T.; Nakajima, N.; Saito, A. Development of a new synthetic strategy for procyanidin dimer condensation using peracetylated electrophiles. Heterocycles 2014, 88, 1595–1602. [Google Scholar] [CrossRef]
- Nakajima, N.; Horikawa, K.; Takekawa, N.; Hamada, M.; Kishimoto, T. Condensation of catechin and epicatechin incorporating a TBS-protecting group. Heterocycles 2012, 84, 349–354. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 4, 8, 14, 15, 20 and 21 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Okamoto, S.; Ishihara, S.; Okamoto, T.; Doi, S.; Harui, K.; Higashino, Y.; Kawasaki, T.; Nakajima, N.; Saito, A. Inhibitory Activity of Synthesized Acetylated Procyanidin B1 Analogs against HeLa S3 Cells Proliferation. Molecules 2014, 19, 1775-1785. https://doi.org/10.3390/molecules19021775
Okamoto S, Ishihara S, Okamoto T, Doi S, Harui K, Higashino Y, Kawasaki T, Nakajima N, Saito A. Inhibitory Activity of Synthesized Acetylated Procyanidin B1 Analogs against HeLa S3 Cells Proliferation. Molecules. 2014; 19(2):1775-1785. https://doi.org/10.3390/molecules19021775
Chicago/Turabian StyleOkamoto, Syuhei, Sayaka Ishihara, Taisuke Okamoto, Syoma Doi, Kota Harui, Yusuke Higashino, Takashi Kawasaki, Noriyuki Nakajima, and Akiko Saito. 2014. "Inhibitory Activity of Synthesized Acetylated Procyanidin B1 Analogs against HeLa S3 Cells Proliferation" Molecules 19, no. 2: 1775-1785. https://doi.org/10.3390/molecules19021775
APA StyleOkamoto, S., Ishihara, S., Okamoto, T., Doi, S., Harui, K., Higashino, Y., Kawasaki, T., Nakajima, N., & Saito, A. (2014). Inhibitory Activity of Synthesized Acetylated Procyanidin B1 Analogs against HeLa S3 Cells Proliferation. Molecules, 19(2), 1775-1785. https://doi.org/10.3390/molecules19021775