2. Results and Discussion
Phytochemical investigation of the ethyl acetate-methanol extract obtained from the mixture of leaves and twigs of
P. trimera led to the isolation of two alkaloids, namely 8-hydroxy-1,4,5-trimethoxy-7-oxoaporphine (
1) and 1,2,3-trimethoxy-4,5-dioxo-6a,7-dehydroaporphine (
2). The structures of aporphine alkaloids
1 and
2 (
Figure 1) were elucidated by spectroscopic methods, including
1H-NMR,
13C-NMR, UV, IR, MS.
Figure 1.
Structures of the isolated aporphine alkaloids.
Figure 1.
Structures of the isolated aporphine alkaloids.
Compound
1 crystallized as orange crystals, m.p. 251–252 °C and its molecular formula was determined to be C
19H
15NO
5 by HR-ESIMS. Moreover, its UV spectrum exhibited absorption peaks at 210, 241, 317 and 417 nm indicating the presence of a highly conjugated system. The IR absorptions for OH (3249 cm
−1), C=O (1705 cm
−1), aromatic (1658, 1559, 1508, 1458 cm
−1) and ether moieties (1281, 1211 cm
−1) were also observed. From the UV and IR spectral data it was indicated that
1 was an oxoaporphine derivative [
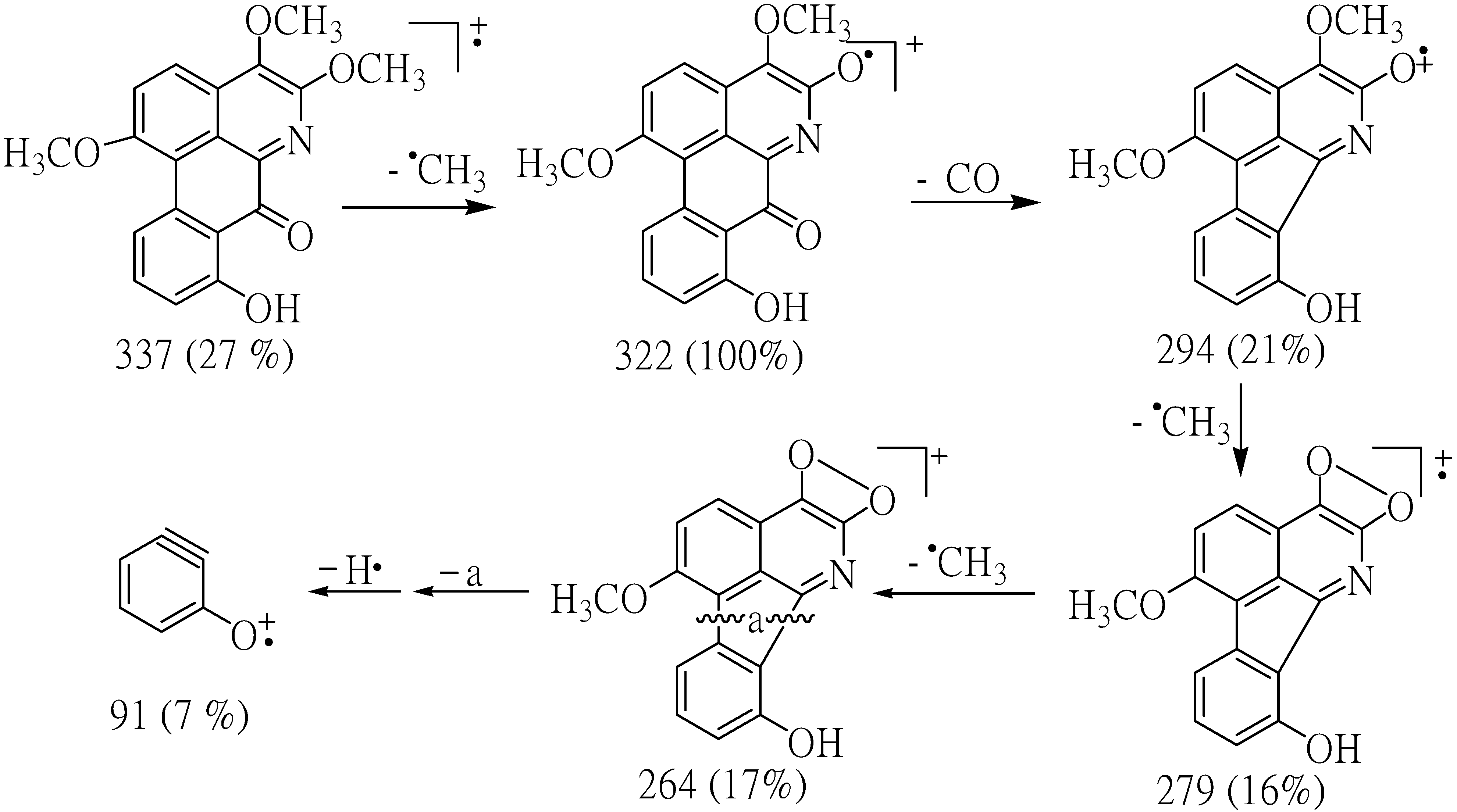
9]. The fragment ions at
m/z 337 (M
+) and 322 (M
+-Me) in the mass spectrum of compound
1 indicated the presence of OMe group in the position next to N in the structure. The results from decarbonylation cleavage at
m/z 294 indicated the presence of carbonyl group in ring C. The ion of
m/z 91 supported that
1 contained one hydroxyl group in ring D (
Figure 2).
Figure 2.
Most important observed fragmentations for compound 1.
Figure 2.
Most important observed fragmentations for compound 1.
The
1H-NMR spectrum of the major alkaloid, in addition to three methoxy (
δ 4.10,
s), (3.99,
s) and 3.97,
s) groups, had three obscured signals at
δ 7.57, 7.60 and 7.62, and two multiplet signals at
δ 7.88 and 9.34 in aromatic ring region, so adding together hydroxyl group (12.02,
s), this accounts for all 15 protons. A downfield singlet proton at
δ 12.02 was assigned to a hydroxyl group at the C-8 position due to the formation of hydrogen bonding with neighboring carbonyl group at C-7. Moreover, the sequential correlations of aromatic proton signals at C-9 (
δ 7.57), C-10 (
δ 7.88) and C-11 (
δ 9.34) on ring D were clearly proven by the COSY spectrum. Additionally, the aromatic proton at C-2 (
δ 7.60) and C-3 (
δ 7.62) are also clearly supported by this COSY spectrum and HMBC correlation with C-1a ,C-3 (strong correlation) and C-1a (weak correlation), C-2, respectively (
Figure 3). Moreover, the identification of H-9, 10, 11 in ring D was determined by NOE difference spectra. Irradiation of H-8 (OH) signal showed enhancement of the H-9, thus indicating that it definitely has one proton next to the OH group. In addition, irradiation at the methoxy signal of position 1 also showed enhancement of H-11. Accordingly, the H-10 was confirmed at this position since there was NOE enhancement with the H-9 (
Figure 3). Correspondingly, the presence of a hydroxyl group in the molecule located in the ring D at C-8 was established on the basis of long-range
1H-
13C correlation of the HO-8 at
δ 12.02 with the carbon C-9 (
δ 113.76), C-7 (
δ 175.55). The
13C-NMR spectrum exhibited the presence of three methoxyl group, five methine, and eleven quaternary carbons (
Table 1). In comparison with the literature data [
10] the keto group at C-7 position usually resonance at
δ 175 indicating the existence of this carbonyl at the peri-position.
Figure 3.
NOE experiment, significant correlations in the COSY and HMBC spectra.
Figure 3.
NOE experiment, significant correlations in the COSY and HMBC spectra.
Table 1.
1H-NMR (500 MHz), 13C-NMR (125 MHz) in DMSO-d6 1 and CDCl3 for 2.
Table 1.
1H-NMR (500 MHz), 13C-NMR (125 MHz) in DMSO-d6 1 and CDCl3 for 2.
| Position | 8-Hydroxy-1,4,5-trimethoxy-7-oxoaporphine (1) | 1,2,3-Trimethoxy-4,5-dioxo-6a,7-dehydroaporphine (2) |
|---|
| *
δ 1H m (J Hz) | δ 13C(DEPT) | *
δ 1H m (J Hz) | δ 13C(DEPT) |
|---|
| 1 | - | 159.15 (C) | - | 158.66 (C) |
| 1a | - | 131.58 (C) | - | 116.26 (C) |
| 1b | - | 120.21 (C) | - | 120.31 (C) |
| 2 | 7.60
obsc. | 127.51 (CH) | - | 147.54 (C) |
| 3 | 7.62
obsc. | 126.87 (CH) | - | 160.44 (C) |
| 3a | - | 117.65 (C) | - | 117.62 (C) |
| 4 | - | 147.02 (C) | - | 175.41 (C) |
| 5 | - | 157.49 (C) | - | 157.63 (C) |
| 6a | - | 129.82 (C) | - | 128.38 (C) |
| 7 | - | 175.55 (C) | 7.83
s | 116.0 (CH) |
| 7a | - | 119.21 (C) | - | 131.77 (C) |
| 8 | - | 155.80 (C) | 7.98
m | 128.48 (CH) |
| 9 | 7.57
obsc. | 113.76 (CH) | 7.65
obsc. | 127.57 (CH) |
| 10 | 7.88
m | 128.17 (CH) | 7.66
obsc. | 127.44 (CH) |
| 11 | 9.34
m | 126.81 (CH) | 9.5
m | 127.24 (CH) |
| 11a | - | 125.91 (C) | - | 121.23 (C) |
| 1-OMe | 4.10
s | 61.09 (CH3) | 4.17
s | 62.08 (CH3) |
| 2-OMe | - | - | 4.10
s | 61.74 (CH3) |
| 3-OMe | - | - | 4.21
s | 61.17 (CH3) |
| 4-OMe | 3.99
s | 61.59 (CH3) | - | - |
| 5-OMe | 3.97
s | 61.65 (CH3) | - | - |
| 8-OH | 12.02
s | - | - | - |
| N-H | - | - | 11.77
s | - |
In addition, the HMBC spectra gave further support for carbonyl group by correlations with hydroxy proton. The methoxy group at C-1 position of ring A was confirmed by NOE enhancement with H-11. In addition, the methoxy group at C-4 and 5 position of ring B confirmed by direct comparison of those chemical shifts with the
13C-NMR of artabonatine C, a compound isolated from
Artabotrys uncinatus [
10]. According to the
1H and
13C-NMR 1D/2D data this compound was identified as the oxoaporphine alkaloid. On the basis of the above data, the structure for
1 was formulated as 8-hydroxy-1,4,5-trimethoxy-7-oxoaporphine. It is a new alkaloid and does not appear to have been previously isolated from this species. Chromatographic separation of the ethyl acetate extract afforded compound
2. This compound is isomeric with compound
1. The structure was established by comparison of their UV, IR, EIMS, and 1D, 2D NMR data with the literature data [
4,
7,
11].
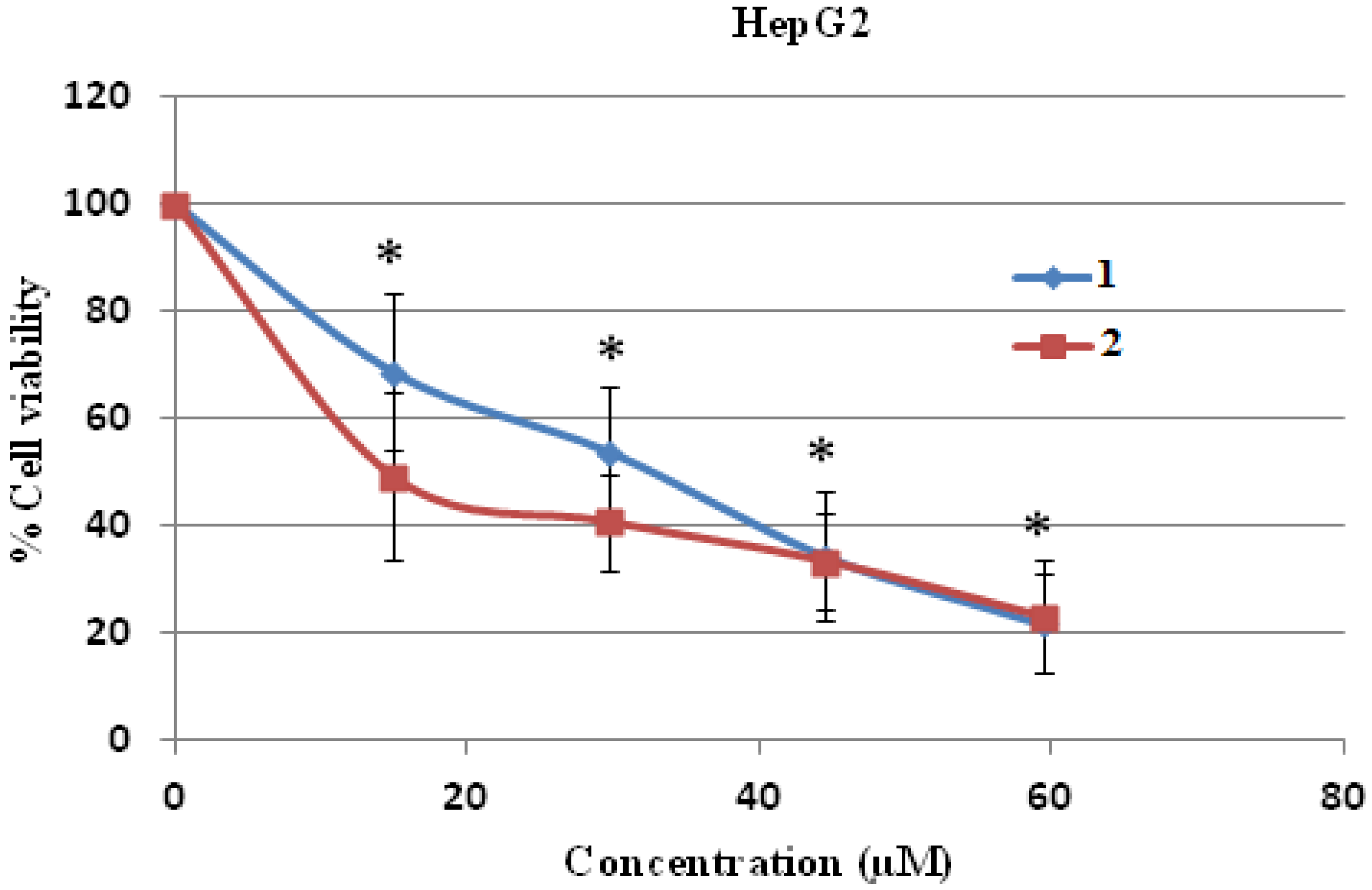
The cytotoxicity test was performed by the MTT assay and it was found that compounds
1 and
2 were toxic to both human hepatocellular carcinoma HepG2 and human breast cancer MDA-MB231 cell lines dose dependently, and these toxicities were statistically and significantly different at the IC
10, IC
20 and IC
50 levels when compared to control (without treatment) (
Figure 4 and
Figure 5). Compound
2 was more cytotoxic to HepG2 cells than compound
1, with an IC
50 concentration of 12.88 ± 2.49 μM compared to 26.36 ± 5.18 μM, respectively, and the IC
50 values of both compounds were significantly different when compared to each other (
Table 2). Both IC
50 levels of compound
1 and
2 were statistically different compared to those of doxorubicin in both cells (
Table 2 and
Table 3).
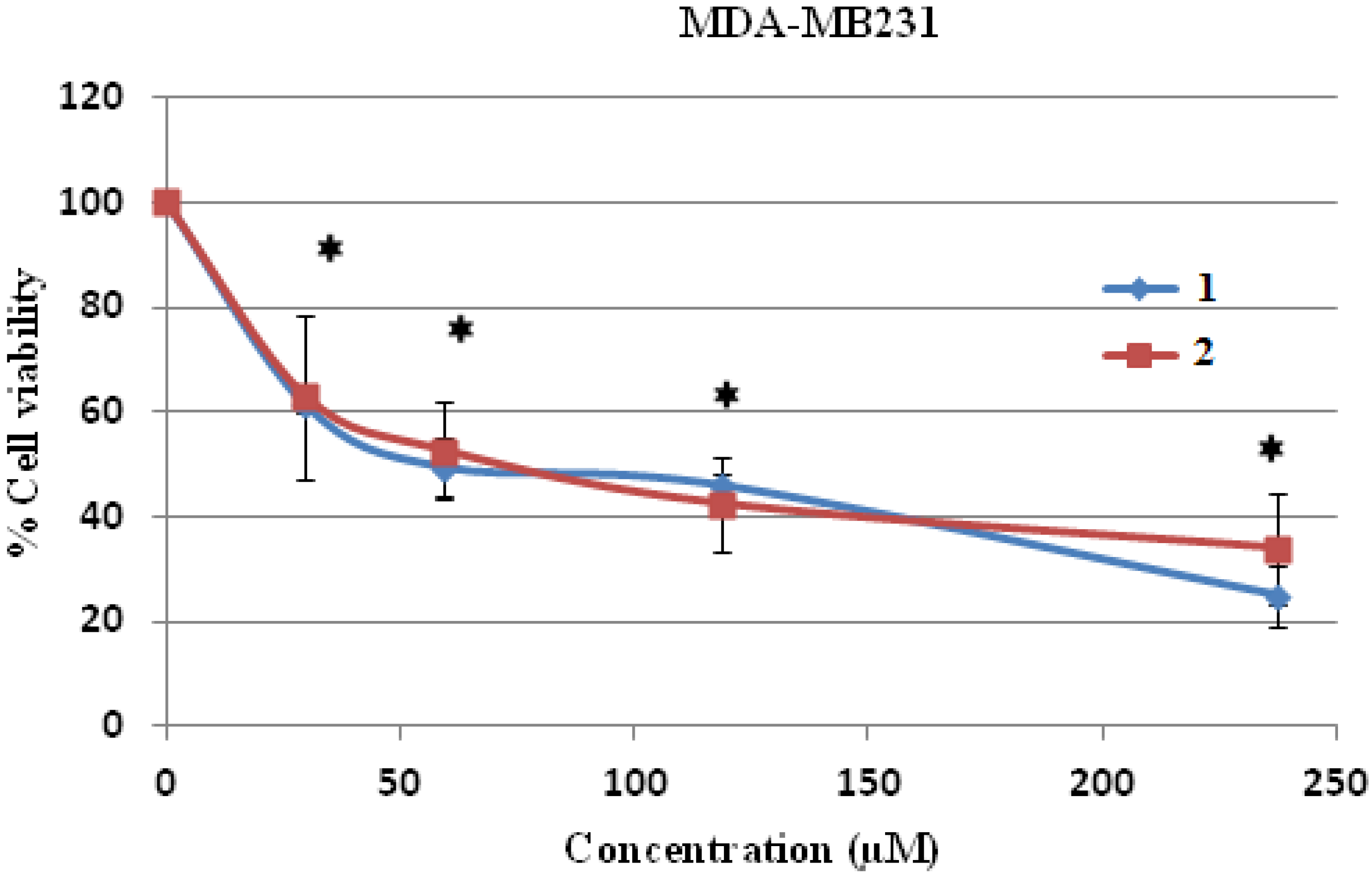
However, when human breast cancer MDA-MB231 cells were treated with both compounds, it was shown that both compound
1 and
2 were cytotoxic to the MDA-MB231 cells up to the tested concentration of 240 μM (
Figure 5). The IC
50 levels of both compound
1 and
2 on human breast cancer MDA-MB231 cells were 64.75 ± 4.45 and 67.06 ± 3.5 μM, respectively (
Table 3). It could be concluded that human hepatocellular carcinoma HepG2 cells were more sensitive to both compounds than human breast cancer MD-MB231 cells at the lower IC
50 value level.
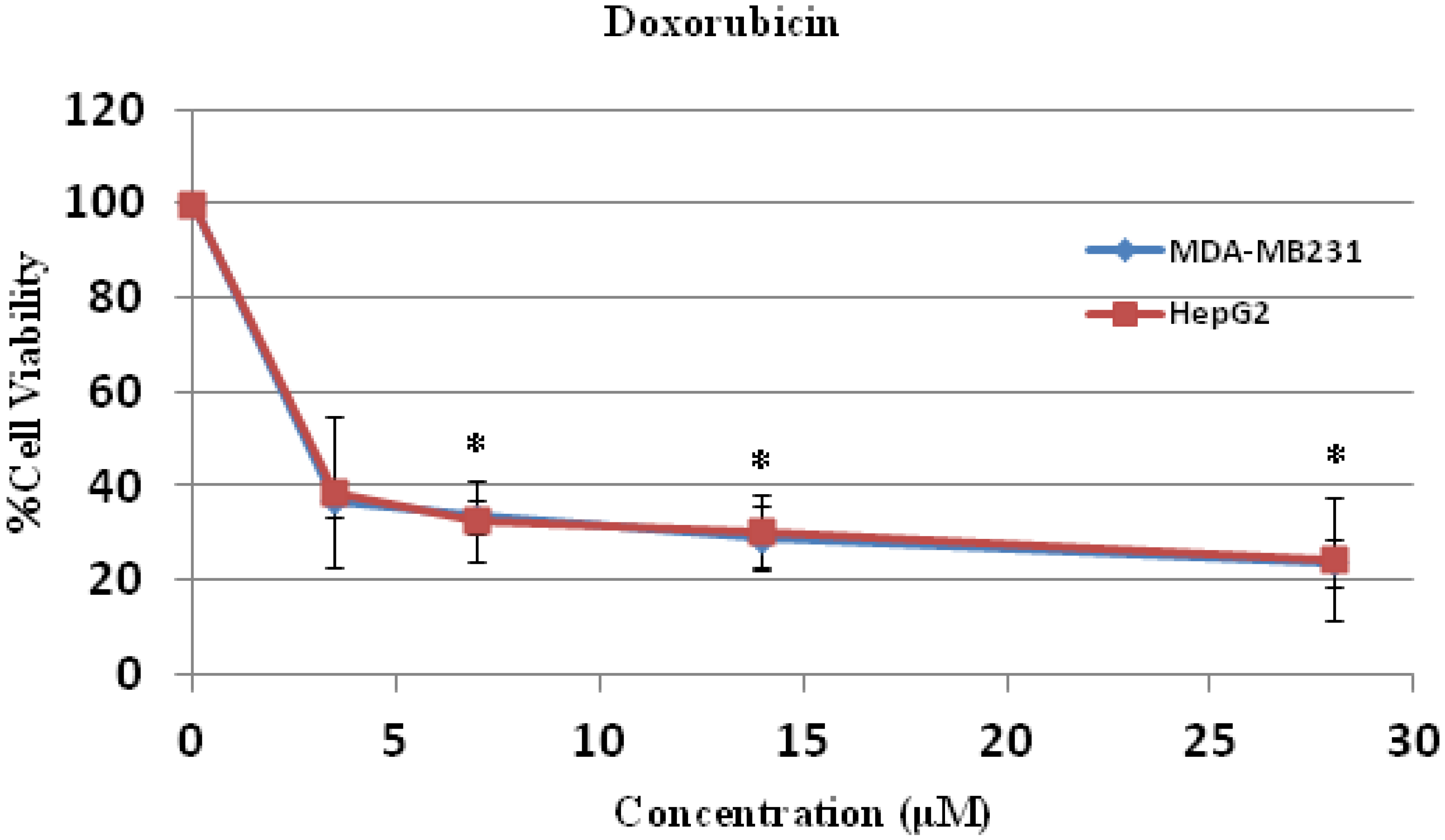
The anti-cancer activities of both compounds in both cell lines were less potent when compared to doxorubicin (
Figure 6,
Table 2 and
Table 3). The mechanisms of action of doxorubicin are DNA intercalation [
12], increased free radical production [
13], and inhibition of topoisomerase II progression [
14]. Doxorubicin is used for treatment of leukemia, lymphoma and several solid tumors, such as osteosarcoma. It also induces apoptosis and necrosis in healthy tissue of the brain, liver, kidney and heart. The drug influences Bcl-2/Bax apoptosis pathway and caspase activation [
15]. However, further study of the mode and mechanism of cell death of compounds
1 and
2 will provide more information of anti-cancer activities of these two compounds and an
in vivo assay in animal model is required before safe application for cancer treatment in human-beings.
Figure 4.
Cell cytotoxicity of 1 and 2 on human hepatocellular carcinoma HepG2 cells. HepG2 cells were treated with 1 and 2 at various concentrations for 24 h and the cell viability was determined by MTT assay. * p < 0.05, compared to control of both compound 1 and 2.
Figure 4.
Cell cytotoxicity of 1 and 2 on human hepatocellular carcinoma HepG2 cells. HepG2 cells were treated with 1 and 2 at various concentrations for 24 h and the cell viability was determined by MTT assay. * p < 0.05, compared to control of both compound 1 and 2.
Figure 5.
Cell cytotoxicity of 1 and 2 on human breast cancer MDA-MB231cells. MDA-MB231 cells were treated with 1 and 2 at various concentrations for 24 h and the cell viability was determined by MTT assay. * p < 0.05, compared to control of both compound 1 and 2.
Figure 5.
Cell cytotoxicity of 1 and 2 on human breast cancer MDA-MB231cells. MDA-MB231 cells were treated with 1 and 2 at various concentrations for 24 h and the cell viability was determined by MTT assay. * p < 0.05, compared to control of both compound 1 and 2.
Table 2.
IC10, IC20 and IC50 levels of compound 1 and 2 in human hepatocellular carcinoma HepG2 cells.
Table 2.
IC10, IC20 and IC50 levels of compound 1 and 2 in human hepatocellular carcinoma HepG2 cells.
| Compound | HepG2 |
|---|
| IC10 (μM) | IC20 (μM) | IC50 (μM) * |
|---|
| MEAN | SD | MEAN | SD | MEAN | SD |
|---|
| 1 | 4.21 *a | ±1.60 | 8.74 *a | ±2.22 | 26.36 *a | ±5.18 |
| 2 | 1.74 *b | ±0.23 | 3.46 *b | ±1.10 | 12.88 *b,c | ±2.49 |
| Doxorubicin | - | - | - | - | 2.21 | ±1.72 |
Table 3.
IC10, IC20 and IC50 levels of compound 1 and 2 in human breast cancer MDA-MB231 cells.
Table 3.
IC10, IC20 and IC50 levels of compound 1 and 2 in human breast cancer MDA-MB231 cells.
| Compound | MDA-MB231 |
|---|
| IC10 (μM) | IC20 (μM) | IC50 (μM) * |
|---|
| MEAN | SD | MEAN | SD | MEAN | SD |
|---|
| 1 | 5.02 *a | ±0.53 | 15.12 *a | ±1.24 | 64.75 *a,c | ±4.45 |
| 2 | 3.37 *b | ±0.88 | 8.20 *b | ±2.45 | 67.06 *b | ±3.5 |
| Doxorubicin | - | - | - | - | 1.83 | ±0.09 |
Figure 6.
Cell cytotoxicity of doxorubicin on human hepatocellular carcinoma HepG2 and breast cancer MDA-MB231 cells. HepG2 and MDA-MB231 cells were treated with doxorubicin at various concentrations for 24 h and the cell viability were determined by MTT assay. * p < 0.05, compared to control.
Figure 6.
Cell cytotoxicity of doxorubicin on human hepatocellular carcinoma HepG2 and breast cancer MDA-MB231 cells. HepG2 and MDA-MB231 cells were treated with doxorubicin at various concentrations for 24 h and the cell viability were determined by MTT assay. * p < 0.05, compared to control.
3. Experimental
3.2. Plant Material
The mixture leaves and twigs of P. trimera (Annonaceae) were collected in August 2013 from Chiang Rai Province, the North Thailand. The species was identified by Mr. Narong Nantasean, from The Forest Herbarium, Department of National Park, Wildlife and Plant Conservation, Ministry of Natural Resources and Environment, Bangkok, Thailand. A voucher specimen (BKF.158070) was deposited in the herbarium of this institute.
3.3. Extraction and Isolation
The milled dried mixture of leaves and twigs (500 g) was extracted exhaustively in turn with hexane and ethyl acetate-methanol (1:2). The ethyl acetate-methanol extract, on removal of solvent under reduced pressure, gave a black residue (100 g). The extract of the ethyl acetate-methanol extract was fractionated by CC, silica gel, Merck No. 7734, Mesh 70–230 ASTM (600 g, 11 × 20 cm, hexane, hexane-ethyl acetate, ethyl acetate, ethyl acetate-methanol, methanol in order of increasing polarity) to give five fractions (F1–F5). Fraction F4 (31.2 g) was further subjected to CC (silica gel, 200 g, 5 × 16 cm, hexane-ethyl acetate in order of increasing polarity). Eight fractions (A1–A8) were ultimately obtained on combining the eluates on the basis of TLC. Further, the fraction A6 (3.7g) was rechromatographed over silica gel, eluting with dichloromethane, dichloromethane-methanol. The solvents were evaporated to dryness to afford seven subfractions (B1–B7). The subfraction B5 (0.61 g) yielded yellow solid which was recrystallized from methanol/dichlormethane (2:1) to afford orange crystals (26 mg) and identified as 6a,7-dehydro-1,4,5-trimethoxy-7-oxoaporphine (1). Further, the mother liquor of B5 was recrystallized from dichloromethane (2:1) to give yellow crystals (6.3 mg) of ouregidione (2, 6.3 mg).
8-Hydroxy-1,4,5-trimethoxy-7-oxoaporphine (
1): Orange crystals, m.p. 251–252 °C. UV (MeOH) λ
max (log ε): 417(4.58), 317(4.76), 241(5.12), 210(5.08) nm. IR (KBr) v
max 3384, 3249, 2936, 1705, 1658, 1559, 1508, 1458, 1281, 1211 cm
−1.
1H-NMR (DMSO-
d6, 500 MHz) data see
Table 1,
13C-NMR (DMSO-
d6, 125 MHz) data see
Table 1. EI-MS
m/z: 337 [M]
+(26), 322(100), 307(6), 294(21), 297(16), 264(17), 91(15). COSY correlations H/H: 2/3, 3/2, 9/10, 10/9, 11, 11/10. HMBC correlations H/C:2/1a, 3; 3/1a, 2; 9/7a, 10, 10a; 10/9, 11, 11a; 11/1a, 7a, 11a; 1OC
H3/1; 4OC
H3/4; 5OC
H3/5; 8O
H/ 8. HR-ESI-MS (pos.)
m/z: 360.0846 ([M+Na]
+, C
19H
15NO
5Na. calcd. 360.0848.
1,2,3-Trimethoxy-4,5-dioxo-6a,7-dehydroaporphine (
2): Yellow crystals, m.p. 242–243 °C. UV (MeOH) λ
max (log ε): 416(4.15), 317(4.34), 214(4.70), 210(4.66) nm. IR (KBr) v
max 3420, 2853, 1689, 1658, 1559, 1458, 1211 cm
−1.
1H-NMR (CDCl
3, 500 MHz) data see
Table 1,
13C-NMR (CDCl
3, 125 MHz) data see
Table 1. EI-MS
m/z: 337 [M]
+(48), 322(100), 306(3), 294(29), 279(23), 264(15), 251(52), 236(21), 180(25), 152(23). COSY correlations H/H: 8/9, 9/10, 10/11. HMBC correlations H/C:7/1b, 8; 8/7, 10; 9/11; 10/9; 11/7a, 9; 1OC
H3/1; 2OC
H3/2; 3OC
H3/3; N
H/1b, 4. HR-ESI-MS (pos.)
m/z: 360.0846 ([M+Na]
+, C
19H
15NO
5Na. calcd. 360.0849.
3.4. Cell Culture
The human hepatocellular carcinoma HepG2 and breast cancer MDA-MB231 cells were cultured in Leibovitz’s L-15 and Dulbecco’s modified eagle media (DMEM), respectively, with 25 mM NaHCO3, 20 mM HEPES, 100 units/mL penicillin, 100 μg/mL streptomycin and supplemented with 10% foetal bovine serum. The cell lines were grown at 37 °C in a 5% CO2 atmosphere. The cells (5 × 103) were treated with each compound at indicated concentrations for 24 h. The compound 1 and 2 was dissolved in dimethyl sulfoxide (DMSO) as a vehicle and the maximum volume used did not exceed 10 μL/mL of media to avoid the cytotoxicity of DMSO.
3.5. MTT Assay
Cell viability was measured by MTT assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) [
16]. In brief, human hepatocellular carcinoma HepG2 and human breast cancer MDA-MB231 cells were seeded in 96-well plates at density of 5,000 cells/well. After 24 h incubation, HepG2 cells and MDA-MB231 cells were treated with or without compound
1 and
2 at various concentrations for 24 h at 37 °C in a humidified 5% CO
2 atmosphere. Doxorubicin, a chemotherapeutic drug, was used as positive control. Non-treated cells were used as negative control. Then 15 μL (sterile stock solution of 5 mg/mL MTT dye) were added to each well and the solution was incubated for 4 h at 37 °C in a humidified 5% CO
2 atmosphere. The medium was removed and 100 μL of DMSO were added to each well, and mixed to dissolve the blue crystal formazan. The plate was read at 540 nm, with a reference wavelength of 630 nm, using a microplate reader (Biotek, Winooski, VT, USA) The percentage of cell viability was determined as follows:





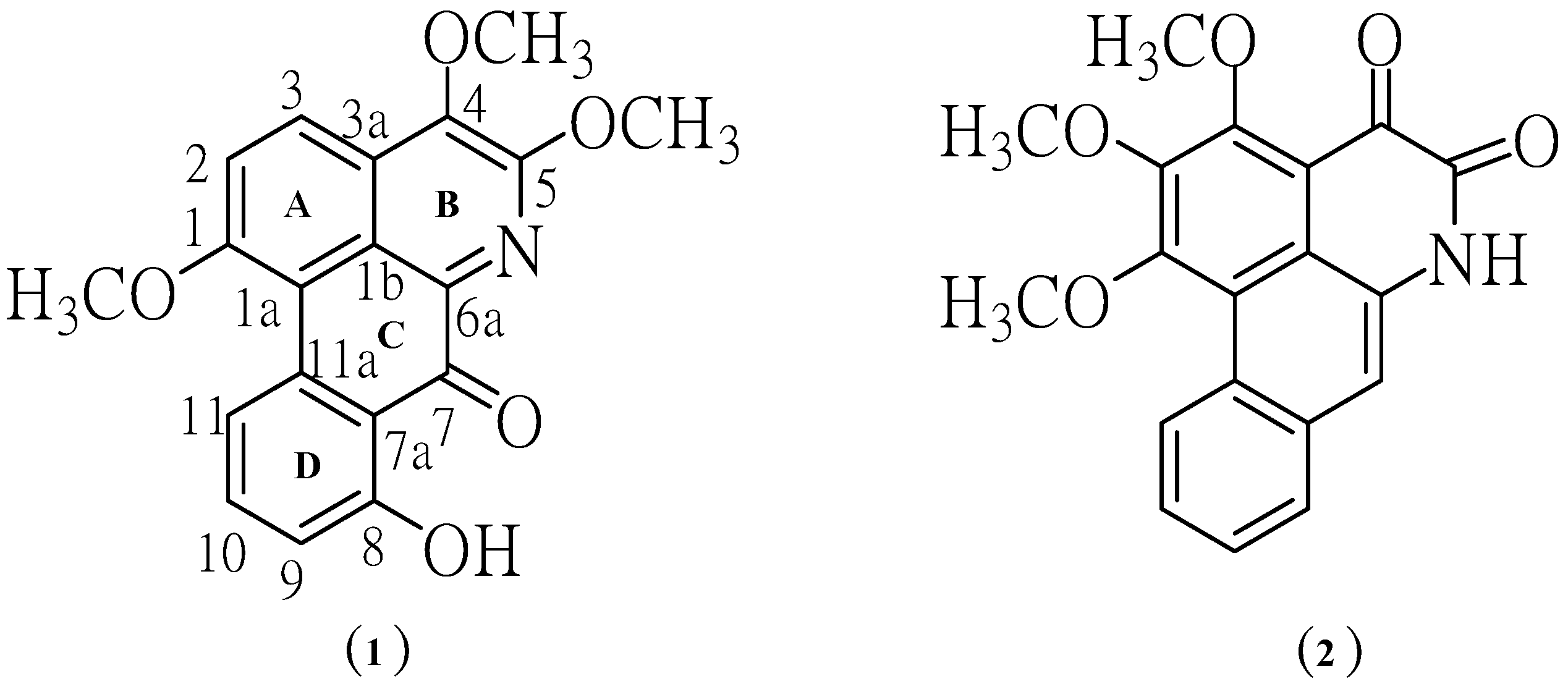{kind=link}
{kind=link}
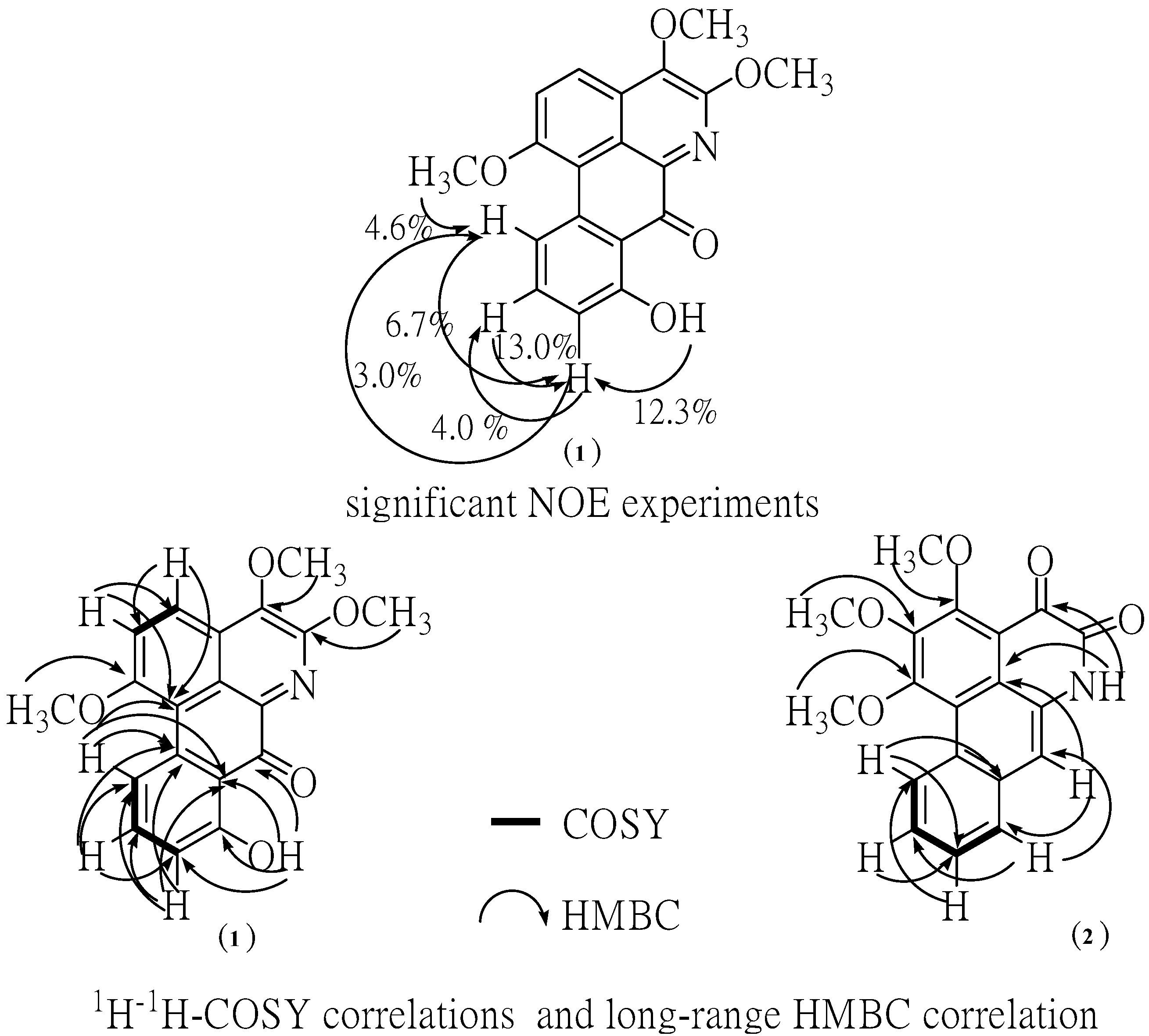{kind=link}
{kind=link}
{kind=link}
{kind=link}



