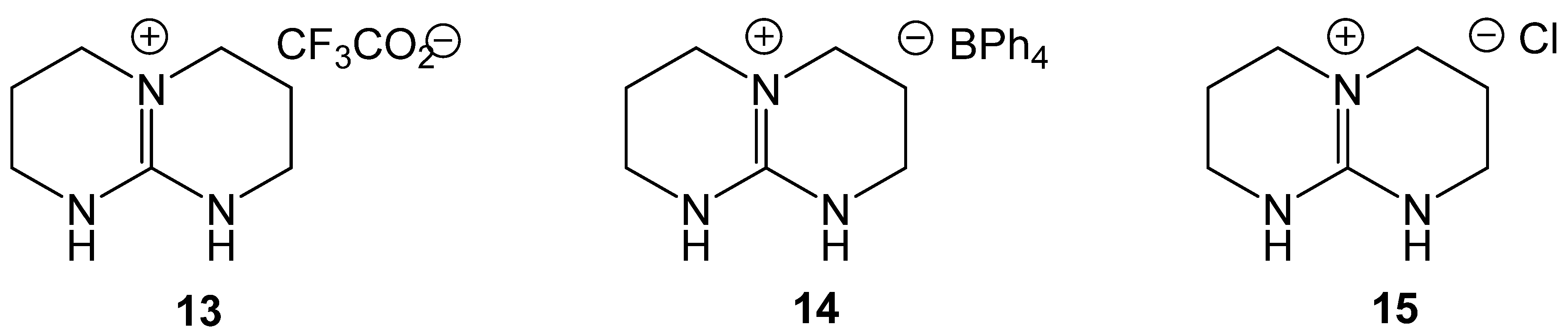
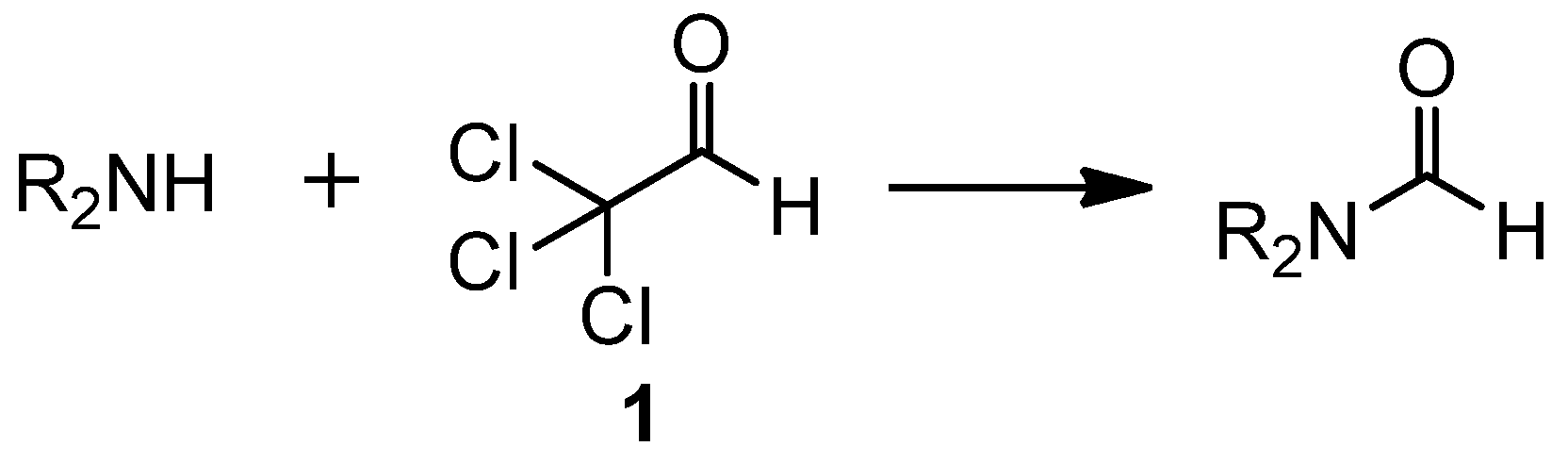
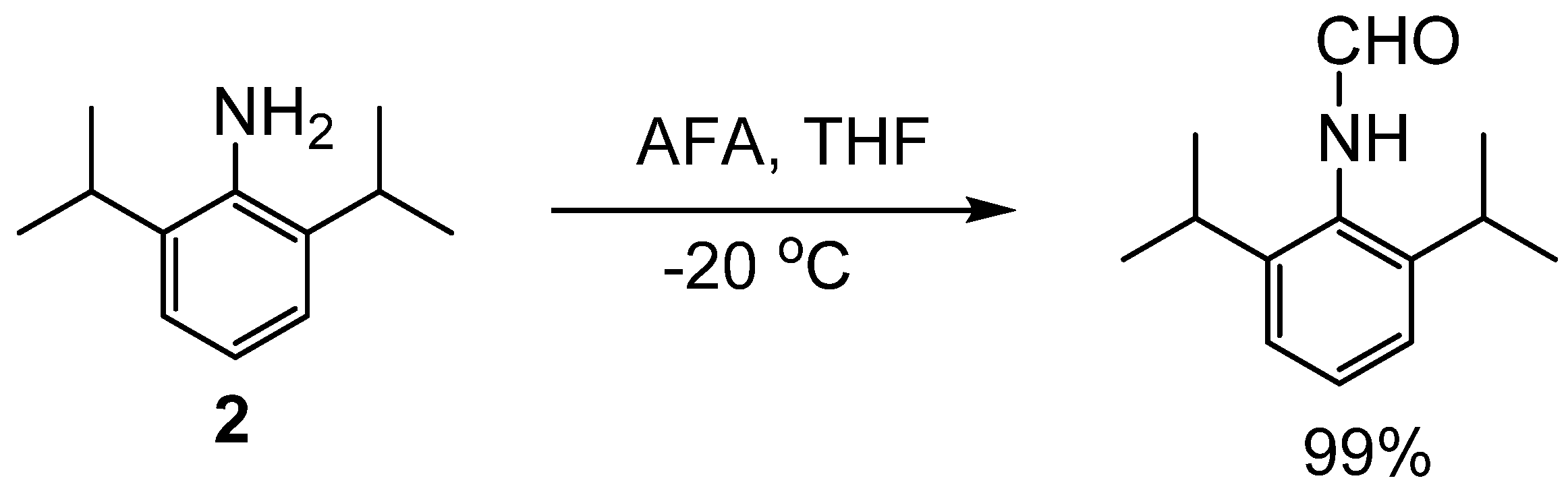
Formylation of Amines

{kind=link}
{kind=link}
{kind=link}
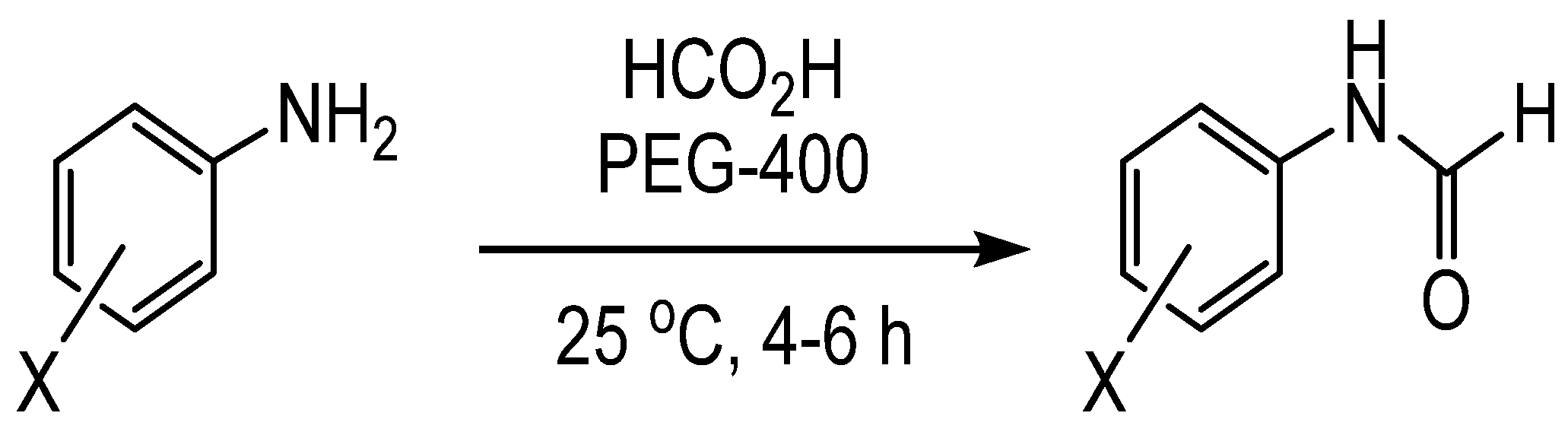{kind=link}
{kind=link}
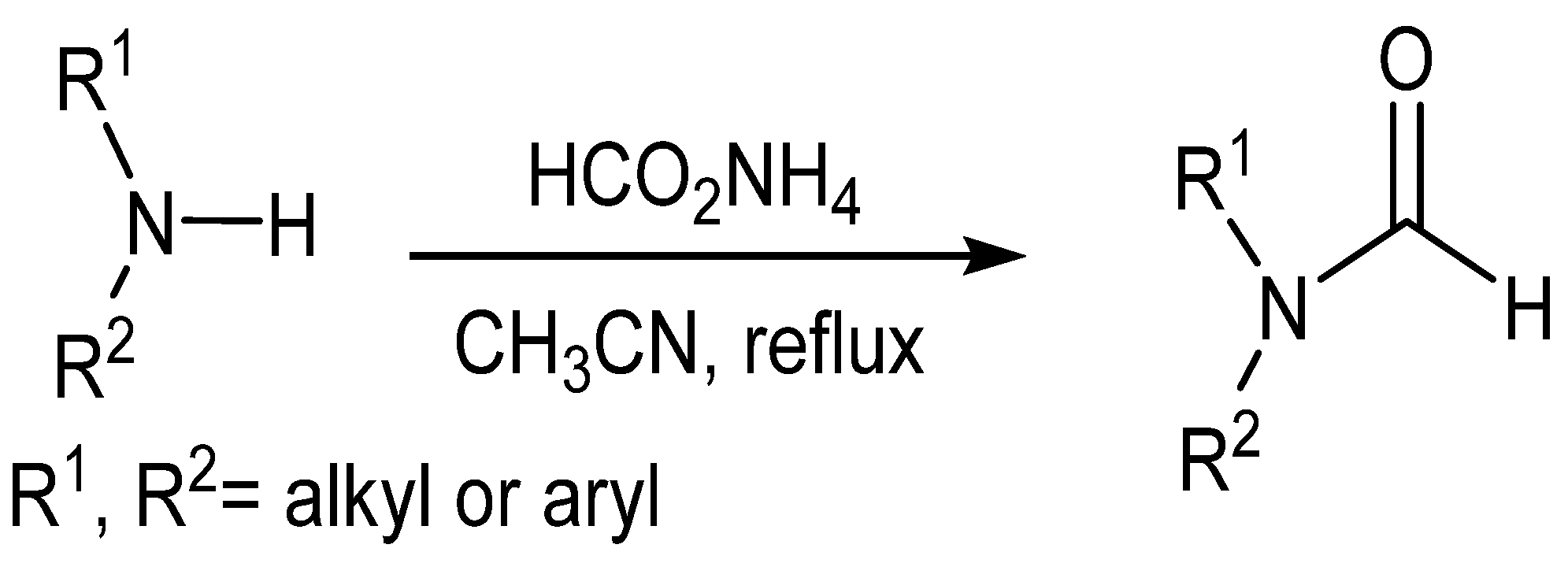{kind=link}
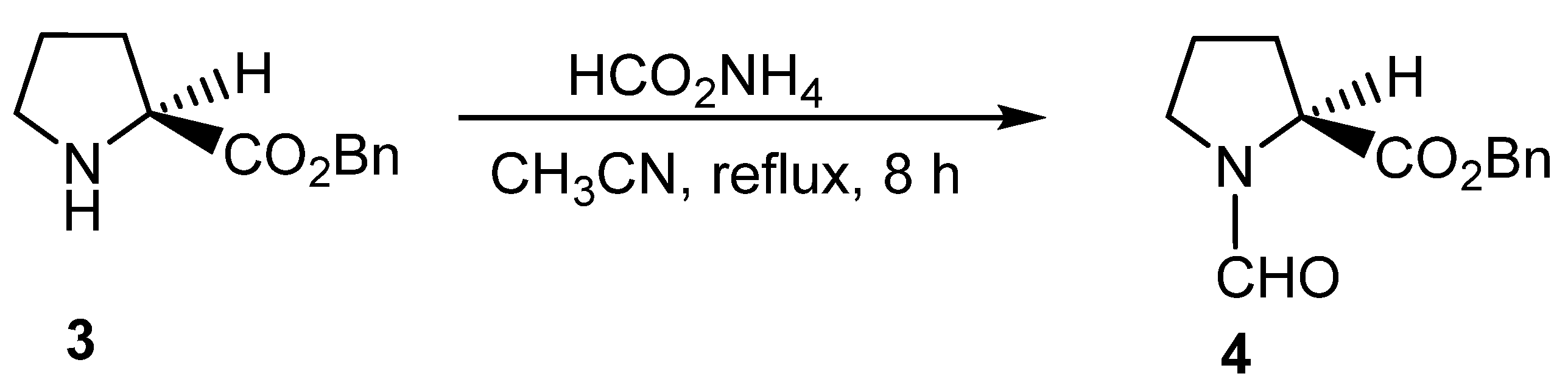{kind=link}
{kind=link}
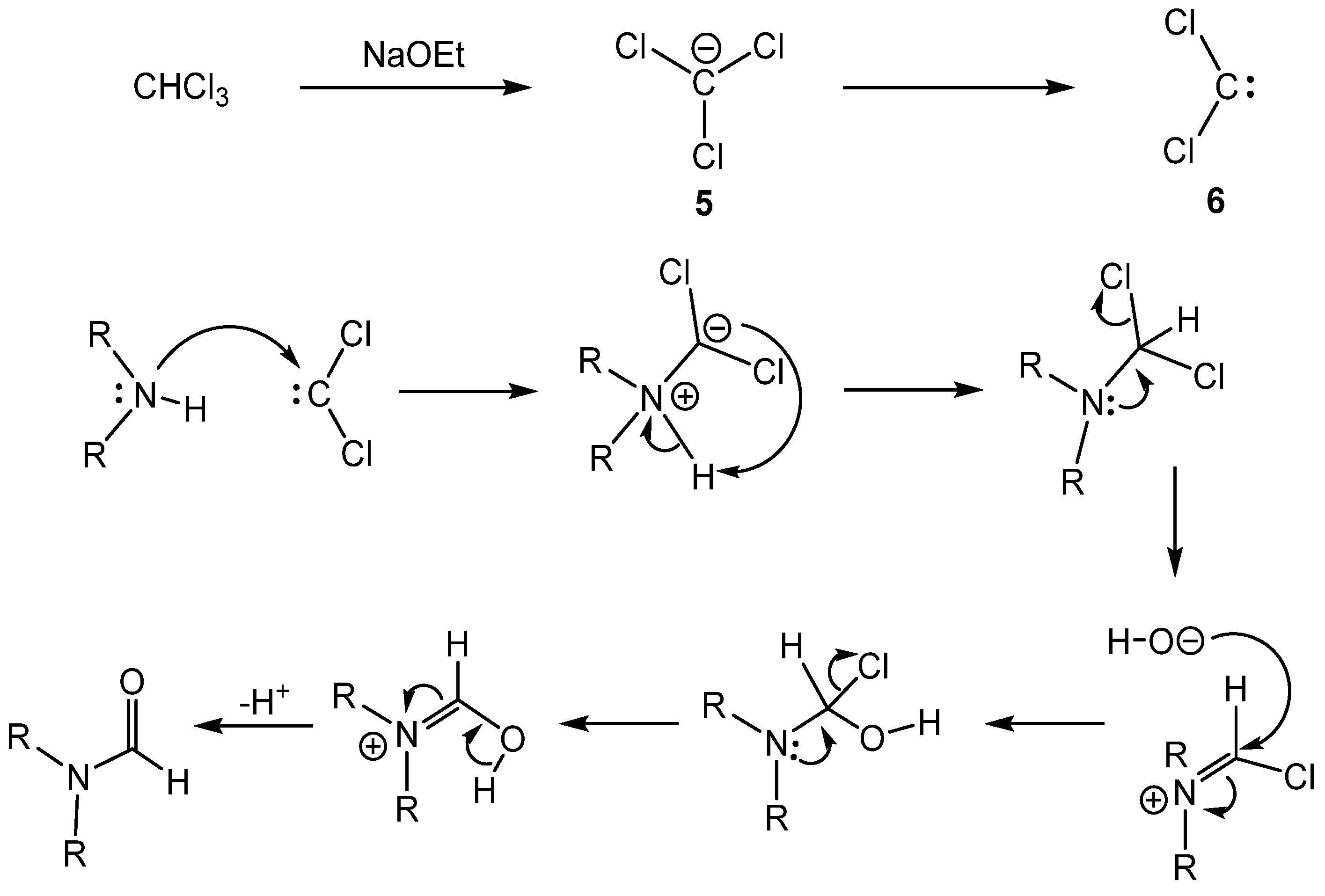{kind=link}
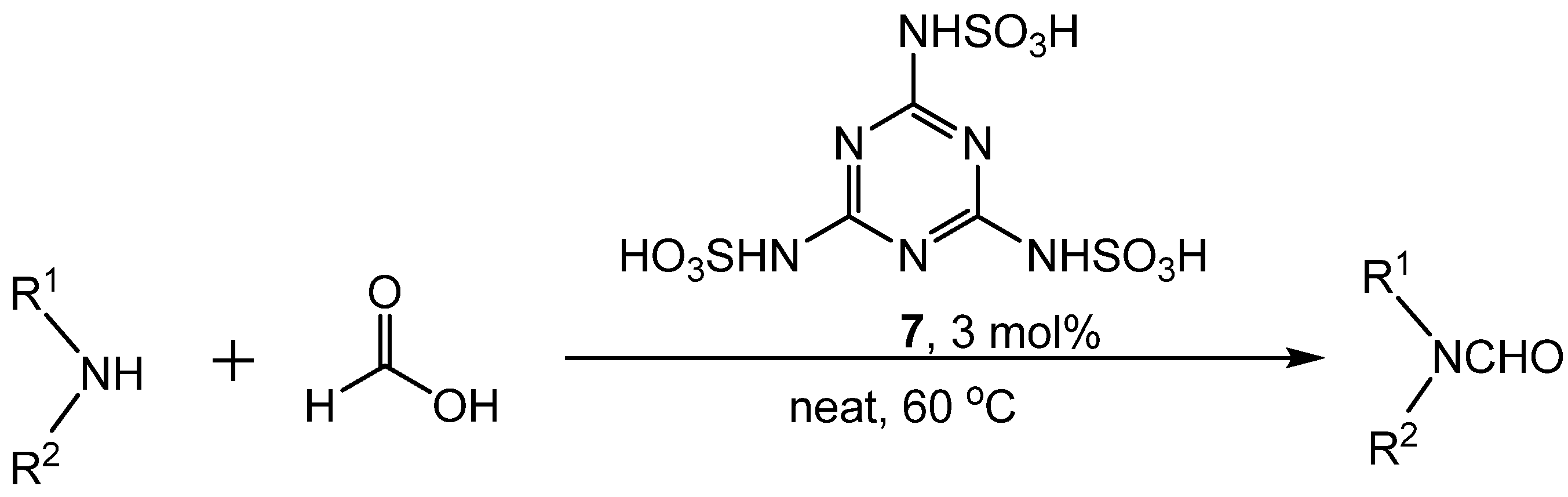{kind=link}
{kind=link}
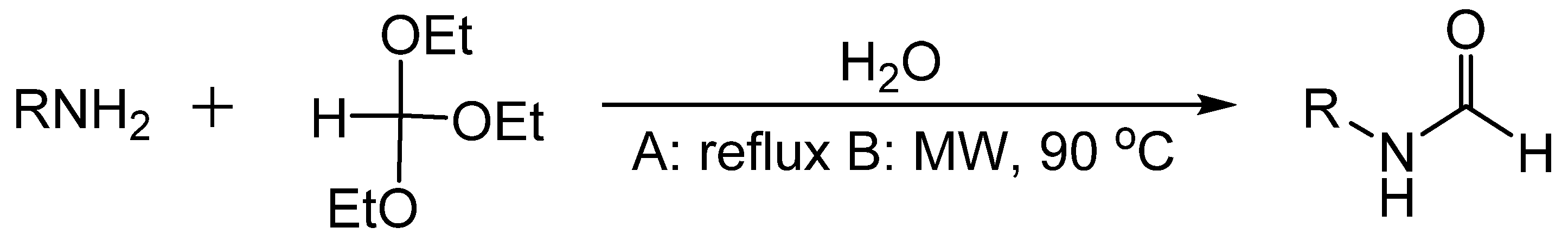{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
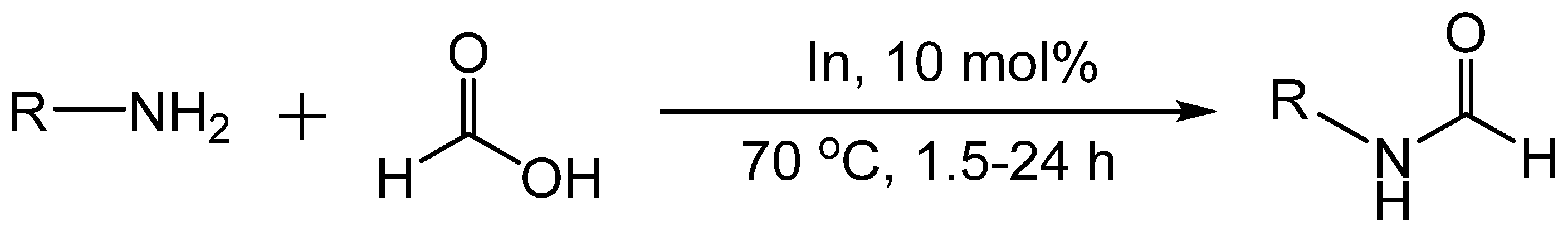{kind=link}
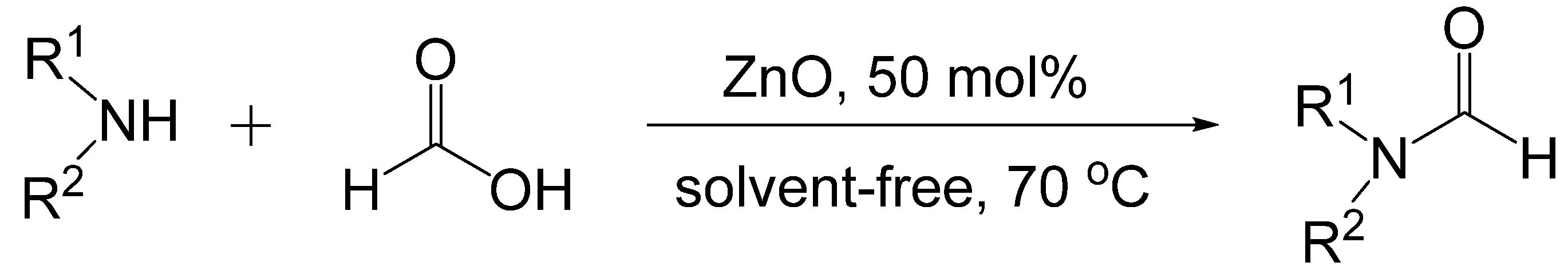{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
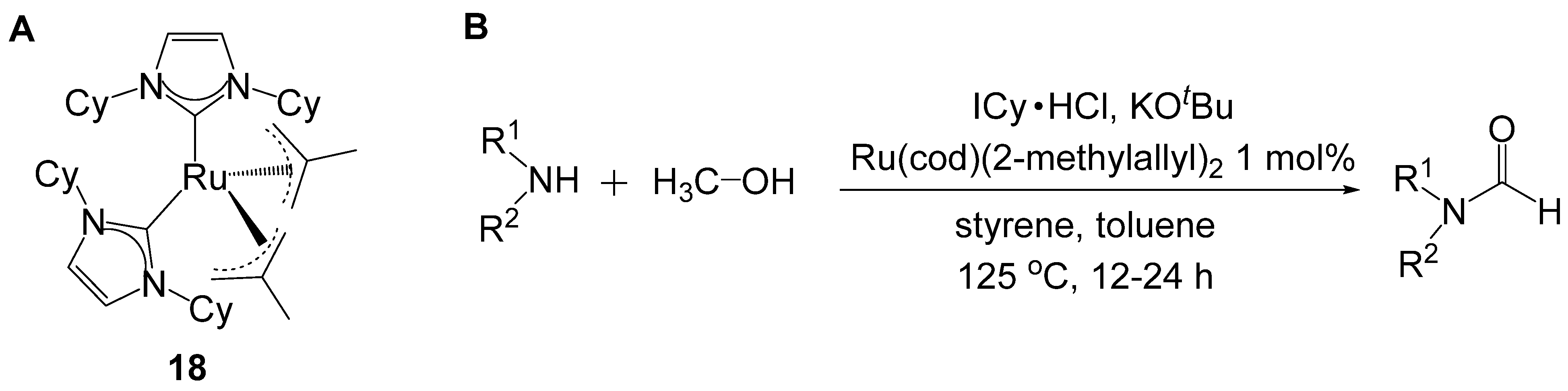{kind=link}
{kind=link}
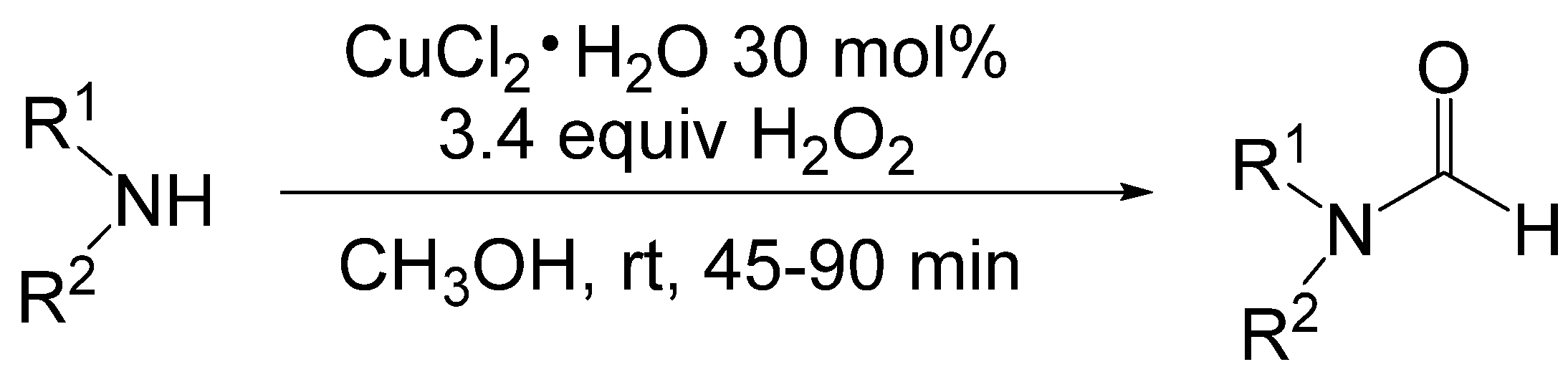{kind=link}
{kind=link}
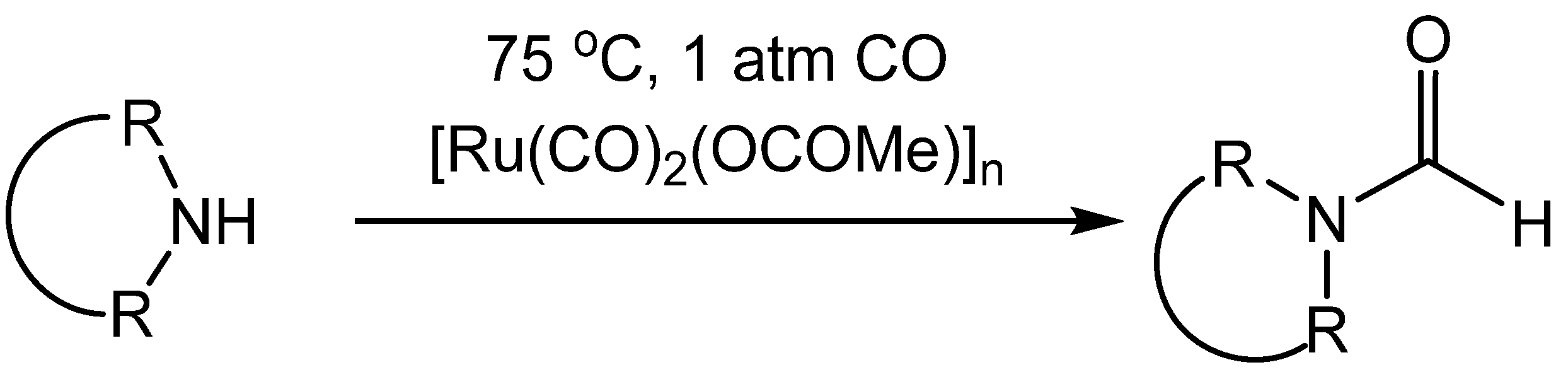{kind=link}
{kind=link}
{kind=link}
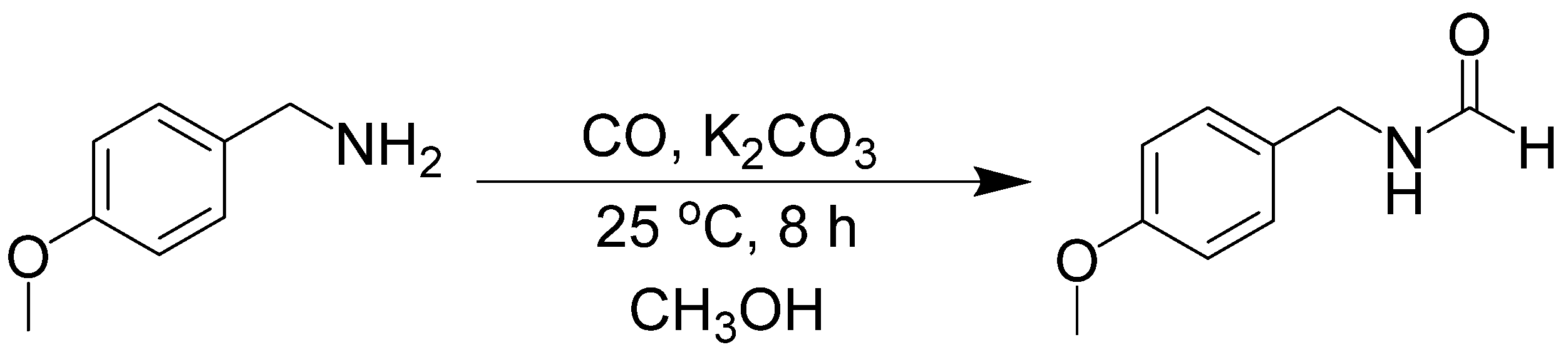{kind=link}
Abstract
:1. Introduction
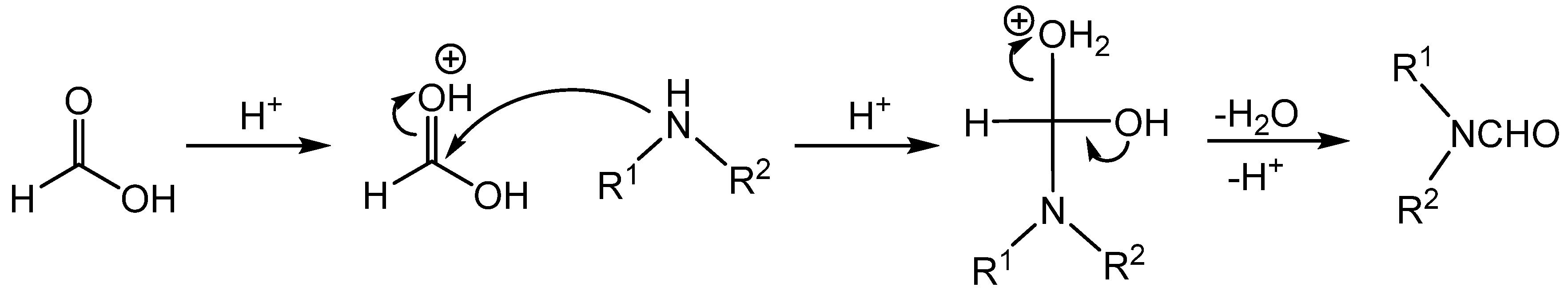
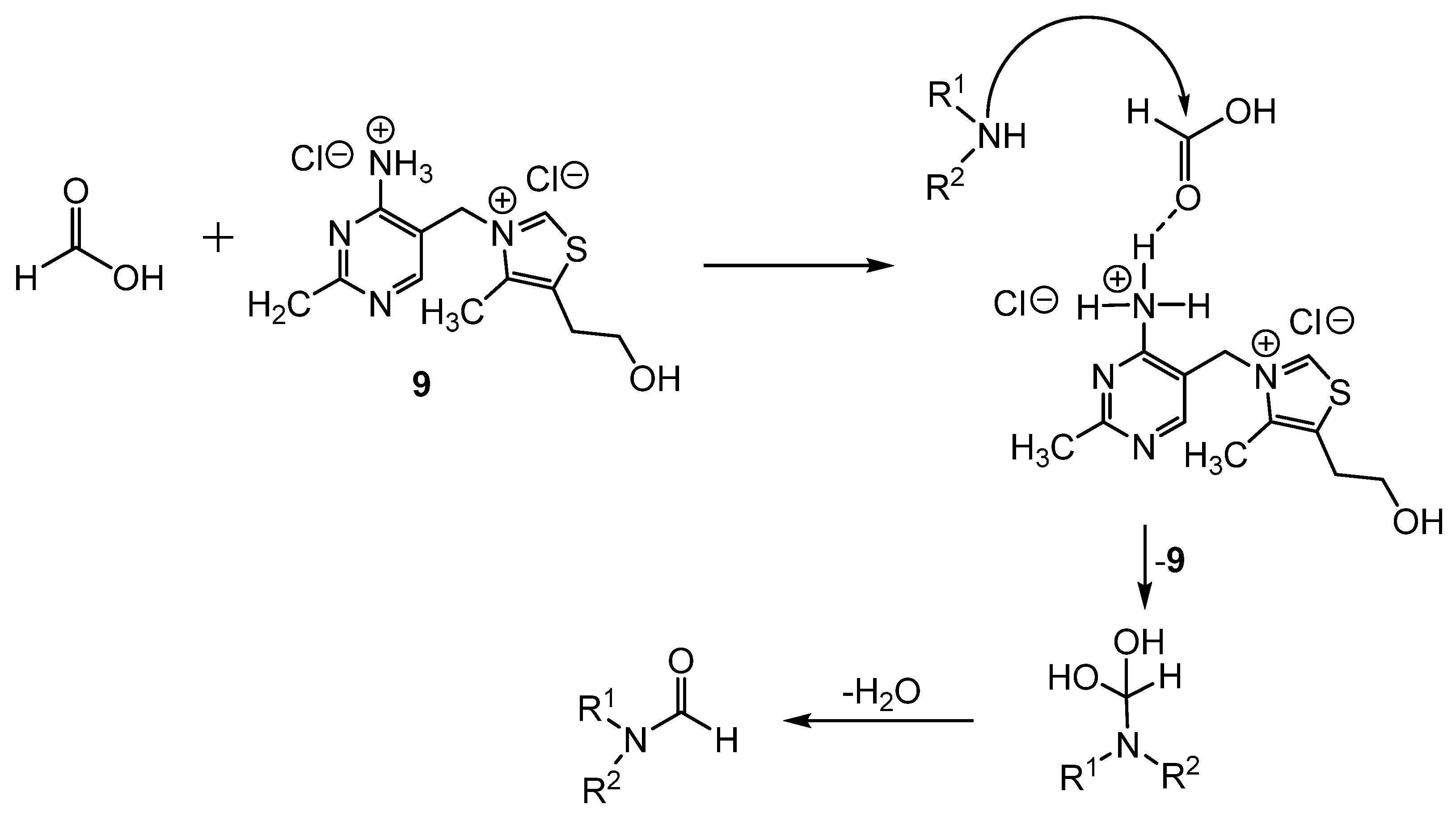
2. Stoichiometric Formylating Agents








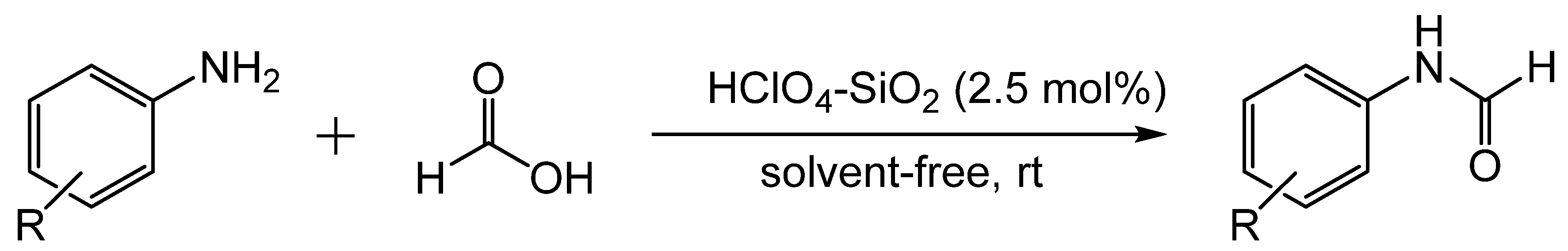
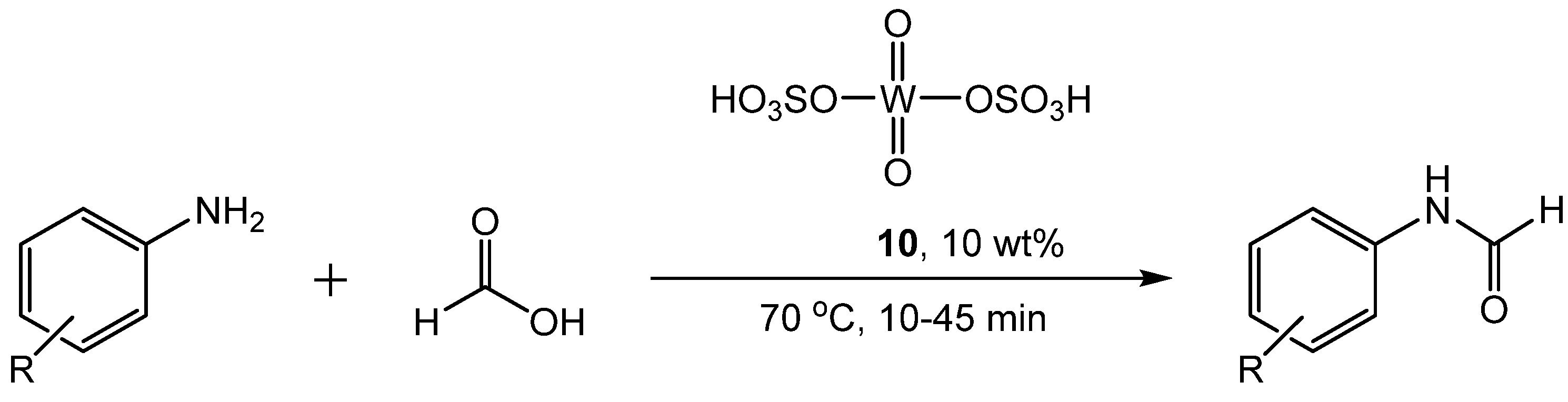
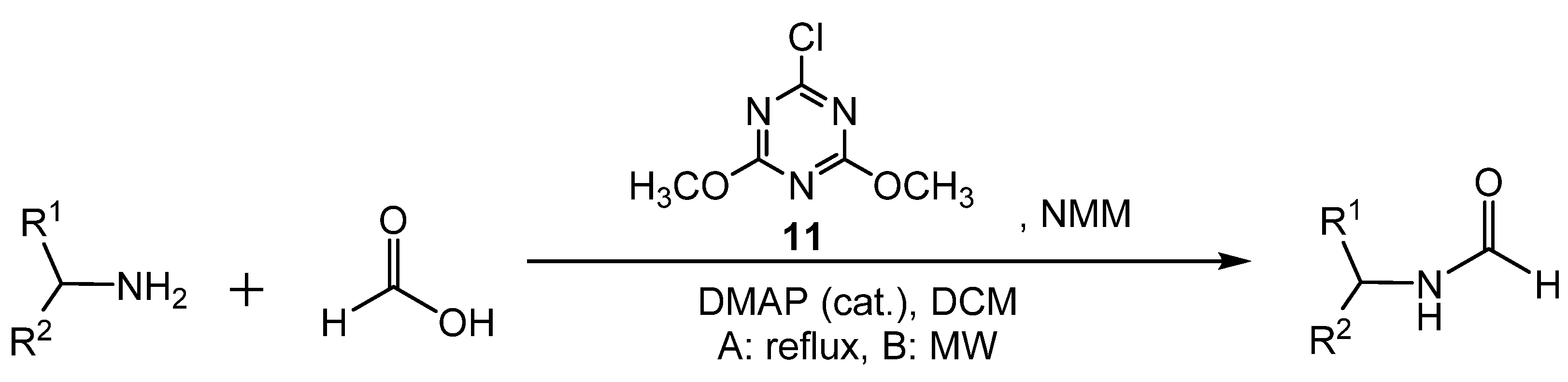
3. Catalytic Formylation with Acid Catalysts










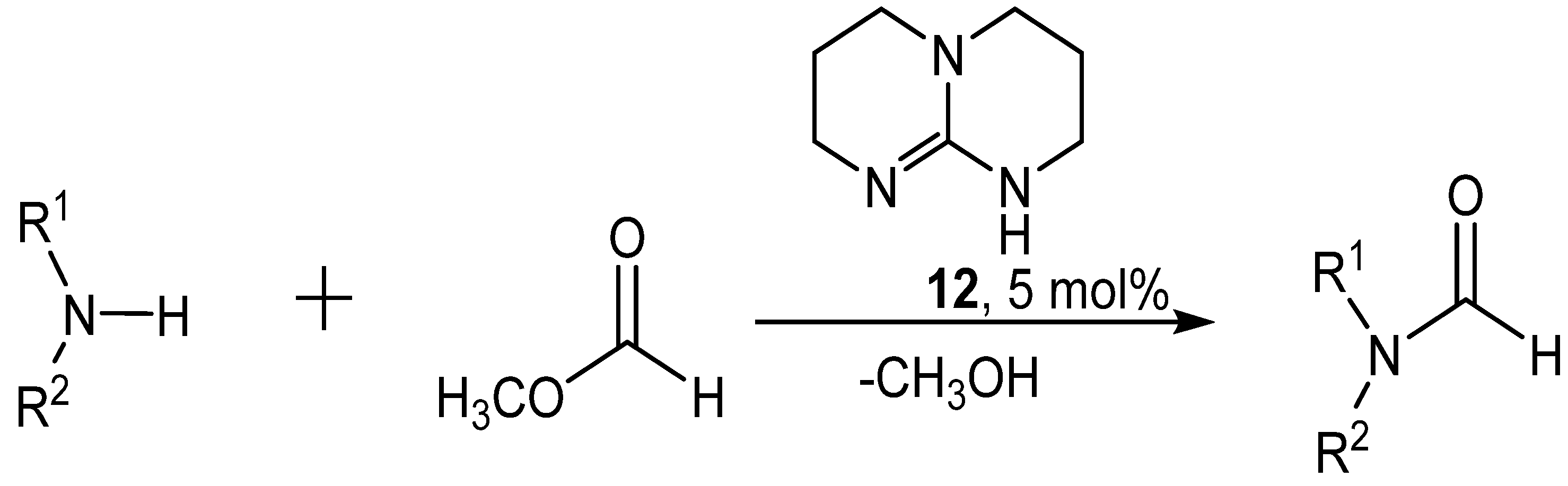
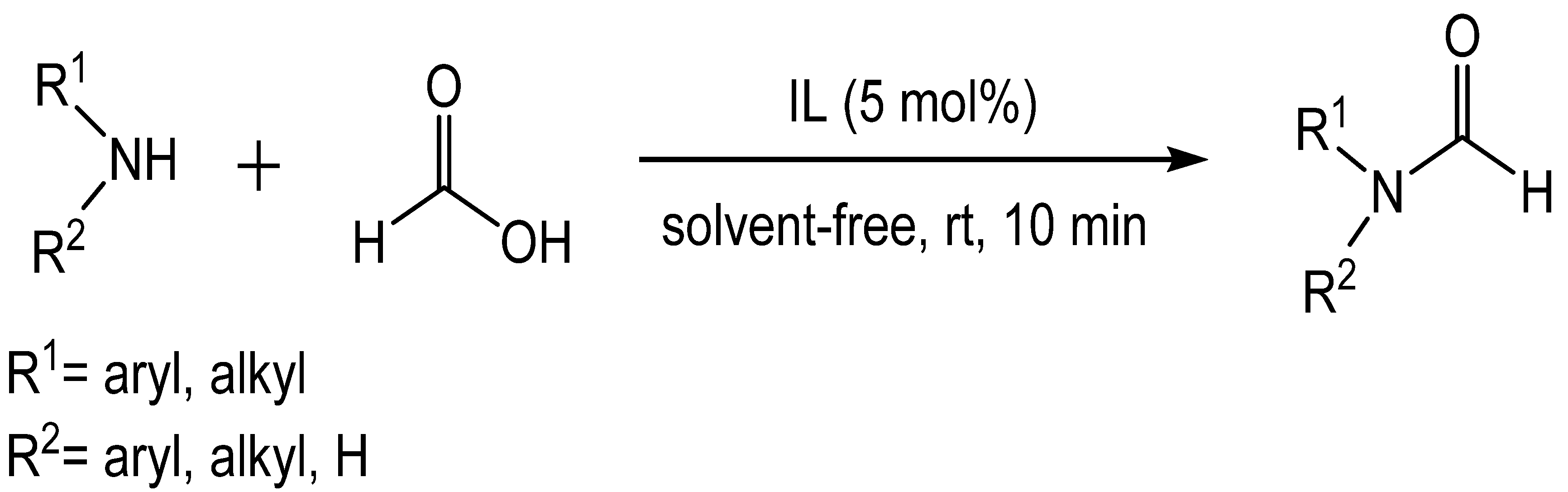
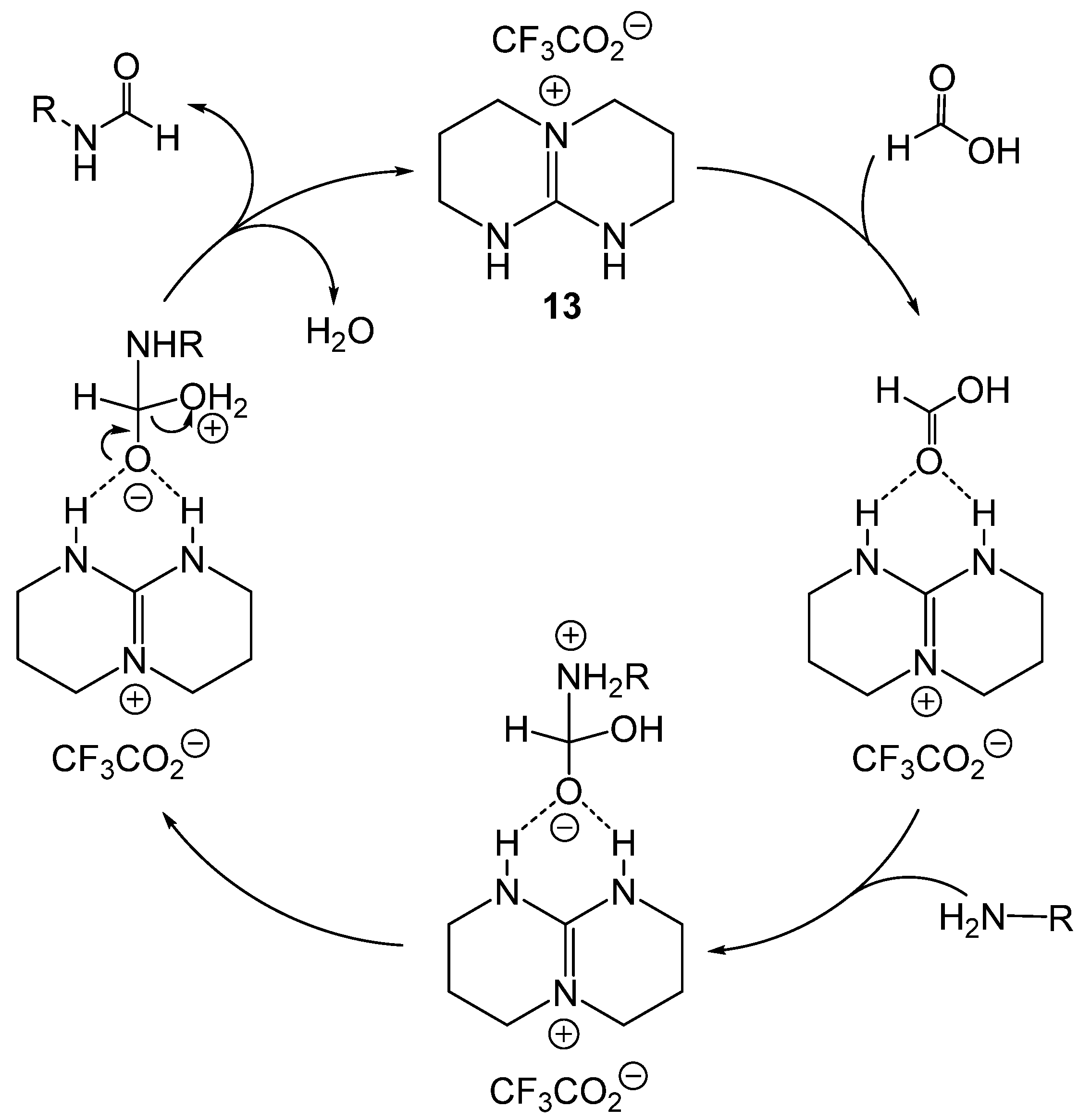
4. Catalytic Formylation with Organic Catalysts






5. Catalytic Formylation with Metal Catalysts













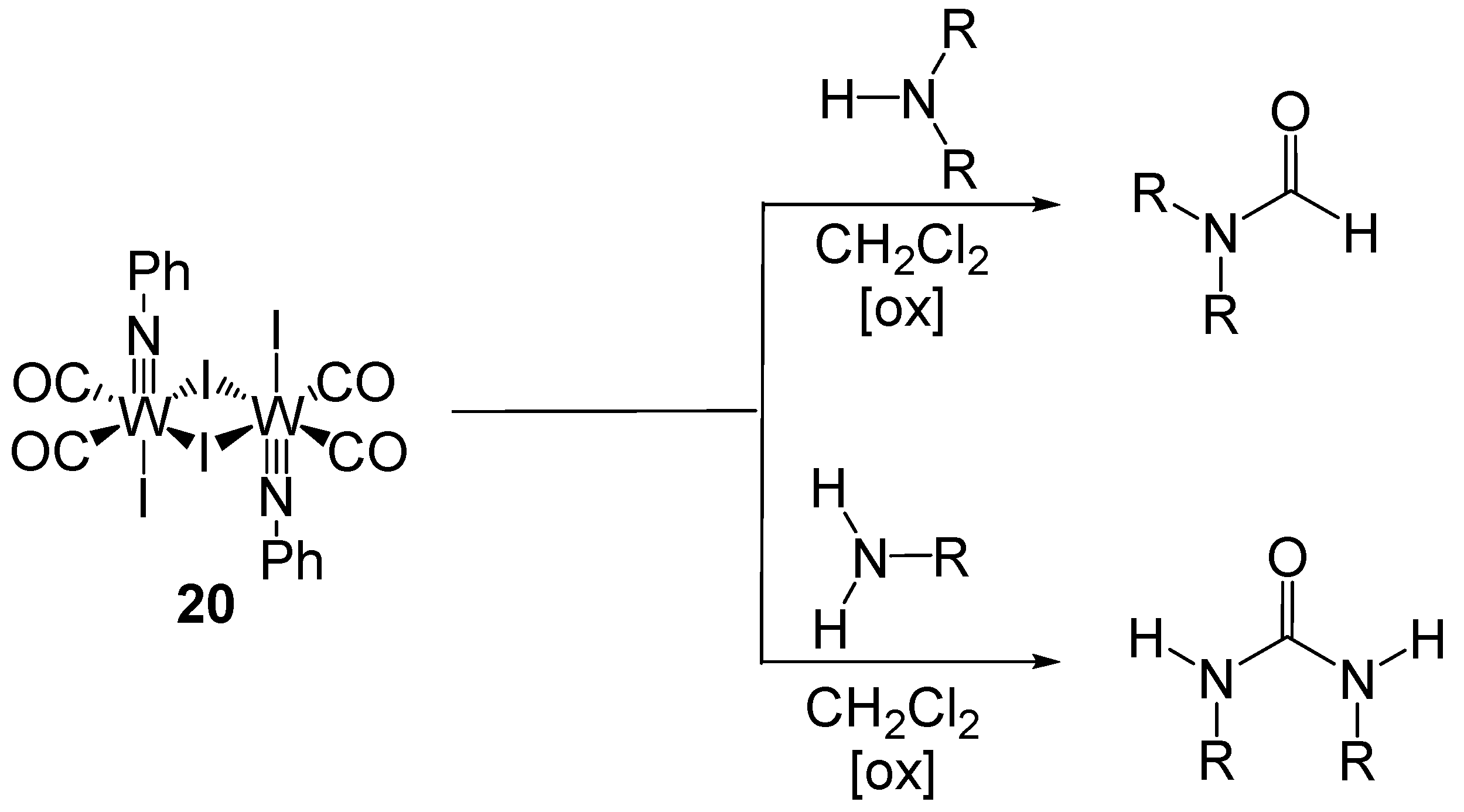
6. Formylation by Catalytic Carbonylation





7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chen, B.C.; Bednarz, M.S.; Zhao, R.; Sundeen, J.E.; Chen, P.; Shen, Z.; Skoumbourdis, A.P.; Barrish, J.C. A new facile method for the synthesis of 1-arylimidazole-5-carboxylates. Tetrahedron Lett. 2000, 41, 5453–5456. [Google Scholar] [CrossRef]
- Grant, H.G.; Summers, L.A. Synthesis of N-methyl-N-(2,2,2-trichloro-1-arylaminoethyl)tormamides and related-compounds as potential fungicides. Aust. J. Chem. 1980, 33, 613–617. [Google Scholar] [CrossRef]
- Kobayashi, K.; Nagato, S.; Kawakita, M.; Morikawa, O.; Konishi, H. Synthesis of 1-formyl-1,2-dihydroquinoline derivatives by a lewis acid-catalyzed cyclization of o-(1-hydroxy-2-alkenyl)phenyl isocyanides. Chem. Lett. 1995, 24, 575–576. [Google Scholar]
- Jackson, A.; Meth-Cohn, O. A new short and efficient strategy for the synthesis of quinolone antibiotics. J. Chem. Soc. Chem. Commun. 1995, 1319–1319. [Google Scholar] [CrossRef]
- Pettit, G.; Kalnins, M.; Liu, T.; Thomas, E.; Parent, K. Notes- potential cancerocidal agents. III. Formanilides. J. Org. Chem. 1961, 26, 2563–2566. [Google Scholar] [CrossRef]
- Faraj, M.K. Synthesis of Isocyanate Precursors from Primary Formamides. U.S. Patent 5,686,645, 1997. [Google Scholar]
- Han, Y.; Cai, L. An efficient and convenient synthesis of formamidines. Tetrahedron Lett. 1997, 38, 5423–5426. [Google Scholar] [CrossRef]
- Arlt, D.; Klein, G. Preparation of Nitriles from Formamides. U.S. Patent 4,419,297, 1983. [Google Scholar]
- Ding, S.; Jiao, N. N,N-dimethylformamide: A multipurpose building block. Angew. Chem. Int. Ed. 2012, 51, 9226–9237. [Google Scholar] [CrossRef]
- Downie, I.M.; Earle, M.J.; Heaney, H.; Shuhaibar, K.F. Vilsmeier formylation and glyoxylation reactions of nucleophilic aromatic compounds using pyrophosphoryl chloride. Tetrahedron 1993, 49, 4015–4034. [Google Scholar] [CrossRef]
- Kobayashi, S.; Nishio, K. Facile and highly stereoselective synthesis of homoallylic alcohols using organosilicon intermediates. J. Org. Chem. 1994, 59, 6620–6628. [Google Scholar] [CrossRef]
- Kobayashi, S.; Yasuda, M.; Hachiya, I. Trichlorosilane-dimethylformamide (Cl3SiH-DMF) as an efficient reducing agent. Reduction of aldehydes and imines and reductive amination of aldehydes under mild conditions using hypervalent hydridosilicates. Chem. Lett. 1996, 25, 407–408. [Google Scholar] [CrossRef]
- Blicke, F.F.; Lu, C.-J. Formylation of amines with chloral and reduction of the N-formyl derivatives with lithium aluminum hydride. J. Am. Chem. Soc. 1952, 74, 3933–3934. [Google Scholar] [CrossRef]
- Jung, S.H.; Ahn, J.H.; Park, S.K.; Choi, J.-K. A practical and convenient procedure for the N-formylation of amines using formic acid. Bull. Korean Chem. Soc. 2002, 23, 149–150. [Google Scholar] [CrossRef]
- Rahman, M.; Kundu, D.; Hajra, A.; Majee, A. Formylation without catalyst and solvent at 80 °C. Tetrahedron Lett. 2010, 51, 2896–2899. [Google Scholar] [CrossRef]
- Das, B.; Krishnaiah, M.; Balasubramanyam, P.; Veeranjaneyulu, B.; Nandan Kumar, D. A remarkably simple N-formylation of anilines using polyethylene glycol. Tetrahedron Lett. 2008, 49, 2225–2227. [Google Scholar]
- Strazzolini, P.; Giumanini, A.G.; Cauci, S. Acetic formic anhydride a review. Tetrahedron 1990, 46, 1081–1118. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Bringmann, G.; Lamotte, G.; Motherwell, W.B.; Motherwell, R.S.H.; Porter, A.E.A. Reactions of relevance to the chemistry of aminoglycoside antibiotics. Part 14. A useful radical-deamination reaction. J. Chem. Soc. Perkin 1 1980, 2657–2664. [Google Scholar]
- Evans, D.; Eastwood, F. Synthesis of an arylhydroxytetronimide and of 3-hydroxy-4(1H)-quinolone derivatives. Aust. J. Chem. 1974, 27, 537–542. [Google Scholar] [CrossRef]
- Terashima, S.; Takashima, K.; Sato, T.; Yamada, S.I. Stereochemical studies. XXII. Thermal rearrangement of s(−)-1-phenylethyl isocyanide. Chem. Pharm. Bull. 1973, 21, 1135–1139. [Google Scholar] [CrossRef]
- Johannsen, F.; Jorgensen, A.; Pedersen, E.B. Reactions of heterocyclic o-aminonitriles with acetic formic anhydride. Chem. Scr. 1986, 26, 347–351. [Google Scholar]
- Van Dort, M.; Neubig, R.; Counsell, R.E. Radioiodinated p-iodoclonidine. A high-affinity probe for the α2-adrenergic receptor. J. Med. Chem. 1987, 30, 1241–1244. [Google Scholar] [CrossRef]
- Nolte, R.J.M.; van Zomeren, J.A.J.; Zwikker, J.W. Poly(iminomethylenes). 6. Synthesis and polymerization of α- and β-d-glucopyranosyl isocyanide. J. Org. Chem. 1978, 43, 1972–1975. [Google Scholar] [CrossRef]
- Krishnamurthy, S. A highly efficient and general N-monomethylation of functionalized primary amines via formylation--borane:methyl sulfide reduction. Tetrahedron Lett. 1982, 23, 3315–3318. [Google Scholar]
- Du Vigneaud, V.; Dorfmann, R.; Loring, H.S. A comparison of the growth-promoting properties of d- and l-cystine. J. Biol. Chem. 1932, 98, 577–589. [Google Scholar]
- Sheehan, J.C.; Yang, D.D.H. The use of N-formylamino acids in peptide synthesis. J. Am. Chem. Soc. 1958, 80, 1154–1158. [Google Scholar] [CrossRef]
- Waki, M.; Meienhofer, J. Efficient preparation of nα-formylamino acid tert-butyl esters. J. Org. Chem. 1977, 42, 2019–2020. [Google Scholar] [CrossRef]
- Chen, F.M.F.; Benoiton, N.L. A general method for formylating sensitive amino acid esters. Synthesis 1979, 1979, 709–710. [Google Scholar] [CrossRef]
- Duczek, W.; Deutsch, J.; Vieth, S.; Niclas, H.J. A simple and convenient synthesis of N-formyl amino acid esters under mild conditions. Synthesis 1996, 1996, 37–38. [Google Scholar]
- Ganapati Reddy, P.; Kishore Kumar, G.D.; Baskaran, S. A convenient method for the N-formylation of secondary amines and anilines using ammonium formate. Tetrahedron Lett. 2000, 41, 9149–9151. [Google Scholar] [CrossRef]
- Shastri, L.A.; Shastri, S.L.; Bathula, C.D.; Basanagouda, M.; Kulkarni, M.V. Mild, simple, and efficient method for N-formylation of secondary amines via Reimer-Tiemann reaction. Synth. Commun. 2009, 41, 476–484. [Google Scholar]
- Yang, X.; Zhang, Y. Melamine trisulfonic acid-catalyzed N-formylation of amines under solvent-free conditions. Res. Chem. Intermed. 2012, 1–6. [Google Scholar]
- Swaringen, R.A.; Eaddy, J.F.; Henderson, T.R. Reaction of ortho esters with secondary amines. J. Org. Chem. 1980, 45, 3986–3989. [Google Scholar] [CrossRef]
- Kaboudin, B.; Khodamorady, M. Organic reactions in water: A practical and convenient method for the N-formylation of amines in water. Synlett 2010, 2010, 2905–2907. [Google Scholar] [CrossRef]
- Brahmachari, G.; Laskar, S. A very simple and highly efficient procedure for N-formylation of primary and secondary amines at room temperature under solvent-free conditions. Tetrahedron Lett. 2010, 51, 2319–2322. [Google Scholar] [CrossRef]
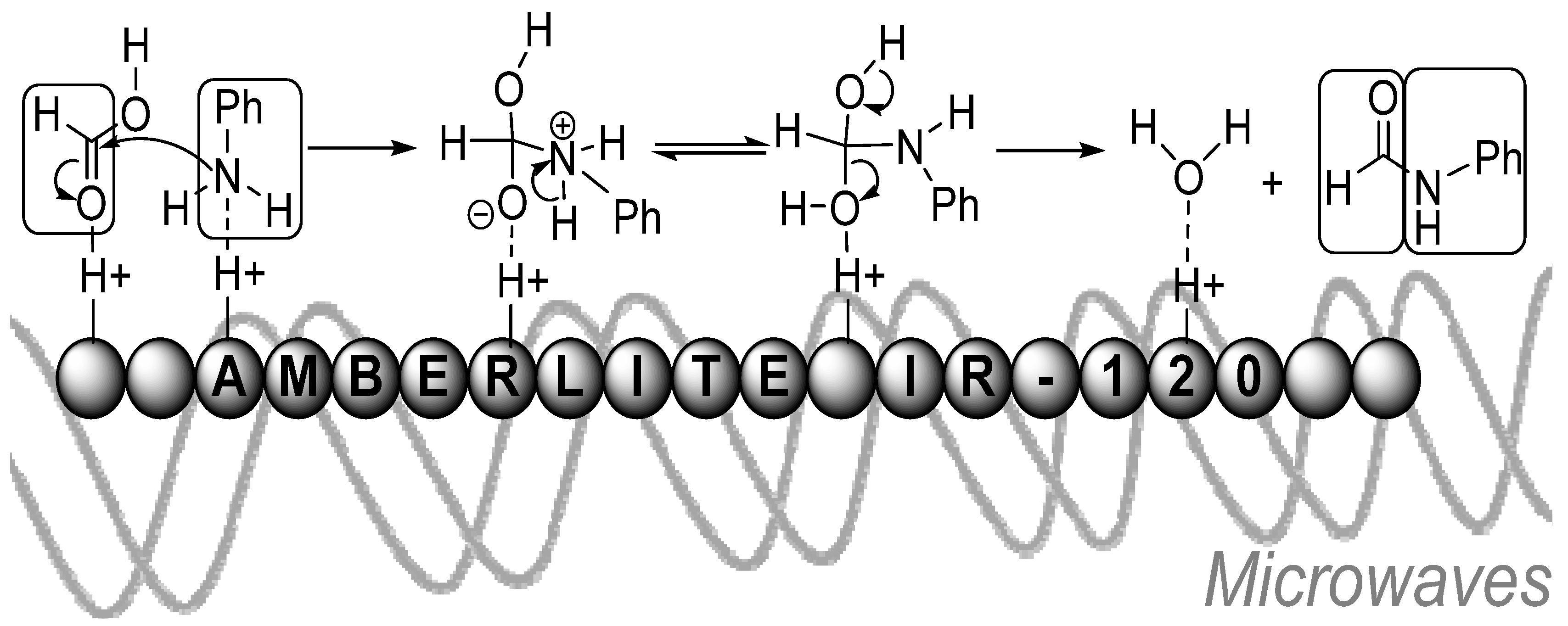
- Muthukur Bhojegowd, M.R.; Nizam, A.; Pasha, M.A. Amberlite IR-120: A reusable catalyst for N-formylation of amines with formic acid using microwaves. Chin. J. Catal. 2010, 31, 518–520. [Google Scholar] [CrossRef]
- Kim, J.G.; Jang, D.O. Facile and highly efficient N-formylation of amines using a catalytic amount of iodine under solvent-free conditions. Synlett 2010, 2010, 2093–2096. [Google Scholar] [CrossRef]
- Hammick, D.L.; Zvegintzov, M. CXLIV.-The rate of reaction between formic acid and iodine in aqueous solution. J. Chem. Soc. 1926, 129, 1105–1108. [Google Scholar] [CrossRef]
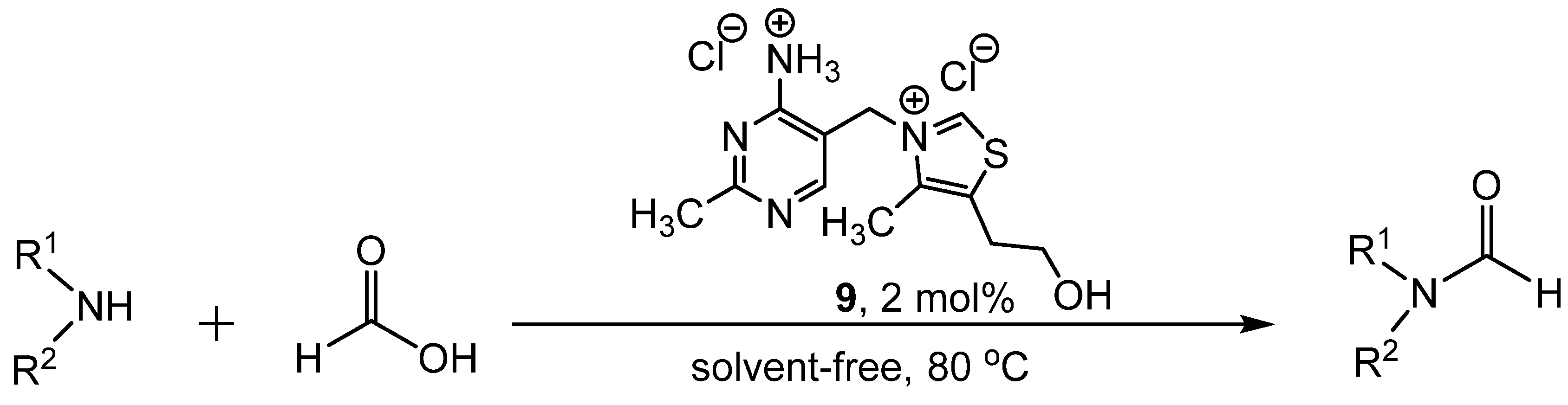
- Lei, M.; Ma, L.; Hu, L. A convenient one-pot synthesis of formamide derivatives using thiamine hydrochloride as a novel catalyst. Tetrahedron Lett. 2010, 51, 4186–4188. [Google Scholar] [CrossRef]
- Ansari, M.I.; Hussain, M.K.; Yadav, N.; Gupta, P.K.; Hajela, K. Silica supported perchloric acid catalyzed rapid N-formylation under solvent-free conditions. Tetrahedron Lett. 2012, 53, 2063–2065. [Google Scholar] [CrossRef]
- Ma'mani, L.; Sheykhan, M.; Heydari, A.; Faraji, M.; Yamini, Y. Sulfonic acid supported on hydroxyapatite-encapsulated-γ-Fe2O3 nanocrystallites as a magnetically Brønsted acid for N-formylation of amines. Appl. Catal. A 2010, 377, 64–69. [Google Scholar] [CrossRef]
- Pathare, S.P.; Sawant, R.V.; Akamanchi, K.G. Sulfated tungstate catalyzed highly accelerated N-formylation. Tetrahedron Lett. 2012, 53, 3259–3263. [Google Scholar] [CrossRef]
- De Luca, L.; Giacomelli, G.; Porcheddu, A.; Salaris, M. A new, simple procedure for the synthesis of formyl amides. Synlett 2004, 2004, 2570–2572. [Google Scholar]
- Deutsch, J.; Eckelt, R.; Köckritz, A.; Martin, A. Catalytic reaction of methyl formate with amines to formamides. Tetrahedron 2009, 65, 10365–10369. [Google Scholar] [CrossRef]
- Baghbanian, S.M.; Farhang, M. Protic [TBD][TFA] ionic liquid as a reusable and highly efficient catalyst for N-formylation of amines using formic acid under solvent-free condition. J. Mol. Liq. 2013, 183, 45–49. [Google Scholar] [CrossRef]
- Kim, J.-G.; Jang, D.O. Indium-catalyzed N-formylation of amines under solvent-free conditions. Synlett 2010, 2010, 1231–1234. [Google Scholar] [CrossRef]
- Hosseini-Sarvari, M.; Sharghi, H. ZnO as a new catalyst for N-formylation of amines under solvent-free conditions. J. Org. Chem. 2006, 71, 6652–6654. [Google Scholar] [CrossRef]
- Shekhar, A.C.; Kumar, A.R.; Sathaiah, G.; Paul, V.L.; Sridhar, M.; Rao, P.S. Facile N-formylation of amines using lLewis acids as novel catalysts. Tetrahedron Lett. 2009, 50, 7099–7101. [Google Scholar] [CrossRef]
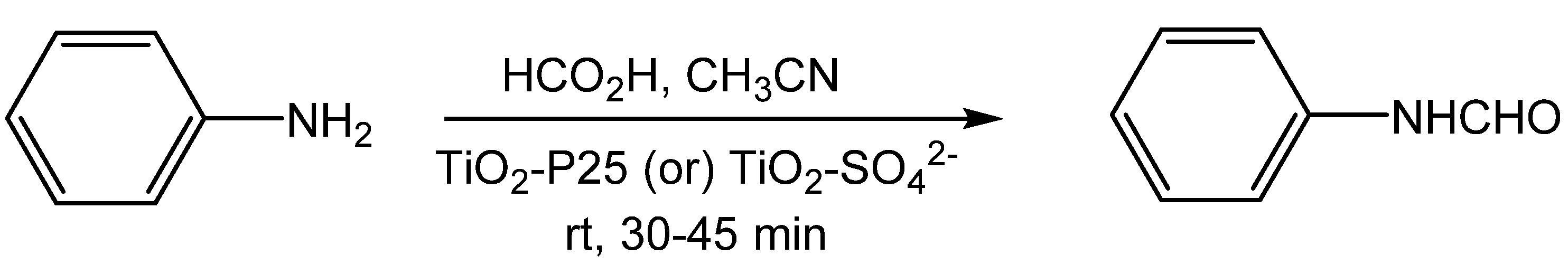
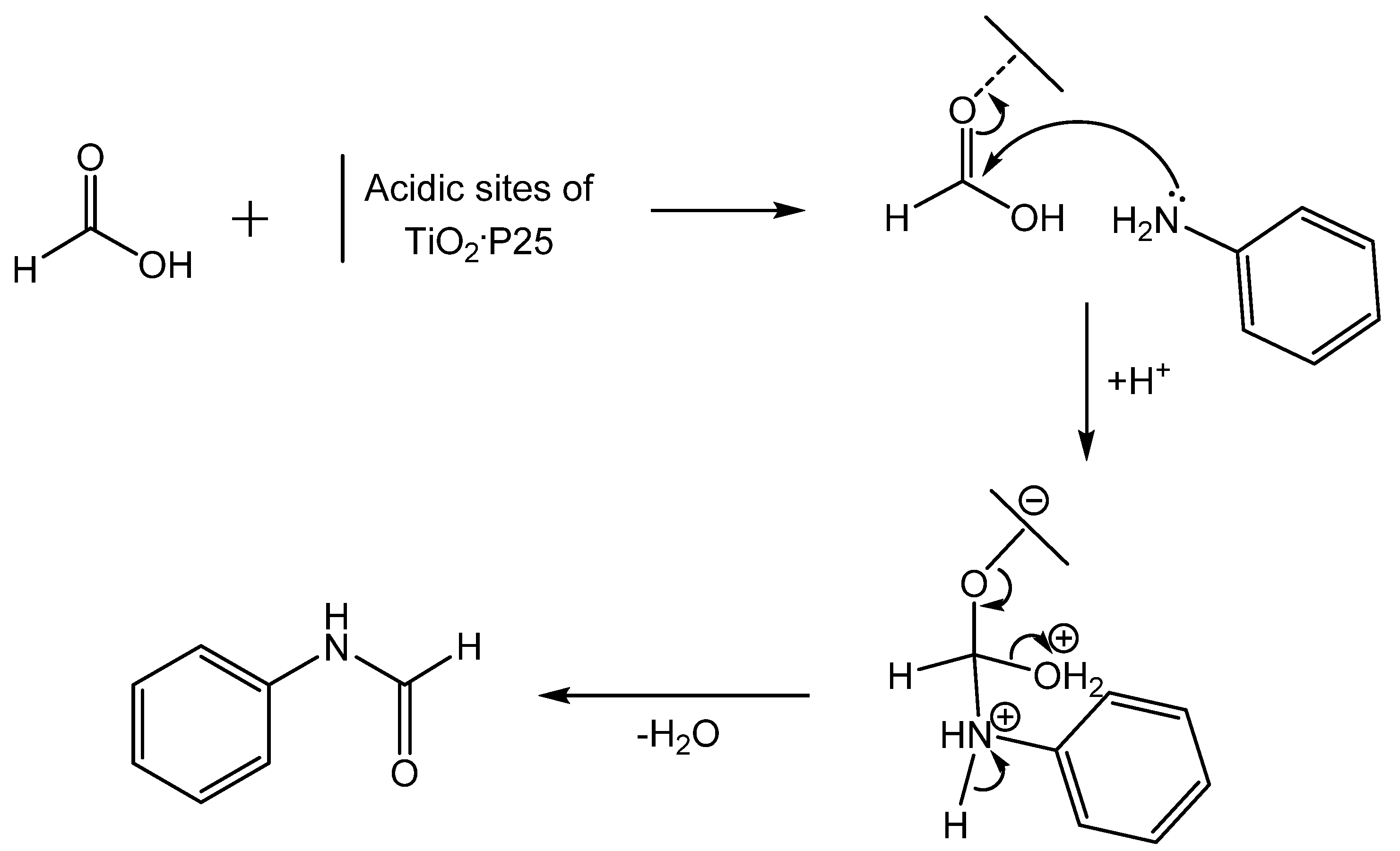
- Krishnakumar, B.; Swaminathan, M. A convenient method for the N-formylation of amines at room temperature using TiO2-P25 or sulfated titania. J. Mol. Catal. A Chem. 2011, 334, 98–102. [Google Scholar]
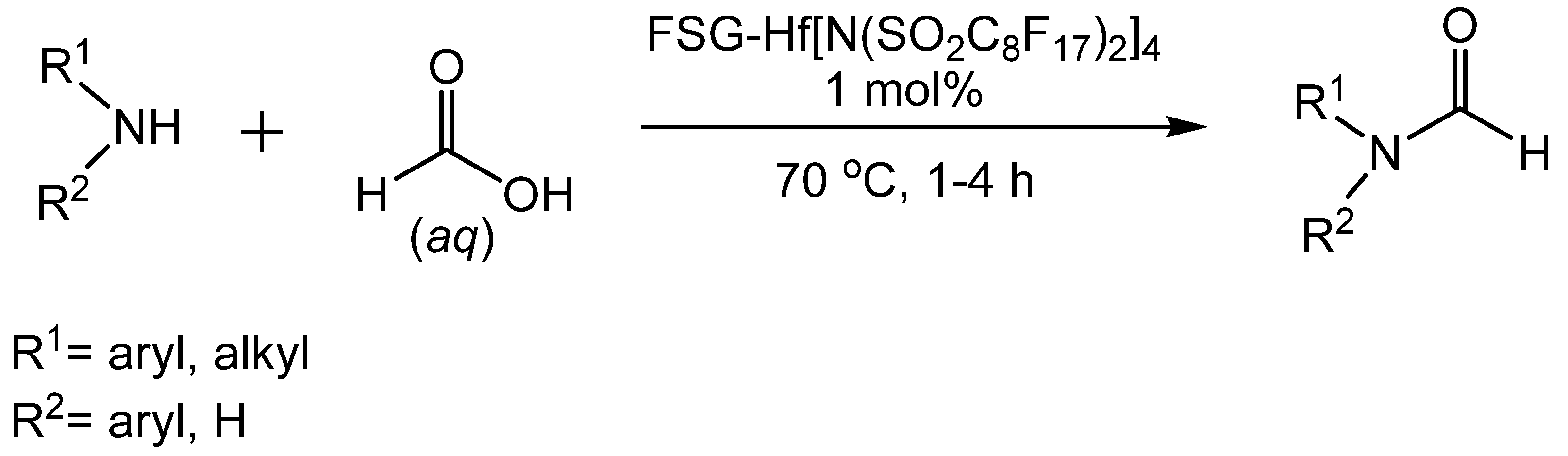
- Hong, M.; Xiao, G. Hafnium(IV) bis(perfluorooctanesulfonyl)imide complex supported on fluorous silica gel catalyzed N-formylation of amines using aqueous formic acid. J. Fluorine Chem. 2013, 146, 11–14. [Google Scholar] [CrossRef]
- Saidi, O.; Bamford, M.J.; Blacker, A.J.; Lynch, J.; Marsden, S.P.; Plucinski, P.; Watson, R.J.; Williams, J.M.J. Iridium-catalyzed formylation of amines with paraformaldehyde. Tetrahedron Lett. 2010, 51, 5804–5806. [Google Scholar] [CrossRef]
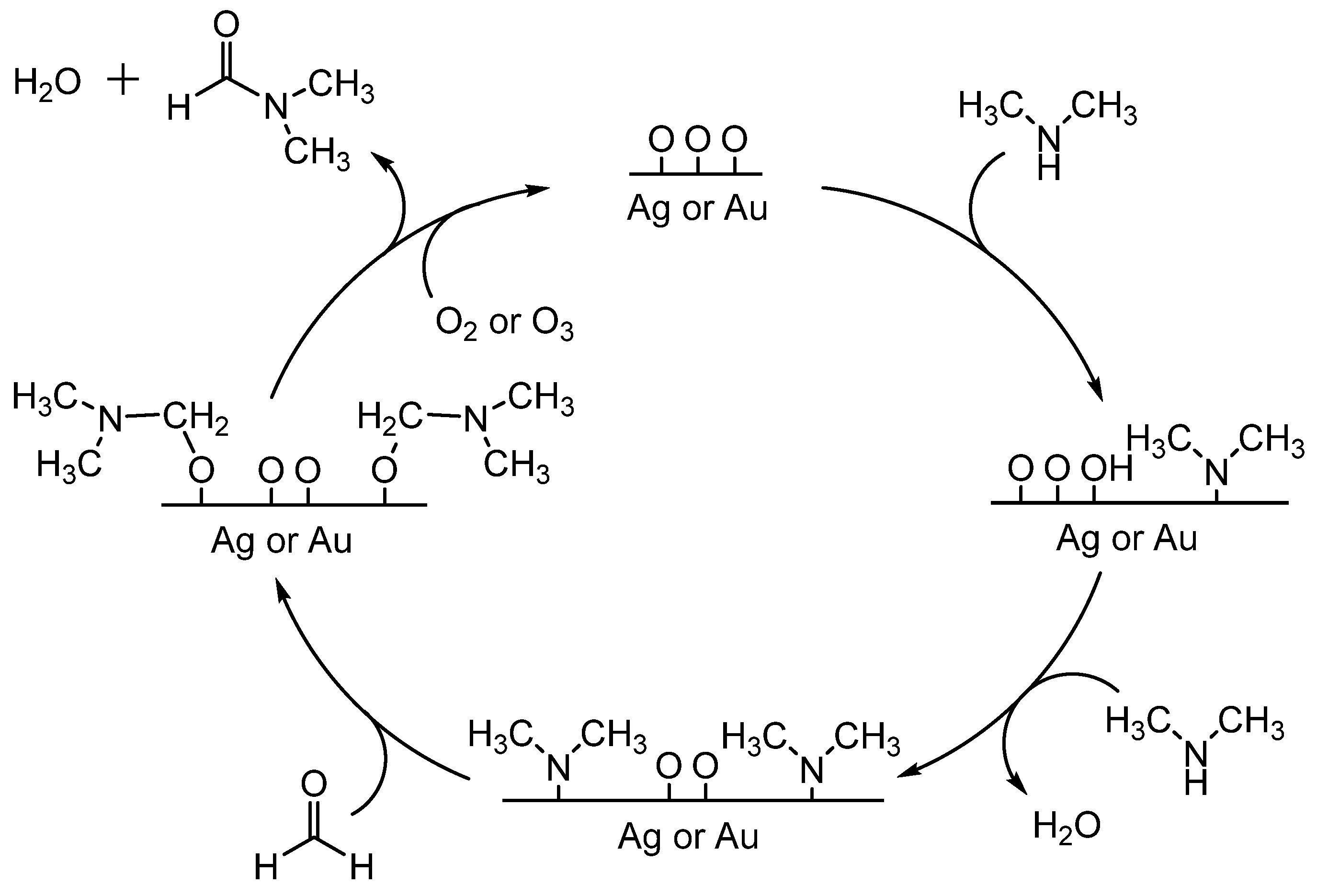
- Zhou, L.; Freyschlag, C.G.; Xu, B.; Friend, C.M.; Madix, R.J. Direct selective oxygen-assisted acylation of amines driven by metallic silver surfaces: Dimethylamine with formaldehyde. Chem. Commun. 2010, 46, 704–706. [Google Scholar] [CrossRef]
- Xu, B.; Zhou, L.; Madix, R.J.; Friend, C.M. Highly selective acylation of dimethylamine mediated by oxygen atoms on metallic gold surfaces. Angew. Chem. Int. Ed. 2010, 49, 394–398. [Google Scholar] [CrossRef]
- Ishida, T.; Haruta, M. N-formylation of amines via the aerobic oxidation of methanol over supported gold nanoparticles. ChemSusChem 2009, 2, 538–541. [Google Scholar] [CrossRef]
- Preedasuriyachai, P.; Kitahara, H.; Chavasiri, W.; Sakurai, H. N-formylation of amines catalyzed by nanogold under aerobic oxidation conditions with MeOH or formalin. Chem. Lett. 2010, 39, 1174–1176. [Google Scholar] [CrossRef]
- Ortega, N.; Richter, C.; Glorius, F. N-formylation of amines by methanol activation. Org. Lett. 2013, 15, 1776–1779. [Google Scholar] [CrossRef]
- Tumma, H.; Nagaraju, N.; Reddy, K.V. A facile method for the N-formylation of primary and secondary amines by liquid phase oxidation of methanol in the presence of hydrogen peroxide over basic copper hydroxyl salts. J. Mol. Catal. A Chem. 2009, 310, 121–129. [Google Scholar]
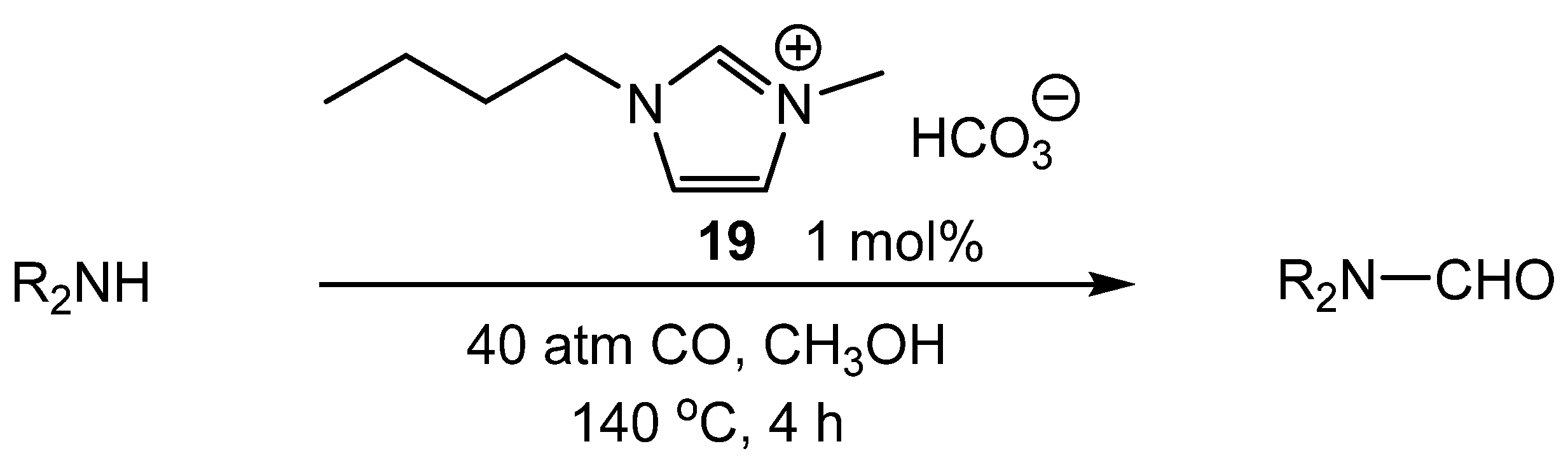
- Choi, Y.S.; Shim, Y.N.; Lee, J.; Yoon, J.H.; Hong, C.S.; Cheong, M.; Kim, H.S.; Jang, H.G.; Lee, J.S. Ionic liquids as benign catalysts for the carbonylation of amines to formamides. Appl. Catal. A 2011, 404, 87–92. [Google Scholar] [CrossRef]
- Jenner, G.; Bitsi, G. Ruthenium-cobalt and ruthenium-cobalt-promoted carbonylation of primary and secondary-amines. Appl. Catal. 1987, 32, 293–304. [Google Scholar] [CrossRef]
- Bitsi, G.; Jenner, G. Solvent effect in the ruthenium catalyzed carbonylation of amines. Selective synthesis of dialkylformamides. J. Organomet. Chem. 1987, 330, 429–435. [Google Scholar] [CrossRef]
- Byerley, J.J.; Rempel, G.L.; Takebe, N.; James, B.R. Catalytic carbonylation of amines using ruthenium complexes under mild conditions. J. Chem. Soc. D 1971, 1482–1483. [Google Scholar]
- Ovchinnikov, M.V.; Guzei, I.A.; Angelici, R.J. Amine attack on the carbonyl ligands of the protonated dicyclopentadienyl-bridged diruthenium complex [{(η5-C5H3)2(SiMe2)2}Ru2(Co)4(μ-H)]+. Organometallics 2001, 20, 691–696. [Google Scholar] [CrossRef]
- McCusker, J.E.; Abboud, K.A.; McElwee-White, L. Carbonylation of amines with a tungsten(IV) carbonyl complex. Organometallics 1997, 16, 3863–3866. [Google Scholar] [CrossRef]
- Süss-Fink, G.; Langenbahn, M.; Jenke, T. Rutheniumcluster als Katalysatoren für die Carbonylierung von cyclischen Aminen. J. Organomet. Chem. 1989, 368, 103–109. [Google Scholar] [CrossRef]
- Jenner, G.; Bitsi, G.; Schleiffer, E. Ruthenium-catalyzed carbonylation of cyclic amines. J. Mol. Catal. 1987, 39, 233–236. [Google Scholar]
- Benedini, F.; Nali, M.; Rindone, B.; Tollari, S.; Cenini, S.; Lamonica, G.; Porta, F. The bis(salicylaldehyde)ethylenediimine cobalt(II)-catalyzed oxidative carbonylation of primary and secondary-amines. J. Mol. Catal. 1986, 34, 155–161. [Google Scholar] [CrossRef]
- Dombek, B.D.; Angelici, R.J. Pentacarbonyliron-catalyzed carbonylation of amines to formamides. J. Catal. 1977, 48, 433–435. [Google Scholar] [CrossRef]
- Gabriele, B.; Salerno, G.; Costa, M. Oxidative carbonylations. In Catalytic Carbonylation Reactions; Beller, M., Ed.; Springer: Heidelberg, Germany, 2006; pp. 239–272. [Google Scholar]
- Díaz, D.J.; Darko, A.K.; McElwee-White, L. Transition metal-catalyzed oxidative carbonylation of amines to ureas. Eur. J. Org. Chem. 2007, 4453–4465. [Google Scholar]
- Ragaini, F. Away from phosgene: Reductive carbonylation of nitroarenes and oxidative carbonylation of amines, understanding the mechanism to improve performance. Dalton Trans. 2009, 2009, 6251–6266. [Google Scholar] [CrossRef]
- Gabriele, B.; Mancuso, R.; Salerno, G. Oxidative carbonylation as a powerful tool for the direct synthesis of carbonylated heterocycles. Eur. J. Org. Chem. 2012, 6825–6839. [Google Scholar] [CrossRef]
- Saegusa, T.; Kobayashi, S.; Hirota, K.; Ito, Y. Synthetic reactions by complex catalysts. XIII. Carbonylation of amines by group IB and IIB metal compound catalysts. Bull. Chem. Soc. Jap. 1969, 42, 2610–2614. [Google Scholar] [CrossRef]
- Tsuji, Y.; Ohsumi, T.; Kondo, T.; Watanabe, Y. Dodecacarbonyltriruthenium catalyzed carbonylation of amines and hydroamidation of olefins. J. Organomet. Chem. 1986, 309, 333–344. [Google Scholar] [CrossRef]
- McCusker, J.E.; Logan, J.; McElwee-White, L. Oxidative carbonylation of primary amines to ureas using tungsten carbonyl catalysts. Organometallics 1998, 17, 4037–4041. [Google Scholar] [CrossRef]
- McCusker, J.E.; Main, A.D.; Johnson, K.S.; Grasso, C.A.; McElwee-White, L. W(CO)6-catalyzed oxidative carbonylation of primary amines to N,N'-disubstituted ureas in single or biphasic solvent systems. Optimization and functional group compatibility studies. J. Org. Chem. 2000, 65, 5216–5222. [Google Scholar] [CrossRef]
- McCusker, J.E.; Qian, F.; McElwee-White, L. Catalytic oxidative carbonylation of aliphatic secondary amines to tetrasubstituted ureas. J. Mol. Catal. A Chem. 2000, 159, 11–17. [Google Scholar] [CrossRef]
- McCusker, J.E.; Grasso, C.A.; Main, A.D.; McElwee-White, L. Catalytic oxidative carbonylation of primary and secondary α, ω-diamines to cyclic ureas. Org. Lett. 1999, 1, 961–964. [Google Scholar] [CrossRef]
- Qian, F.; McCusker, J.E.; Zhang, Y.; Main, A.D.; Chlebowski, M.; Kokka, M.; McElwee-White, L. Catalytic oxidative carbonylation of primary and secondary diamines to cyclic ureas. Optimization and substituent studies. J. Org. Chem. 2002, 67, 4086–4092. [Google Scholar]
- Hylton, K.G.; Main, A.D.; McElwee-White, L. Catalytic carbonylation of functionalized diamines: Application to the core structure of DMP 323 and DMP 450. J. Org. Chem. 2003, 68, 1615–1617. [Google Scholar] [CrossRef]
- Darko, A.K.; Curran, F.C.; Copin, C.; McElwee-White, L. Carbonylation of functionalized diamine diols to cyclic ureas: Application to derivatives of DMP 450. Tetrahedron 2011, 67, 3976–3983. [Google Scholar] [CrossRef]
- Zhang, L.; Darko, A.K.; Johns, J.I.; McElwee-White, L. Catalytic oxidative carbonylation of arylamines to ureas with W(CO)6/I2 as catalyst. Eur. J. Org. Chem. 2011, 6261–6268. [Google Scholar]
- Díaz, D.J.; Hylton, K.G.; McElwee-White, L. Selective catalytic oxidative carbonylation of amino alcohols to ureas. J. Org. Chem. 2006, 71, 734–738. [Google Scholar] [CrossRef]
- Dumbris, S.M.; Díaz, D.J.; McElwee-White, L. Preparation of hydantoins by catalytic oxidative carbonylation of α-amino amides. J. Org. Chem. 2009, 74, 8862–8865. [Google Scholar] [CrossRef]
- Shelton, P.A.; Zhang, Y.; Nguyen, T.H.H.; McElwee-White, L. NaIO4-oxidized carbonylation of amines to ureas. Chem. Commun. 2009, 2009, 947–949. [Google Scholar]
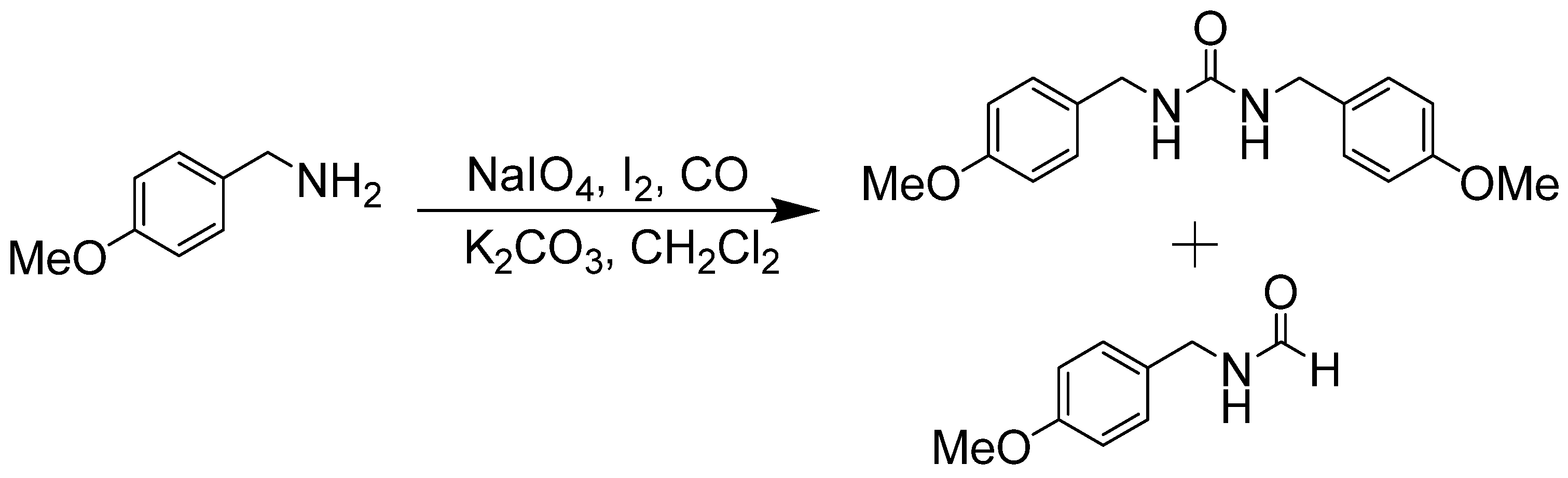
- Gerack, C.J.; McElwee-White, L. Oxidative carbonylation of amines to formamides using NaIO4. Chem. Commun. 2012, 48, 11310–11312. [Google Scholar] [CrossRef]
- Gerack, C.J.; Johns, J.I.; McElwee-White, L. Base-mediated carbonylation of amines to formamides. Department of Chemistry, University of Florida, Gainesville, FL 32611-7200, USA. 2014; Unpublished work. [Google Scholar]
© 2014 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gerack, C.J.; McElwee-White, L. Formylation of Amines. Molecules 2014, 19, 7689-7713. https://doi.org/10.3390/molecules19067689
Gerack CJ, McElwee-White L. Formylation of Amines. Molecules. 2014; 19(6):7689-7713. https://doi.org/10.3390/molecules19067689
Chicago/Turabian StyleGerack, Ciera J., and Lisa McElwee-White. 2014. "Formylation of Amines" Molecules 19, no. 6: 7689-7713. https://doi.org/10.3390/molecules19067689
APA StyleGerack, C. J., & McElwee-White, L. (2014). Formylation of Amines. Molecules, 19(6), 7689-7713. https://doi.org/10.3390/molecules19067689