
Heparin/Heparan Sulfate Proteoglycans Glycomic Interactome in Angiogenesis: Biological Implications and Therapeutical Use



Abstract
:1. The Process of Neovascularization

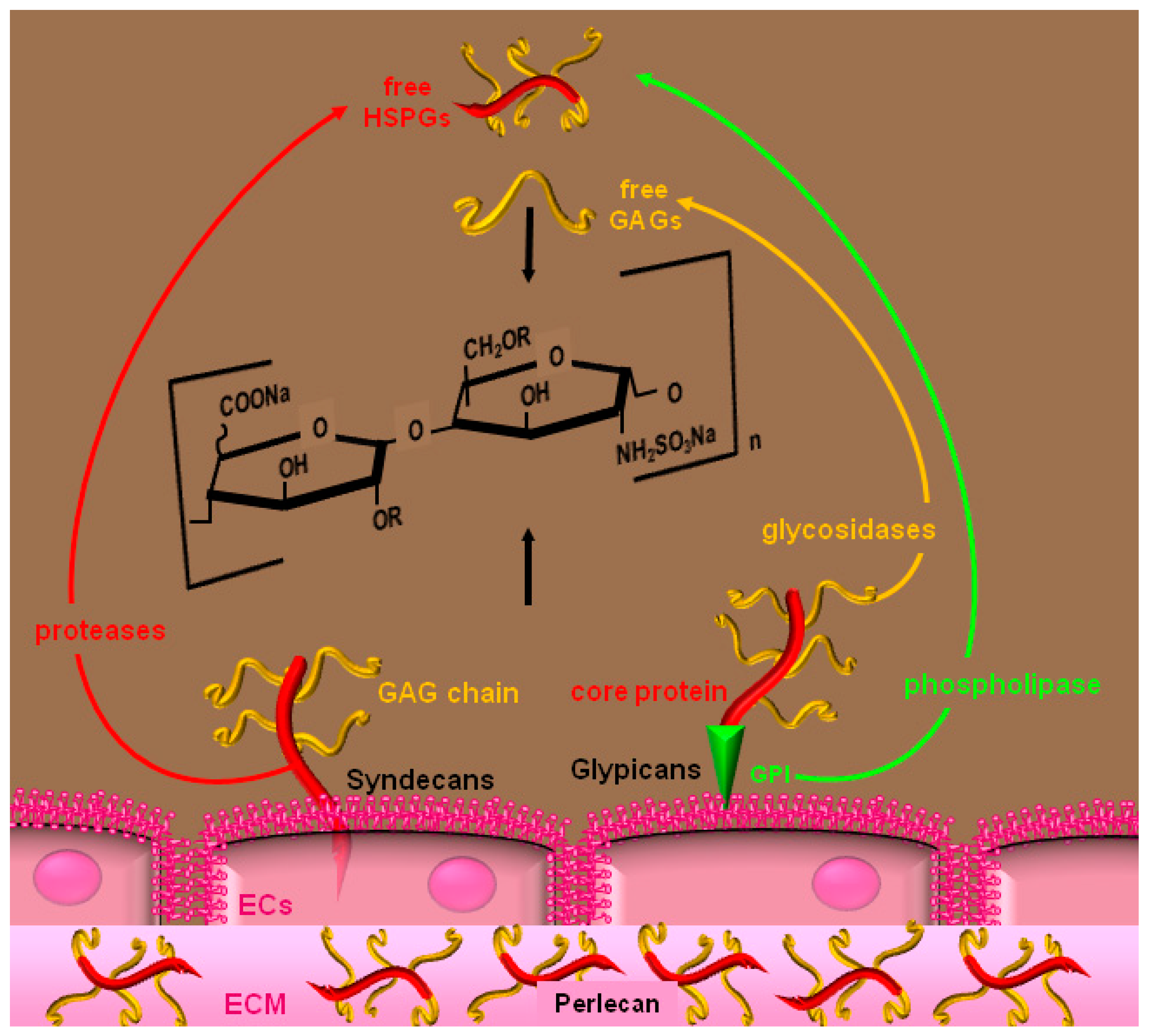
2. Heparin and HSPGs

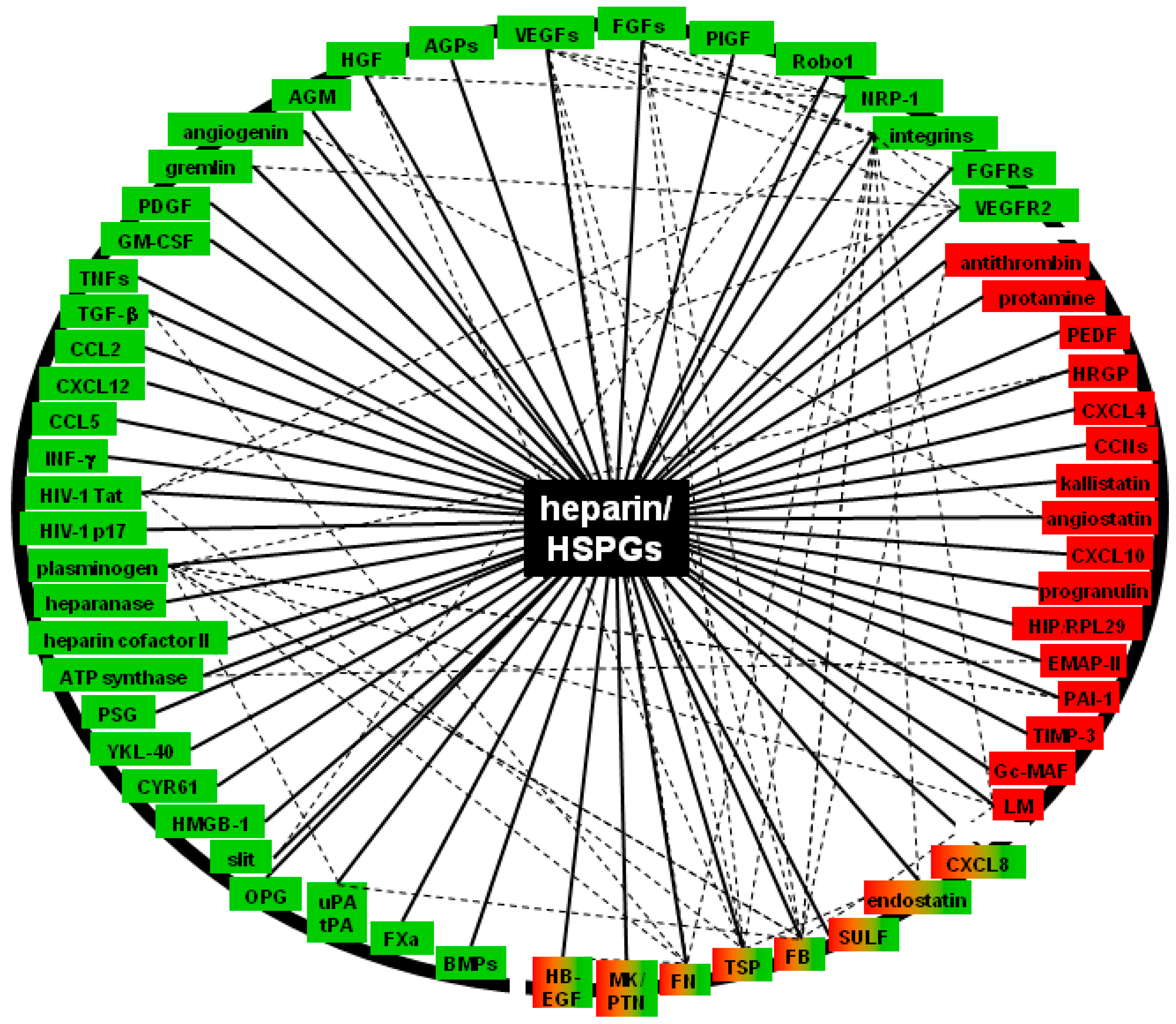
3. Molecular Bases and Biological Sequences of the Interaction of Heparin/HSPGs with Angiogenic Modulators

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Canonical AGFs | Reference |
|---|---|
| VEGF-A | [51] |
| FGFs | [52] |
| angiopoietins | [53] |
| angiogenin | [54] |
| PlGF | [55] |
| platelet-derived growth factor (PDGF) | [56] |
| midkine/pleiotrophin | [57] |
| heparin-binding EGF-like growth factor (HB-EGF) | [58] |
| angiomodulin (AGM/TAF/mac25) | [59] |
| Non Canonical AGFs and Other Regulators | |
| gremlin | [60] |
| transforming growth factor (TGF)-β | [61] |
| hepatocyte growth factor (HGF) | [62] |
| bone morphogenetic proteins (BMPs) | [63] |
| interferon (IFN)-γ | [64] |
| TNFs | [65] |
| granulocyte monocyte colony stimulating factor (GM-CSF) | [66] |
| CXCL8 | [67] |
| CCL2 | [68] |
| CCL5 | [69] |
| CXCL12 | [70] |
| HIV-1 Tat | [71] |
| HIV-1 p17 | [72] |
| pregnancy-specific β1 glycoproteins (PSGs) | [73] |
| α-ATP synthase | [74] |
| HMGB-1 | [17] |
| CYR61 | [75] |
| YKL-40 | [76] |
| osteoprotegerin (OPG) | [77] |
| FN | [18] |
| fibrinogen/fibrin (FB) | [78] |
| heparin cofactor II | [79] |
| FXa | [20] |
| Pro-Angiogenic Receptors | |
| VEGFR2 | [80] |
| FGFR1, 2, 3,4 | [81,82,83,84,85] |
| neuropilin (NPR)-1 | [10] |
| Robo | [86] |
| integrin α5β1 | [87] |
| integrin αvβ3 | [88] |
| Angiogenic Inhibitors | Reference |
| thrombospondin-1 (TSP-1) | [89] |
| endostatin | [87] |
| CXCL4 | [90] |
| histidine rich glycoprotein (HRGP) | [91] |
| protamine | [92] |
| CXCL10 | [93] |
| pigment epithelium-derived factor (PEDF) | [94] |
| endothelial monocyte-activating polypeptide-II (EMAP II) | [74] |
| tissue inhibitor of metallo proteinases (TIMP)-3 | [95] |
| laminin (LM) | [96] |
| serpin protease nexin-1 (PN-1) | [97] |
| plasminogen activator inhibitor type 1 (PAI-1) | [98] |
| HS-binding protein HIP/RPL29 | [99] |
| antithrombin | [100] |
| Effectors | |
| sulfatase SULF-1 | [101] |
| heparanase | [102] |
| tissue and urokinase-like plasminogen activators | [103] |
| plasminogen | [104] |
3.1. Positive Regulators (Canonical, non-Canonical AGFs, Their Receptors and Effectors)
| AGF | Sulfate Groups | Reference | ||
|---|---|---|---|---|
| VEGF-A | 6-OSO3 | [51,113] | ||
| FGF2 | 2-OSO3 | NSO3 | [45,114] | |
| PlGF | 2-OSO | 6-OSO3 | [115] | |
| HGF | 6-OSO3 | [116] | ||
| TGF-β | NSO3 | [117] | ||
| PDGF | 2-OSO | 6-OSO3 | NSO3 | [56] |
| midkine | NSO3 | [118] | ||
| angiomodulin | 2-OSO3 | < 6-OSO3 | <NSO3 | [119] |
| HB-EGF | 6-OSO3 | [120] | ||
| gremlin | 2-OSO3 | 6-OSO3 | NSO3 | [60] |
| HIV-1 Tat | 2-OSO3 | 6-OSO3 | NSO3 | [121] |
| HIV-1 p17 | 2-OSO3 | 6-OSO3 | NSO3 | [72] |
| CXCL8 | 2-OSO | 6-OSO3 | NSO3 | [122] |
| CXCL12 | 2-OSO3 | NSO3 | [123] | |
| IFN-γ | NSO3 | [124] | ||
| CCL2 | 6-OSO3 | [125] | ||
| CCL3 | 2-OSO | 6-OSO3 | [126] | |
| CCL21 | 2-OSO3 | 6-OSO3 | [127] | |
| Pro-Angiogenic Receptors | ||||
| FGFR1, FGFR4 | 6-OSO3 | [85,128] | ||
| NRP-1 | 6-OSO3 | [129] | ||
| Natural Angiogenic Inhibitors | ||||
| TSP-1 | 6-OSO3 | NSO3 | [130] | |
| endostatin | 6-OSO3 | [131,132] | ||
| TIMP-3 | 2-OSO3 | NSO3 | [95] | |
| Effectors | ||||
| heparanase | NSO3 | [133] | ||
| FN | 2-OSO | >>6-OSO3 | >NSO3 | [134] |
| AGF | Basic Domain Sequences | Reference | |
|---|---|---|---|
| VEGF-A | R123R124R159 | [150] | |
| FGF2 | K35R53K128R129K134K138K144K26N27R81K119R120T121Q123K125K129Q134K135 | [151,152] | |
| FGF1 | N18K112K113N114 | [153] | |
| midkine | K79R81K86 K87R89K102 | [154] | |
| angiomodulin | K89SRKRRKGK97 | [59] | |
| HGF | K60K62R73R76 K78 R512-R-K516 H645HR-K649 | [155,156,157] | |
| angiogenin | R31RR33 | [54] | |
| CXCL8 | H23K25K28K59R65K69K72 | [158] | |
| INF-γ | K128RKR131 | [159] | |
| HIV-1 Tat | R46KKRRQRRR61 | [71] | |
| HIV-1 p17 | K26KKYKLKH33 | [72] | |
| TGF-β1 | R18R25K26K31H34K37 K60R94K97R107K110 | [117,160] | |
| GM-CSF | H15H83H87 | [161] | |
| HB-EGF | K21RKKKGK27 K31KR33 R38KYK41 | [162] | |
| CCL2 | K5H66 | [163] | |
| Slit | R461R462K466R467K472K475 | [164] | |
| Pro-Angiogenic Receptors | |||
| FGFR1 | K160K163K164H166K172H201K225 | [165] | |
| FGFR2 | K161MEKRLHAVPAANTVKFR178 | [153] | |
| integrin αvβ3 | αvsubunit: | R65K446K489K520K535K645K646 K151 | [88] |
| β3 subunit: | |||
| Angiogenic Inhibitors | |||
| CXCR4 | K77NGR80 R51PRH54 K62 K92KIIKK97 | [166] | |
| endostatin | R27R139 | [167] | |
| antithrombin | K115 K125 | [43] | |
| Effectors | |||
| heparanase | K158KFKN162 | [168] | |

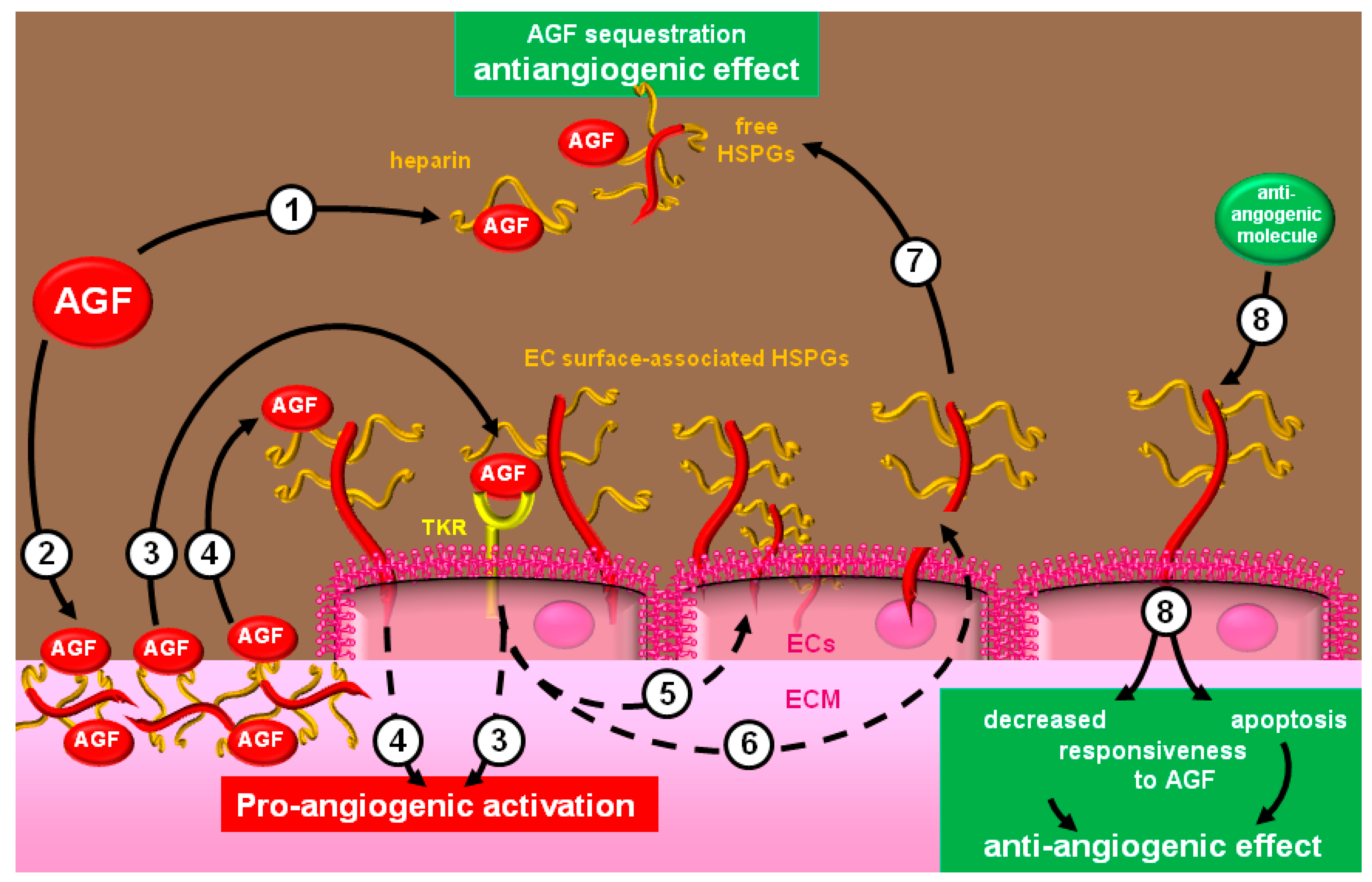
3.2. Modulators with Opposite Effects
3.3. Natural Angiogenic Inhibitors
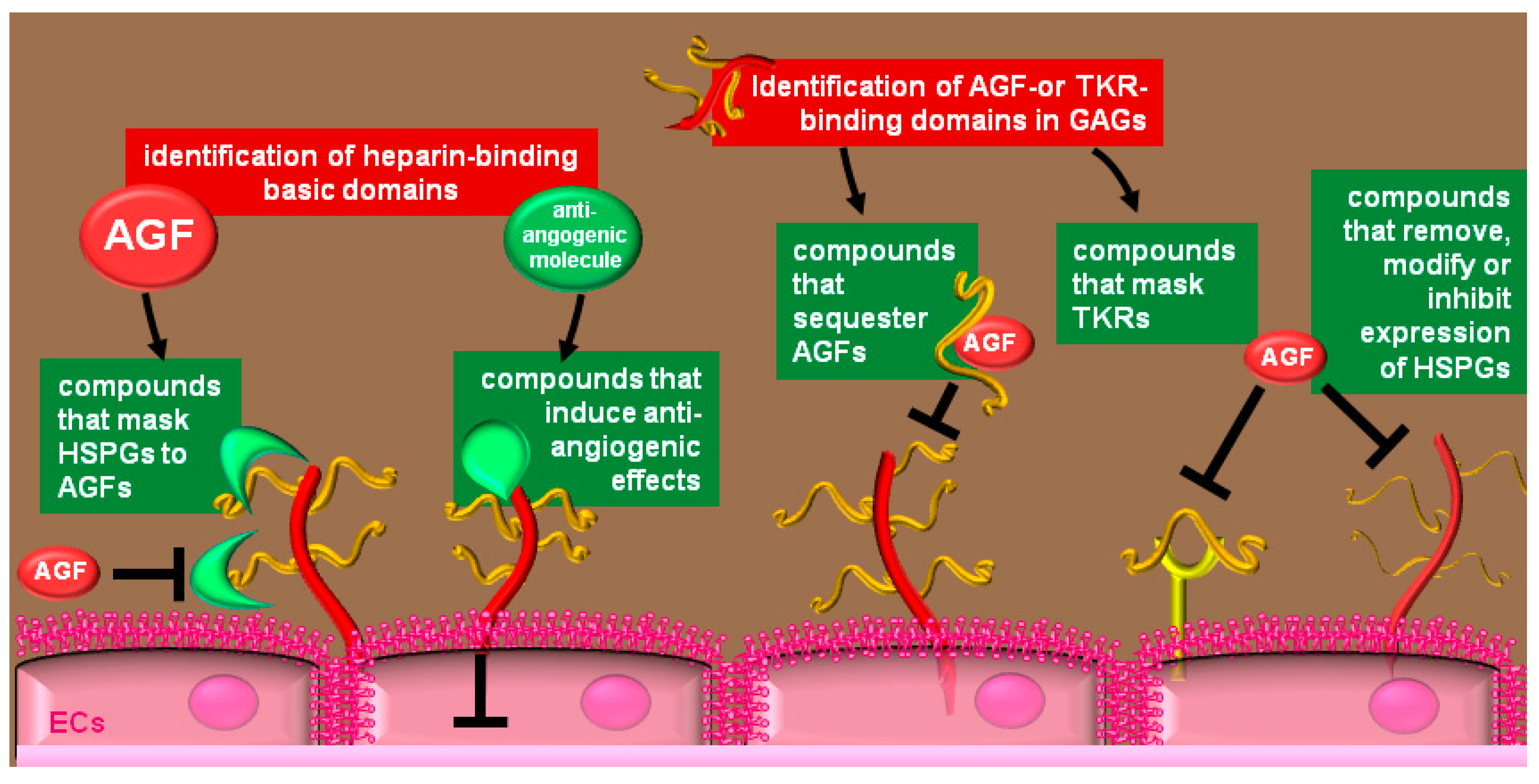
4. Therapeutical Exploitation of the Heparin/HSPGs Glycomic Interactome

4.1. Compounds that Bind to HSPGs

4.2. Heparin-Like Compounds that Bind AGFs
| AGF Inhibited | Heparin-Like Inhibitor | Reference |
|---|---|---|
| VEGF | chemically modified heparins | [106,110,113,248,249,250,251,252,253] |
| oligosaccharides from seaweed alginic acid | [112] | |
| polysaccharides from Antrodia cinnamomea | [254] | |
| fucoidan | [255] | |
| dextran derivatives | [256] | |
| sucrose octasulfate | [107] | |
| HS mimetic compounds | [257] | |
| heparin-mimetic peptide SY(SO3)DY(SO3)G | [258] | |
| phenylacetate carboxymethyl benzylamide dextran | [209] | |
| phosphosulfomannan (PI-88) and derivatives | [107] | |
| defined GAG sequences from chondroitin sulfate | [106] | |
| low molecular weight fucoidan | [259] | |
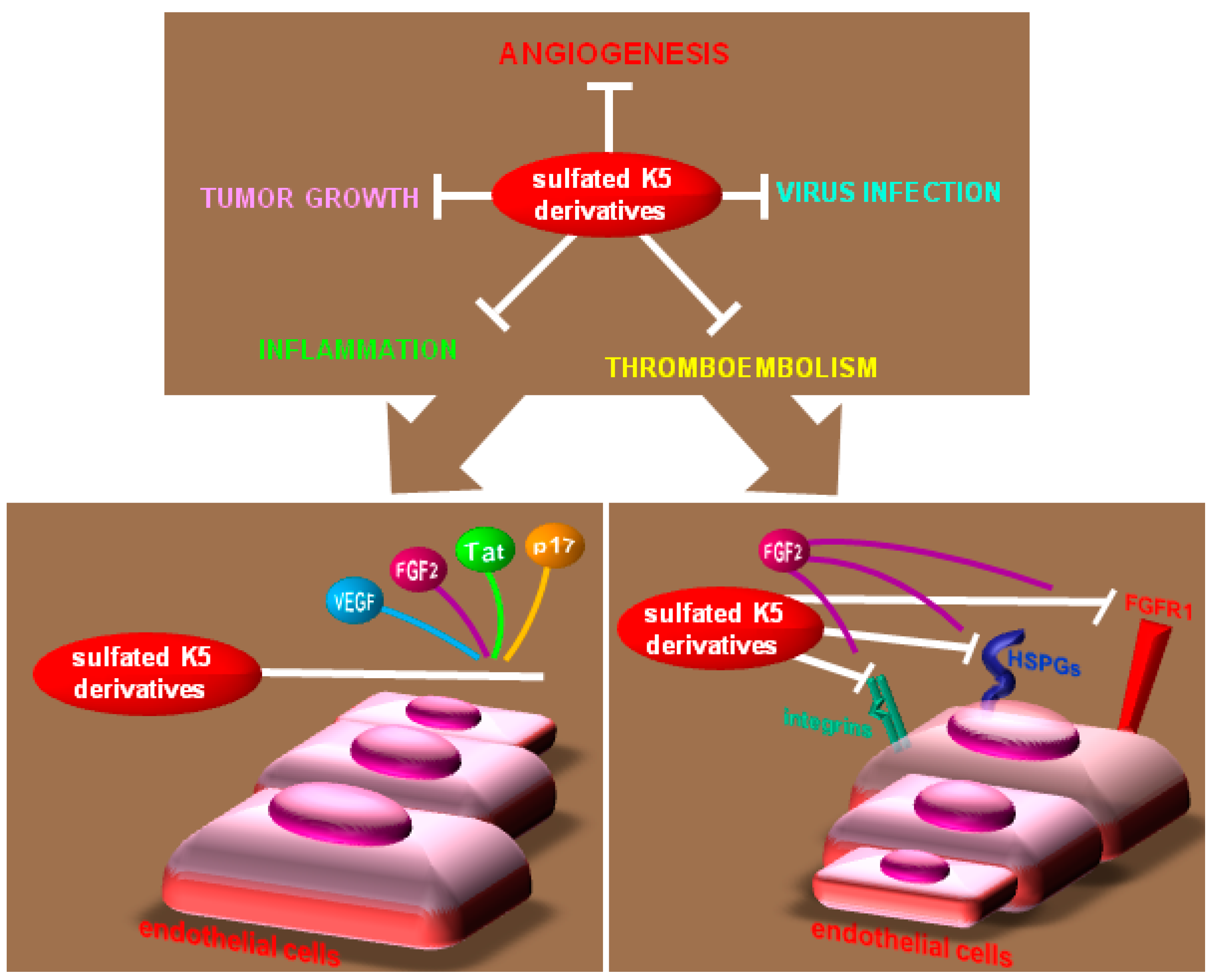
| K5 derivatives | [260] | |
| FGFs | chemically modified heparins | [37,147,248,250,261,262] |
| sulfated beta-(1->4)-galacto oligosaccharides | [263] | |
| sulfated malto oligosaccharides | [264] | |
| Fucoidan | [265] | |
| pentosan polysulfate | [266] | |
| sulfated K5 derivatives | [267] | |
| suleparoide (HS analogue) | [268] | |
| β-cyclodextrin polysulfate | [269] | |
| Carrageenan | [270] | |
| HS mimetic M402 | [271] | |
| synthetic HS | [272] | |
| sucrose octasulfate | [107] | |
| oligomannurarate sulfate JG3 | [102] | |
| marine sulfated polymannuroguluronate | [273] | |
| sulfated glycoconjugates | [274] | |
| PI-88 and derivatives | [107] | |
| linked sulfated tetracyclitols | [275] | |
| disulfated methyl 6-azido-6-deoxy-a-dmannopyranosides | [257] | |
| Gremlin | chemically modified heparins, K5 derivatives | [60] |
| HS mimetic M402 | [271] | |
| chemically modified heparins | [276] | |
| SDF-1α | HS mimetic M402 | [271] |
| chemically modified heparins | [248] | |
| IL-8 | chemically modified heparins, PI-88 | [107] |
| HIV-1 Tat | K5 derivatives | [277] |
| pentosan polysulfate | [183] | |
| dextrin-2-sulphate | [278] | |
| sulfated polymannuroguluronate | [279] | |
| HIV-1 p17 | chemically modified heparins, K5 derivatives | [72] |
| CXCL8 | Fucoidan | [280] |
| CCL2 | Fucoidan | |
| cyclodextrin sulfate | [196] | |
| sucrose octasulfate | ||
| PDGF | heparin-derived angiogenesis inhibitor LHT7 | [251] |
| low molecular weight heparins | [188] | |
| TGF-β1 | Fucoidan | [281] |
| IFN-γ | HS-derived glycoconjugate mimetics | [282] |
| BMPs | HS mimetic WSS25 | [63] |
| heparanase | N-acetylated glycol split heparin SST0001 | [283] |
4.3. Heparin-Like Molecules that Bind and Mask Pro-Angiogenic Receptors
4.4. Inhibition of EC-Surface HSPGs Expression
4.5. Removal of EC-Surface HSPGs
5. Conclusions

Acknowledgments
Author Contributions
Abbreviations
| AGFs | angiogenic growth factors |
| ECs | endothelial cells |
| ECM | extracellular matrix |
| FGFs | fibroblast growth factors |
| FGFRs | fibroblast growth factor receptors |
| GAGs | glycosaminoglycans |
| GM-CSF | granulocyte monocyte colony stimulating factor |
| GPI | glycosyl-phosphatidylinositol |
| HB-EGF | heparin-binding epidermal growth factor |
| HGF | hepatocyte growth factor |
| HRGP | histidine-rich glycoprotein |
| HS | heparan sulfate |
| HSPGs | heparan sulfate proteoglycans |
| CXCL8 | interleukin 8 |
| INF-γ | interferon γ |
| Kd | dissociation constant |
| VEGFR2 | vascular endothelial growth factor receptor 2 flk-1 |
| CCL2 | monocyte chemoattractant protein 1 |
| PDGF | platelet derived growth factor |
| PF-4 | platelet factor 4 |
| PlGF | placenta growth factor |
| Tat | HIV-1 transactivating factor |
| TGFs | transforming growth factors |
| TKRs | tyrosine kinase receptors |
| TNFs | tumor necrosis factors |
| VEGFs | vascular endothelial growth factors |
| VEGFRs | vascular endothelial growth factor receptors |
Conflicts of Interest
References
- Presta, M.; Dell’Era, P.; Mitola, S.; Moroni, E.; Ronca, R.; Rusnati, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 159–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Plow, E.F.; Meller, J.; Byzova, T.V. Integrin function in vascular biology: A view from 2013. Curr. Opin. Hematol. 2014, 21, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E.; Giampietro, C. Vascular endothelial-cadherin and vascular stability. Curr. Opin. Hematol. 2012, 19, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Presta, M. Extracellular angiogenic growth factor interactions: An angiogenesis interactome survey. Endothelium 2006, 13, 93–111. [Google Scholar] [PubMed]
- Montuori, N.; Ragno, P. Role of upa/upar in the modulation of angiogenesis. Chem. Immunol. Allergy 2014, 99, 105–122. [Google Scholar] [PubMed]
- Kanda, S.; Miyata, Y.; Kanetake, H. Fibroblast growth factor-2-mediated capillary morphogenesis of endothelial cells requires signals via flt-1/vascular endothelial growth factor receptor-1: Possible involvement of c-akt. J. Biol. Chem. 2004, 279, 4007–4016. [Google Scholar] [CrossRef] [PubMed]
- Oommen, S.; Gupta, S.K.; Vlahakis, N.E. Vascular endothelial growth factor a (vegf-a) induces endothelial and cancer cell migration through direct binding to integrin α9β1: Identification of a specific α9β1 binding site. J. Biol. Chem. 2011, 286, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Fuh, G.; Garcia, K.C.; de Vos, A.M. The interaction of neuropilin-1 with vascular endothelial growth factor and its receptor flt-1. J. Biol. Chem. 2000, 275, 26690–26695. [Google Scholar] [PubMed]
- Itoh, N.; Ornitz, D.M. Fibroblast growth factors: From molecular evolution to roles in development, metabolism and disease. J. Biochem. 2011, 149, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.G. Isoforms of receptors of fibroblast growth factors. J. Cell. Physiol. 2014, 229, 1887–1895. [Google Scholar] [CrossRef] [PubMed]
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Tanghetti, E.; Dell’Era, P.; Gualandris, A.; Presta, M. Alphavbeta3 integrin mediates the cell-adhesive capacity and biological activity of basic fibroblast growth factor (fgf-2) in cultured endothelial cells. Mol. Biol. Cell 1997, 8, 2449–2461. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Urbinati, C.; Tanghetti, E.; Dell’Era, P.; Lortat-Jacob, H.; Presta, M. Cell membrane gm1 ganglioside is a functional coreceptor for fibroblast growth factor 2. Proc. Natl. Acad. Sci. USA 2002, 99, 4367–4372. [Google Scholar] [CrossRef] [PubMed]
- West, D.C.; Rees, C.G.; Duchesne, L.; Patey, S.J.; Terry, C.J.; Turnbull, J.E.; Delehedde, M.; Heegaard, C.W.; Allain, F.; Vanpouille, C.; et al. Interactions of multiple heparin binding growth factors with neuropilin-1 and potentiation of the activity of fibroblast growth factor-2. J. Biol. Chem. 2005, 280, 13457–13464. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Mori, S.; Liu, K.; Takahashi, H.K.; Nishibori, M. Histidine-rich glycoprotein inhibited high mobility group box 1 in complex with heparin-induced angiogenesis in matrigel plug assay. Eur. J. Pharmacol. 2009, 623, 89–95. [Google Scholar] [CrossRef]
- Viji, R.I.; Kumar, V.B.; Kiran, M.S.; Sudhakaran, P.R. Angiogenic response of endothelial cells to heparin-binding domain of fibronectin. Int. J. Biochem. Cell Biol. 2008, 40, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Abboud-Jarrous, G.; Elkin, M.; Naggi, A.; Casu, B.; Sasisekharan, R.; Ilan, N. The impact of heparanese and heparin on cancer metastasis and angiogenesis. Pathophysiol. Haemost. Thromb. 2006, 35, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Hollborn, M.; Kohen, L.; Werschnik, C.; Tietz, L.; Wiedemann, P.; Bringmann, A. Activated blood coagulation factor x (fxa) induces angiogenic growth factor expression in human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5930–5939. [Google Scholar] [CrossRef]
- Pakala, R.; Watanabe, T.; Benedict, C.R. Induction of endothelial cell proliferation by angiogenic factors released by activated monocytes. Cardiovasc. Radiat. Med. 2002, 3, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Clauss, M.; Grell, M.; Fangmann, C.; Fiers, W.; Scheurich, P.; Risau, W. Synergistic induction of endothelial tissue factor by tumor necrosis factor and vascular endothelial growth factor: Functional analysis of the tumor necrosis factor receptors. FEBS Lett. 1996, 390, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Charalambous, C.; Pen, L.B.; Su, Y.S.; Milan, J.; Chen, T.C.; Hofman, F.M. Interleukin-8 differentially regulates migration of tumor-associated and normal human brain endothelial cells. Cancer Res. 2005, 65, 10347–10354. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.H.; Ryu, J.; Han, K.H. Monocyte chemoattractant protein-1-induced angiogenesis is mediated by vascular endothelial growth factor-a. Blood 2005, 105, 1405–1407. [Google Scholar] [CrossRef] [PubMed]
- Wempe, F.; Lindner, V.; Augustin, H.G. Basic fibroblast growth factor (bfgf) regulates the expression of the cc chemokine monocyte chemoattractant protein-1 (mcp-1) in autocrine-activated endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2471–2478. [Google Scholar] [CrossRef] [PubMed]
- Vrancken, K.; Vervaeke, P.; Balzarini, J.; Liekens, S. Viruses as key regulators of angiogenesis. Rev. Med. Virol. 2011, 21, 181–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dezube, B.J. The role of human immunodeficiency virus-i in the pathogenesis of acquired immunodeficiency syndrome-related kaposi’s sarcoma: The importance of an inflammatory and angiogenic milieu. Semin. Oncol. 2000, 27, 420–423. [Google Scholar] [PubMed]
- Ensoli, B.; Gendelman, R.; Markham, P.; Fiorelli, V.; Colombini, S.; Raffeld, M.; Cafaro, A.; Chang, H.K.; Brady, J.N.; Gallo, R.C. Synergy between basic fibroblast growth factor and HIV-1 tat protein in induction of kaposi’s sarcoma. Nature 1994, 371, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Eggert, A.; Ikegaki, N.; Kwiatkowski, J.; Zhao, H.; Brodeur, G.M.; Himelstein, B.P. High-level expression of angiogenic factors is associated with advanced tumor stage in human neuroblastomas. Clin. Cancer Res. 2000, 6, 1900–1908. [Google Scholar] [PubMed]
- Barthlen, W.; Flaadt, D.; Girgert, R.; Conzelmann, J.; Schweizer, P.; Zugmaier, G.; Buck, M.; Knabbe, C. Significance of heparin-binding growth factor expression on cells of solid pediatric tumors. J. Pediatr. Surg. 2003, 38, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Bikfalvi, A.; Moenner, M.; Javerzat, S.; North, S.; Hagedorn, M. Inhibition of angiogenesis and the angiogenesis/invasion shift. Biochem. Soc. Trans. 2011, 39, 1560–1564. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Esko, J.D. Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 2014, 83, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, U.; Kjellen, L. Pathophysiology of heparan sulphate: Many diseases, few drugs. J. Intern. Med. 2013, 273, 555–571. [Google Scholar] [CrossRef] [PubMed]
- Esko, J.D.; Lindahl, U. Molecular diversity of heparan sulfate. J. Clin. Investig. 2001, 108, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Aviezer, D.; Hecht, D.; Safran, M.; Eisinger, M.; David, G.; Yayon, A. Perlecan, basal lamina proteoglycan, promotes basic fibroblast growth factor-receptor binding, mitogenesis, and angiogenesis. Cell 1994, 79, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Madri, J.A.; Yurchenco, P.D. Endothelial cells interact with the core protein of basement membrane perlecan through beta 1 and beta 3 integrins: An adhesion modulated by glycosaminoglycan. J. Cell Biol. 1992, 119, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Presta, M. Interaction of angiogenic basic fibroblast growth factor with endothelial cell heparan sulfate proteoglycans. Biological implications in neovascularization. Int. J. Clin. Lab. Res. 1996, 26, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pedersen, L.C. Anticoagulant heparan sulfate: Structural specificity and biosynthesis. Appl. Microbiol. Biotechnol. 2007, 74, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.; Schotz, M.C. The lipase gene family. J. Lipid Res. 2002, 43, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Marcum, J.A.; Rosenberg, R.D. Heparinlike molecules with anticoagulant activity are synthesized by cultured endothelial cells. Biochem. Biophys. Res. Commun. 1985, 126, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Quinsey, N.S.; Greedy, A.L.; Bottomley, S.P.; Whisstock, J.C.; Pike, R.N. Antithrombin: In control of coagulation. Int. J. Biochem. Cell Biol. 2004, 36, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Oreste, P.; Zoppetti, G.; Presta, M. Biotechnological engineering of heparin/heparan sulphate: A novel area of multi-target drug discovery. Curr. Pharm. Des. 2005, 11, 2489–2499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Swanson, R.; Izaguirre, G.; Xiong, Y.; Lau, L.F.; Olson, S.T. The heparin-binding site of antithrombin is crucial for antiangiogenic activity. Blood 2005, 106, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, N.V.; Desai, U.R. Toward a robust computational screening strategy for identifying glycosaminoglycan sequences that display high specificity for target proteins. Glycobiology 2014, 24, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Maccarana, M.; Casu, B.; Lindahl, U. Minimal sequence in heparin/heparan sulfate required for binding of basic fibroblast growth factor. J. Biol. Chem. 1994, 269, 3903–3903. [Google Scholar] [PubMed]
- Mosier, P.D.; Krishnasamy, C.; Kellogg, G.E.; Desai, U.R. On the specificity of heparin/heparan sulfate binding to proteins. Anion-binding sites on antithrombin and thrombin are fundamentally different. PLoS ONE 2012, 7, e48632. [Google Scholar] [CrossRef] [PubMed]
- Kreuger, J.; Spillmann, D.; Li, J.P.; Lindahl, U. Interactions between heparan sulfate and proteins: The concept of specificity. J. Cell Biol. 2006, 174, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Kayitmazer, A.B.; Quinn, B.; Kimura, K.; Ryan, G.L.; Tate, A.J.; Pink, D.A.; Dubin, P.L. Protein specificity of charged sequences in polyanions and heparins. Biomacromolecules 2010, 11, 3325–3331. [Google Scholar] [CrossRef] [PubMed]
- Rullo, A.; Nitz, M. Importance of the spatial display of charged residues in heparin-peptide interactions. Biopolymers 2010, 93, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Nunes, Q.M.; Mournetas, V.; Lane, B.; Sutton, R.; Fernig, D.G.; Vasieva, O. The heparin-binding protein interactome in pancreatic diseases. Pancreatology 2013, 13, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.J.; Mulloy, B.; Gallagher, J.T.; Stringer, S.E. Vegf165-binding sites within heparan sulfate encompass two highly sulfated domains and can be liberated by k5 lyase. J. Biol. Chem. 2006, 281, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Uehara, Y.; Asada, M.; Honda, E.; Nagai, N.; Kimata, K.; Suzuki, M.; Imamura, T. Sulfated glycosaminoglycans are required for specific and sensitive fibroblast growth factor (fgf) 19 signaling via fgf receptor 4 and betaklotho. J. Biol. Chem. 2011, 286, 26418–26423. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, Y.J.; Yu, Q. Angiopoietin-3 is tethered on the cell surface via heparan sulfate proteoglycans. J. Biol. Chem. 2004, 279, 41179–41188. [Google Scholar] [CrossRef] [PubMed]
- Soncin, F.; Strydom, D.J.; Shapiro, R. Interaction of heparin with human angiogenin. J. Biol. Chem. 1997, 272, 9818–9824. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.; Weich, H.A. A heparin-binding form of placenta growth factor (plgf-2) is expressed in human umbilical vein endothelial cells and in placenta. Growth Factors 1993, 9, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Feyzi, E.; Lustig, F.; Fager, G.; Spillmann, D.; Lindahl, U.; Salmivirta, M. Characterization of heparin and heparan sulfate domains binding to the long splice variant of platelet-derived growth factor a chain. J. Biol. Chem. 1997, 272, 5518–5524. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.; Zou, K.; Muramatsu, H.; Ichihara-Tanaka, K.; Habuchi, O.; Ohtake, S.; Ikematsu, S.; Sakuma, S.; Muramatsu, T. Glycosaminoglycan structures required for strong binding to midkine, a heparin-binding growth factor. Glycobiology 2003, 13, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, R.; Mine, N.; Kawaguchi, T.; Minami, S.; Saeki, K.; Mekada, E. Hb-egf function in cardiac valve development requires interaction with heparan sulfate proteoglycans. Development 2010, 137, 2205–2214. [Google Scholar] [CrossRef] [PubMed]
- Sato, J.; Hasegawa, S.; Akaogi, K.; Yasumitsu, H.; Yamada, S.; Sugahara, K.; Miyazaki, K. Identification of cell-binding site of angiomodulin (agm/taf/mac25) that interacts with heparan sulfates on cell surface. J. Cell. Biochem. 1999, 75, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Chiodelli, P.; Mitola, S.; Ravelli, C.; Oreste, P.; Rusnati, M.; Presta, M. Heparan sulfate proteoglycans mediate the angiogenic activity of the vascular endothelial growth factor receptor-2 agonist gremlin. Arterioscler. Thromb. Vasc. Biol. 2011, 31, e116–e127. [Google Scholar] [CrossRef]
- Rider, C.C. Heparin/heparan sulphate binding in the tgf-beta cytokine superfamily. Biochem. Soc. Trans. 2006, 34, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Raiber, E.A.; Wilkinson, J.A.; Manetti, F.; Botta, M.; Deakin, J.; Gallagher, J.; Lyon, M.; Ducki, S.W. Novel heparin/heparan sulfate mimics as inhibitors of hgf/sf-induced met activation. Bioorg. Med. Chem. Lett. 2007, 17, 6321–6325. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Yang, B.; Pei, Z.C.; Zhang, Z.; Ding, K. Wss25 inhibits growth of xenografted hepatocellular cancer cells in nude mice by disrupting angiogenesis via blocking bone morphogenetic protein (bmp)/smad/id1 signaling. J. Biol. Chem. 2010, 285, 32638–32646. [Google Scholar] [CrossRef] [PubMed]
- Fluhr, H.; Spratte, J.; Heidrich, S.; Ehrhardt, J.; Steinmuller, F.; Zygmunt, M. Heparin inhibits interferon-gamma signaling in human endometrial stromal cells by interference with the cellular binding of interferon-gamma. Fertil. Steril. 2011, 95, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Spratte, J.; Meyer zu Schwabedissen, H.; Endlich, N.; Zygmunt, M.; Fluhr, H. Heparin inhibits tnf-alpha signaling in human endometrial stromal cells by interaction with nf-kappab. Mol. Hum. Reprod. 2013, 19, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Liang, A.; Du, Y.; Wang, K.; Lin, B. Quantitative investigation of the interaction between granulocyte-macrophage colony-stimulating factor and heparin by capillary zone electrophoresis. J. Sep. Sci. 2006, 29, 1637–1641. [Google Scholar] [CrossRef] [PubMed]
- Pichert, A.; Samsonov, S.A.; Theisgen, S.; Thomas, L.; Baumann, L.; Schiller, J.; Beck-Sickinger, A.G.; Huster, D.; Pisabarro, M.T. Characterization of the interaction of interleukin-8 with hyaluronan, chondroitin sulfate, dermatan sulfate and their sulfated derivatives by spectroscopy and molecular modeling. Glycobiology 2012, 22, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Meissen, J.K.; Sweeney, M.D.; Girardi, M.; Lawrence, R.; Esko, J.D.; Leary, J.A. Differentiation of 3-o-sulfated heparin disaccharide isomers: Identification of structural aspects of the heparin ccl2 binding motif. J. Am. Soc. Mass Spectrom. 2009, 20, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Suffee, N.; Hlawaty, H.; Meddahi-Pelle, A.; Maillard, L.; Louedec, L.; Haddad, O.; Martin, L.; Laguillier, C.; Richard, B.; Oudar, O.; et al. Rantes/ccl5-induced pro-angiogenic effects depend on ccr1, ccr5 and glycosaminoglycans. Angiogenesis 2012, 15, 727–744. [Google Scholar] [CrossRef] [PubMed]
- Seeger, F.H.; Rasper, T.; Fischer, A.; Muhly-Reinholz, M.; Hergenreider, E.; Leistner, D.M.; Sommer, K.; Manavski, Y.; Henschler, R.; Chavakis, E.; et al. Heparin disrupts the cxcr4/sdf-1 axis and impairs the functional capacity of bone marrow-derived mononuclear cells used for cardiovascular repair. Circ. Res. 2012, 111, 854–862. [Google Scholar] [CrossRef]
- Rusnati, M.; Coltrini, D.; Oreste, P.; Zoppetti, G.; Albini, A.; Noonan, D.M.; d’Adda di Fagagna, F.; Giacca, M.; Presta, M. Interaction of HIV-1 tat protein with heparin. Role of the backbone structure, sulfation, and size. J. Biol. Chem. 1997, 272, 11313–11320. [Google Scholar] [CrossRef] [PubMed]
- Bugatti, A.; Giagulli, C.; Urbinati, C.; Caccuri, F.; Chiodelli, P.; Oreste, P.; Fiorentini, S.; Orro, A.; Milanesi, L.; D’Ursi, P.; et al. Molecular interaction studies of HIV-1 matrix protein p17 and heparin: Identification of the heparin-binding motif of p17 as a target for the development of multitarget antagonists. J. Biol. Chem. 2013, 288, 1150–1161. [Google Scholar] [CrossRef] [PubMed]
- Lisboa, F.A.; Warren, J.; Sulkowski, G.; Aparicio, M.; David, G.; Zudaire, E.; Dveksler, G.S. Pregnancy-specific glycoprotein 1 induces endothelial tubulogenesis through interaction with cell surface proteoglycans. J. Biol. Chem. 2011, 286, 7577–7586. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.Y.; Ko, H.J.; Heo, T.H.; Kang, C.Y. Heparan sulfate regulates the antiangiogenic activity of endothelial monocyte-activating polypeptide-II at acidic pH. Mol. Pharmacol. 2005, 67, 1534–1543. [Google Scholar] [CrossRef] [PubMed]
- Babic, A.M.; Kireeva, M.L.; Kolesnikova, T.V.; Lau, L.F. Cyr61, a product of a growth factor-inducible immediate early gene, promotes angiogenesis and tumor growth. Proc. Natl. Acad. Sci. USA 1998, 95, 6355–6360. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Hamel, K.; Petersen, L.; Cao, Q.J.; Arenas, R.B.; Bigelow, C.; Bentley, B.; Yan, W. Ykl-40, a secreted glycoprotein, promotes tumor angiogenesis. Oncogene 2009, 28, 4456–4468. [Google Scholar] [CrossRef] [PubMed]
- Benslimane-Ahmim, Z.; Poirier, F.; Delomenie, C.; Lokajczyk, A.; Grelac, F.; Galy-Fauroux, I.; Mohamedi, A.; Fischer, A.M.; Heymann, D.; Lutomski, D.; et al. Mechanistic study of the proangiogenic effect of osteoprotegerin. Angiogenesis 2013, 16, 575–593. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, M.W. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 2005, 3, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Aihara, K.; Yoshida, S.; Iwase, T.; Tajima, S.; Izawa-Ishizawa, Y.; Kihira, Y.; Ishizawa, K.; Tomita, S.; Tsuchiya, K.; et al. Heparin cofactor II, a serine protease inhibitor, promotes angiogenesis via activation of the amp-activated protein kinase-endothelial nitric-oxide synthase signaling pathway. J. Biol. Chem. 2012, 287, 34256–34263. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Fuster, M.M.; Lawrence, R.; Esko, J.D. Heparan sulfate regulates vegf165- and vegf121-mediated vascular hyperpermeability. J. Biol. Chem. 2011, 286, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, J.; Dutta, K.; Ilghari, D.; Beenken, A.; Goetz, R.; Eliseenkova, A.V.; Cowburn, D.; Mohammadi, M. The alternatively spliced acid box region plays a key role in fgf receptor autoinhibition. Structure 2012, 20, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Ibrahimi, O.A.; Zhang, F.; Hrstka, S.C.; Mohammadi, M.; Linhardt, R.J. Kinetic model for fgf, fgfr, and proteoglycan signal transduction complex assembly. Biochemistry 2004, 43, 4724–4730. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.K.; Fernig, D.G.; Turnbull, J.E. Fibroblast growth factor receptors 1 and 2 interact differently with heparin/heparan sulfate. Implications for dynamic assembly of a ternary signaling complex. J. Biol. Chem. 2002, 277, 28554–28563. [Google Scholar] [CrossRef] [PubMed]
- Olsen, S.K.; Ibrahimi, O.A.; Raucci, A.; Zhang, F.; Eliseenkova, A.V.; Yayon, A.; Basilico, C.; Linhardt, R.J.; Schlessinger, J.; Mohammadi, M. Insights into the molecular basis for fibroblast growth factor receptor autoinhibition and ligand-binding promiscuity. Proc. Natl. Acad. Sci. USA 2004, 101, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Loo, B.M.; Kreuger, J.; Jalkanen, M.; Lindahl, U.; Salmivirta, M. Binding of heparin/heparan sulfate to fibroblast growth factor receptor 4. J. Biol. Chem. 2001, 276, 16868–16876. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Moniz, H.A.; Walcott, B.; Moremen, K.W.; Linhardt, R.J.; Wang, L. Characterization of the interaction between robo1 and heparin and other glycosaminoglycans. Biochimie 2013, 95, 2345–2353. [Google Scholar] [CrossRef] [PubMed]
- Faye, C.; Moreau, C.; Chautard, E.; Jetne, R.; Fukai, N.; Ruggiero, F.; Humphries, M.J.; Olsen, B.R.; Ricard-Blum, S. Molecular interplay between endostatin, integrins, and heparan sulfate. J. Biol. Chem. 2009, 284, 22029–22040. [Google Scholar] [CrossRef] [PubMed]
- Ballut, L.; Sapay, N.; Chautard, E.; Imberty, A.; Ricard-Blum, S. Mapping of heparin/heparan sulfate binding sites on alphavbeta3 integrin by molecular docking. J. Mol. Recognit. JMR 2013, 26, 76–85. [Google Scholar] [CrossRef]
- Kaur, S.; Kuznetsova, S.A.; Pendrak, M.L.; Sipes, J.M.; Romeo, M.J.; Li, Z.; Zhang, L.; Roberts, D.D. Heparan sulfate modification of the transmembrane receptor cd47 is necessary for inhibition of t cell receptor signaling by thrombospondin-1. J. Biol. Chem. 2011, 286, 14991–15002. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Krauel, K.; Gottschalk, K.E.; Renne, T.; Helm, C.A.; Greinacher, A.; Block, S. Characterisation of the conformational changes in platelet factor 4 induced by polyanions: Towards in vitro prediction of antigenicity. Thromb. Haemost. 2014, 112, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Poon, I.K.; Parish, C.R.; Hulett, M.D. Histidine-rich glycoprotein functions cooperatively with cell surface heparan sulfate on phagocytes to promote necrotic cell uptake. J. Leukoc. Biol. 2010, 88, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.J.; Parish, C.R. Histidine-rich glycoprotein and platelet factor 4 mask heparan sulfate proteoglycans recognized by acidic and basic fibroblast growth factor. Biochemistry 1994, 33, 13918–13927. [Google Scholar] [CrossRef] [PubMed]
- Ranjbaran, H.; Wang, Y.; Manes, T.D.; Yakimov, A.O.; Akhtar, S.; Kluger, M.S.; Pober, J.S.; Tellides, G. Heparin displaces interferon-gamma-inducible chemokines (ip-10, i-tac, and mig) sequestered in the vasculature and inhibits the transendothelial migration and arterial recruitment of t cells. Circulation 2006, 114, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, A.; Okano-Kosugi, H.; Yamazaki, C.M.; Koide, T. Pigment epithelium-derived factor (pedf) shares binding sites in collagen with heparin/heparan sulfate proteoglycans. J. Biol. Chem. 2011, 286, 26364–26374. [Google Scholar] [CrossRef] [PubMed]
- Troeberg, L.; Lazenbatt, C.; Anower, E.K.M.F.; Freeman, C.; Federov, O.; Habuchi, H.; Habuchi, O.; Kimata, K.; Nagase, H. Sulfated glycosaminoglycans control the extracellular trafficking and the activity of the metalloprotease inhibitor timp-3. Chem. Biol. 2014, 21, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Hibino, S.; Shibuya, M.; Hoffman, M.P.; Engbring, J.A.; Hossain, R.; Mochizuki, M.; Kudoh, S.; Nomizu, M.; Kleinman, H.K. Laminin alpha5 chain metastasis- and angiogenesis-inhibiting peptide blocks fibroblast growth factor 2 activity by binding to the heparan sulfate chains of cd44. Cancer Res. 2005, 65, 10494–10501. [Google Scholar] [CrossRef] [PubMed]
- Selbonne, S.; Azibani, F.; Iatmanen, S.; Boulaftali, Y.; Richard, B.; Jandrot-Perrus, M.; Bouton, M.C.; Arocas, V. In vitro and in vivo antiangiogenic properties of the serpin protease nexin-1. Mol. Cell. Biol. 2012, 32, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Rijken, D.C.; de Munk, G.A.; Jie, A.F. Interaction of plasminogen activators and plasminogen with heparin: Effect of ionic strength. Thromb. Haemost. 1993, 70, 867–872. [Google Scholar] [PubMed]
- D’Souza, S.; Yang, W.; Marchetti, D.; Muir, C.; Farach-Carson, M.C.; Carson, D.D. Hip/rpl29 antagonizes vegf and fgf2 stimulated angiogenesis by interfering with hs-dependent responses. J. Cell. Biochem. 2008, 105, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Swanson, R.; Xiong, Y.; Richard, B.; Olson, S.T. Antiangiogenic antithrombin blocks the heparan sulfate-dependent binding of proangiogenic growth factors to their endothelial cell receptors: Evidence for differential binding of antiangiogenic and anticoagulant forms of antithrombin to proangiogenic heparan sulfate domains. J. Biol. Chem. 2006, 281, 37302–37310. [Google Scholar] [CrossRef] [PubMed]
- Frese, M.A.; Milz, F.; Dick, M.; Lamanna, W.C.; Dierks, T. Characterization of the human sulfatase sulf1 and its high affinity heparin/heparan sulfate interaction domain. J. Biol. Chem. 2009, 284, 28033–28044. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Liu, H.; Chen, Y.; Xin, X.; Li, J.; Hou, Y.; Zhang, Z.; Zhang, X.; Xie, C.; Geng, M.; et al. Oligomannurarate sulfate, a novel heparanase inhibitor simultaneously targeting basic fibroblast growth factor, combats tumor angiogenesis and metastasis. Cancer Res. 2006, 66, 8779–8787. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.F.; Li, Y.; Yang, V.C. The potential mechanism for the effect of heparin on tissue plasminogen activator-mediated plasminogen activation. Thromb. Res. 2000, 97, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Goretzki, L.; Lombardo, C.R.; Stallcup, W.B. Binding of the ng2 proteoglycan to kringle domains modulates the functional properties of angiostatin and plasmin(ogen). J. Biol. Chem. 2000, 275, 28625–28633. [Google Scholar] [CrossRef] [PubMed]
- Plouet, J.; Moro, F.; Bertagnolli, S.; Coldeboeuf, N.; Mazarguil, H.; Clamens, S.; Bayard, F. Extracellular cleavage of the vascular endothelial growth factor 189-amino acid form by urokinase is required for its mitogenic effect. J. Biol. Chem. 1997, 272, 13390–13396. [Google Scholar] [CrossRef] [PubMed]
- Wijelath, E.; Namekata, M.; Murray, J.; Furuyashiki, M.; Zhang, S.; Coan, D.; Wakao, M.; Harris, R.B.; Suda, Y.; Wang, L.; et al. Multiple mechanisms for exogenous heparin modulation of vascular endothelial growth factor activity. J. Cell. Biochem. 2010, 111, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Cochran, S.; Li, C.P.; Ferro, V. A surface plasmon resonance-based solution affinity assay for heparan sulfate-binding proteins. Glycoconj. J. 2009, 26, 577–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gitay-Goren, H.; Soker, S.; Vlodavsky, I.; Neufeld, G. The binding of vascular endothelial growth factor to its receptors is dependent on cell surface-associated heparin-like molecules. J. Biol. Chem. 1992, 267, 6093–6098. [Google Scholar] [PubMed]
- Soker, S.; Goldstaub, D.; Svahn, C.M.; Vlodavsky, I.; Levi, B.Z.; Neufeld, G. Variations in the size and sulfation of heparin modulate the effect of heparin on the binding of vegf165 to its receptors. Biochem. Biophys. Res. Commun. 1994, 203, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Norrby, K. 2.5 kda and 5.0 kda heparin fragments specifically inhibit microvessel sprouting and network formation in vegf165-mediated mammalian angiogenesis. Int. J. Exp. Pathol. 2000, 81, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Tessler, S.; Rockwell, P.; Hicklin, D.; Cohen, T.; Levi, B.Z.; Witte, L.; Lemischka, I.R.; Neufeld, G. Heparin modulates the interaction of vegf165 with soluble and cell associated flk-1 receptors. J. Biol. Chem. 1994, 269, 12456–12461. [Google Scholar] [PubMed]
- Kawada, A.; Hiura, N.; Tajima, S.; Takahara, H. Alginate oligosaccharides stimulate vegf-mediated growth and migration of human endothelial cells. Arch. Dermatol. Res. 1999, 291, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Hattori, H.; Takeshita, S.; Kurita, A.; Ishihara, M. Structural features in heparin that interact with vegf165 and modulate its biological activity. Glycobiology 1999, 9, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, J.E.; Fernig, D.G.; Ke, Y.; Wilkinson, M.C.; Gallagher, J.T. Identification of the basic fibroblast growth factor binding sequence in fibroblast heparan sulfate. J. Biol. Chem. 1992, 267, 10337–10341. [Google Scholar] [PubMed]
- Migdal, M.; Huppertz, B.; Tessler, S.; Comforti, A.; Shibuya, M.; Reich, R.; Baumann, H.; Neufeld, G. Neuropilin-1 is a placenta growth factor-2 receptor. J. Biol. Chem. 1998, 273, 22272–22278. [Google Scholar] [CrossRef] [PubMed]
- Lyon, M.; Deakin, J.A.; Mizuno, K.; Nakamura, T.; Gallagher, J.T. Interaction of hepatocyte growth factor with heparan sulfate. Elucidation of the major heparan sulfate structural determinants. J. Biol. Chem. 1994, 269, 11216–11223. [Google Scholar] [PubMed]
- Lyon, M.; Rushton, G.; Gallagher, J.T. The interaction of the transforming growth factor-betas with heparin/heparan sulfate is isoform-specific. J. Biol. Chem. 1997, 272, 18000–18006. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, N.; Talukder, A.H.; Ishihara, M.; Hara, S.; Yoshida, K.; Muramatsu, T. Structural characteristics of heparin-line domain required for interaction of midkine with embryonic neurons. Biochem. Biophys. Res. Commun. 1996, 220, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Kishibe, J.; Yamada, S.; Okada, Y.; Sato, J.; Ito, A.; Miyazaki, K.; Sugahara, K. Structural requirements of heparan sulfate for the binding to the tumor-derived adhesion factor/angiomodulin that induces cord-like structures to ecv-304 human carcinoma cells. J. Biol. Chem. 2000, 275, 15321–15329. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.L.; Rushton, G.; Jayson, G.C.; Avizienyte, E. Ovarian cancer cell heparan sulfate 6-o-sulfotransferases regulate an angiogenic program induced by heparin-binding epidermal growth factor (egf)-like growth factor/egf receptor signaling. J. Biol. Chem. 2014, 289, 10488–10501. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Tulipano, G.; Urbinati, C.; Tanghetti, E.; Giuliani, R.; Giacca, M.; Ciomei, M.; Corallini, A.; Presta, M. The basic domain in HIV-1 tat protein as a target for polysulfonated heparin-mimicking extracellular tat antagonists. J. Biol. Chem. 1998, 273, 16027–16037. [Google Scholar] [CrossRef] [PubMed]
- Spillmann, D.; Witt, D.; Lindahl, U. Defining the interleukin-8-binding domain of heparan sulfate. J. Biol. Chem. 1998, 273, 15487–15493. [Google Scholar] [CrossRef] [PubMed]
- Sadir, R.; Baleux, F.; Grosdidier, A.; Imberty, A.; Lortat-Jacob, H. Characterization of the stromal cell-derived factor-1alpha-heparin complex. J. Biol. Chem. 2001, 276, 8288–8296. [Google Scholar] [CrossRef] [PubMed]
- Lortat-Jacob, H.; Grimaud, J.A. Binding of interferon-gamma to heparan sulfate is restricted to the heparin-like domains and involves carboxylic--but not n-sulfated--groups. Biochim. Biophys. Acta 1992, 1117, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Yu, Y.; Leary, J.A. Effects of sulfate position on heparin octasaccharide binding to ccl2 examined by tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2006, 17, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Stringer, S.E.; Forster, M.J.; Mulloy, B.; Bishop, C.R.; Graham, G.J.; Gallagher, J.T. Characterization of the binding site on heparan sulfate for macrophage inflammatory protein 1alpha. Blood 2002, 100, 1543–1550. [Google Scholar] [PubMed]
- De Paz, J.L.; Moseman, E.A.; Noti, C.; Polito, L.; von Andrian, U.H.; Seeberger, P.H. Profiling heparin-chemokine interactions using synthetic tools. ACS Chem. Biol. 2007, 2, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Coltrini, D.; Caccia, P.; Dell’Era, P.; Zoppetti, G.; Oreste, P.; Valsasina, B.; Presta, M. Distinct role of 2-o-, n-, and 6-o-sulfate groups of heparin in the formation of the ternary complex with basic fibroblast growth factor and soluble fgf receptor-1. Biochem. Biophys. Res. Commun. 1994, 203, 450–458. [Google Scholar] [CrossRef]
- Uniewicz, K.A.; Ori, A.; Ahmed, Y.A.; Yates, E.A.; Fernig, D.G. Characterisation of the interaction of neuropilin-1 with heparin and a heparan sulfate mimetic library of heparin-derived sugars. Peer J. 2014, 2, e461. [Google Scholar] [CrossRef] [PubMed]
- Feitsma, K.; Hausser, H.; Robenek, H.; Kresse, H.; Vischer, P. Interaction of thrombospondin-1 and heparan sulfate from endothelial cells. Structural requirements of heparan sulfate. J. Biol. Chem. 2000, 275, 9396–9402. [Google Scholar] [CrossRef] [PubMed]
- Kreuger, J.; Matsumoto, T.; Vanwildemeersch, M.; Sasaki, T.; Timpl, R.; Claesson-Welsh, L.; Spillmann, D.; Lindahl, U. Role of heparan sulfate domain organization in endostatin inhibition of endothelial cell function. EMBO J. 2002, 21, 6303–6311. [Google Scholar] [CrossRef] [PubMed]
- Blackhall, F.H.; Merry, C.L.; Lyon, M.; Jayson, G.C.; Folkman, J.; Javaherian, K.; Gallagher, J.T. Binding of endostatin to endothelial heparan sulphate shows a differential requirement for specific sulphates. Biochem. J. 2003, 375, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Naggi, A.; Casu, B.; Perez, M.; Torri, G.; Cassinelli, G.; Penco, S.; Pisano, C.; Giannini, G.; Ishai-Michaeli, R.; Vlodavsky, I. Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded n-acetylation, and glycol splitting. J. Biol. Chem. 2005, 280, 12103–12113. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, M.; Denys, A.; Allain, F.; Vergoten, G. Molecular docking of heparin oligosaccharides with hep-ii heparin-binding domain of fibronectin reveals an interplay between the different positions of sulfate groups. Glycoconj. J. 2014, 31, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Presta, M. Angiogenic growth factors interactome and drug discovery: The contribution of surface plasmon resonance. Cytokine Growth Factor Rev. 2014, in press. [Google Scholar]
- Eriksson, A.E.; Cousens, L.S.; Weaver, L.H.; Matthews, B.W. Three-dimensional structure of human basic fibroblast growth factor. Proc. Natl. Acad. Sci. USA 1991, 88, 3441–3445. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, M.; Tyrrell, D.J.; Stauber, G.B.; Brown, S.; Cousens, L.S.; Stack, R.J. Preparation of affinity-fractionated, heparin-derived oligosaccharides and their effects on selected biological activities mediated by basic fibroblast growth factor. J. Biol. Chem. 1993, 268, 4675–4683. [Google Scholar] [PubMed]
- Guimond, S.; Maccarana, M.; Olwin, B.B.; Lindahl, U.; Rapraeger, A.C. Activating and inhibitory heparin sequences for fgf-2 (basic fgf). Distinct requirements for fgf-1, fgf-2, and fgf-4. J. Biol. Chem. 1993, 268, 23906–23914. [Google Scholar] [PubMed]
- Presta, M.; Leali, D.; Stabile, H.; Ronca, R.; Camozzi, M.; Moroni, E.; Nicoli, S.; Liekens, S.; Rusnati, M. Heparin derivatives and semisynthetic biotechnological heparins as angiogenesis inhibitors. Front. Med. Chem. 2005, 2, 371–391. [Google Scholar]
- Horowitz, A.; Tkachenko, E.; Simons, M. Fibroblast growth factor-specific modulation of cellular response by syndecan-4. J. Cell Biol. 2002, 157, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Urbinati, C.; Presta, M. Internalization of basic fibroblast growth factor (bfgf) in cultured endothelial cells: Role of the low affinity heparin-like bfgf receptors. J. Cell. Physiol. 1993, 154, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Presta, M.; Maier, J.A.; Rusnati, M.; Ragnotti, G. Basic fibroblast growth factor is released from endothelial extracellular matrix in a biologically active form. J. Cell. Physiol. 1989, 140, 68–74. [Google Scholar] [PubMed]
- Ribatti, D.; Leali, D.; Vacca, A.; Giuliani, R.; Gualandris, A.; Roncali, L.; Nolli, M.L.; Presta, M. In vivo angiogenic activity of urokinase: Role of endogenous fibroblast growth factor-2. J. Cell Sci. 1999, 112, 4213–4221. [Google Scholar] [PubMed]
- Gospodarowicz, D.; Cheng, J. Heparin protects basic and acidic fgf from inactivation. J. Cell. Physiol. 1986, 128, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.; Rifkin, D.B. Interaction of heparin with human basic fibroblast growth factor: Protection of the angiogenic protein from proteolytic degradation by a glycosaminoglycan. J. Cell. Physiol. 1989, 138, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Flaumenhaft, R.; Moscatelli, D.; Rifkin, D.B. Heparin and heparan sulfate increase the radius of diffusion and action of basic fibroblast growth factor. J. Cell Biol. 1990, 111, 1651–1659. [Google Scholar] [CrossRef] [PubMed]
- Casu, B.; Guerrini, M.; Naggi, A.; Perez, M.; Torri, G.; Ribatti, D.; Carminati, P.; Giannini, G.; Penco, S.; Pisano, C.; et al. Short heparin sequences spaced by glycol-split uronate residues are antagonists of fibroblast growth factor 2 and angiogenesis inhibitors. Biochemistry 2002, 41, 10519–10528. [Google Scholar] [CrossRef] [PubMed]
- Klagsbrun, M.; Baird, A. A dual receptor system is required for basic fibroblast growth factor activity. Cell 1991, 67, 229–231. [Google Scholar] [CrossRef] [PubMed]
- Coltrini, D.; Rusnati, M.; Zoppetti, G.; Oreste, P.; Grazioli, G.; Naggi, A.; Presta, M. Different effects of mucosal, bovine lung and chemically modified heparin on selected biological properties of basic fibroblast growth factor. Biochem. J. 1994, 303, 583–590. [Google Scholar] [PubMed]
- Krilleke, D.; Ng, Y.S.; Shima, D.T. The heparin-binding domain confers diverse functions of vegf-a in development and disease: A structure-function study. Biochem. Soc. Trans. 2009, 37, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.D.; Pantoliano, M.W.; Springer, B.A. Energetic characterization of the basic fibroblast growth factor-heparin interaction: Identification of the heparin binding domain. Biochemistry 1994, 33, 3831–3840. [Google Scholar] [CrossRef] [PubMed]
- Rose, K.; Kriha, D.; Pallast, S.; Junker, V.; Klumpp, S.; Krieglstein, J. Basic fibroblast growth factor: Lysine 134 is essential for its neuroprotective activity. Neurochem. Int. 2007, 51, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L.; Burke, D.F.; von Delft, F.; Mulloy, B.; Blundell, T.L. Crystal structure of fibroblast growth factor receptor ectodomain bound to ligand and heparin. Nature 2000, 407, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, W.; Nagata, K.; Hatanaka, H.; Inui, T.; Kimura, T.; Muramatsu, T.; Yoshida, K.; Tasumi, M.; Inagaki, F. Solution structure of midkine, a new heparin-binding growth factor. EMBO J. 1997, 16, 6936–6946. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Casas-Finet, J.R.; Heath Coats, R.; Kaufman, J.D.; Stahl, S.J.; Wingfield, P.T.; Rubin, J.S.; Bottaro, D.P.; Byrd, R.A. Identification and dynamics of a heparin-binding site in hepatocyte growth factor. Biochemistry 1999, 38, 14793–14802. [Google Scholar] [CrossRef] [PubMed]
- Merkulova-Rainon, T.; England, P.; Ding, S.; Demerens, C.; Tobelem, G. The n-terminal domain of hepatocyte growth factor inhibits the angiogenic behavior of endothelial cells independently from binding to the c-met receptor. J. Biol. Chem. 2003, 278, 37400–37408. [Google Scholar] [CrossRef] [PubMed]
- Fazekas, K.; Janovics, A.; Dome, B.; Koska, P.; Albini, A.; Timar, J. Effect of hgf-like basic hexapeptides on angiogenesis. Microvasc. Res. 2001, 62, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Mobius, K.; Nordsieck, K.; Pichert, A.; Samsonov, S.A.; Thomas, L.; Schiller, J.; Kalkhof, S.; Teresa Pisabarro, M.; Beck-Sickinger, A.G.; Huster, D. Investigation of lysine side chain interactions of interleukin-8 with heparin and other glycosaminoglycans studied by a methylation-nmr approach. Glycobiology 2013, 23, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Lortat-Jacob, H.; Grimaud, J.A. Interferon-gamma binds to heparan sulfate by a cluster of amino acids located in the c-terminal part of the molecule. FEBS Lett. 1991, 280, 152–154. [Google Scholar] [CrossRef] [PubMed]
- McCaffrey, T.A.; Falcone, D.J.; Du, B. Transforming growth factor-beta 1 is a heparin-binding protein: Identification of putative heparin-binding regions and isolation of heparins with varying affinity for tgf-beta 1. J. Cell. Physiol. 1992, 152, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Sebollela, A.; Cagliari, T.C.; Limaverde, G.S.; Chapeaurouge, A.; Sorgine, M.H.; Coelho-Sampaio, T.; Ramos, C.H.; Ferreira, S.T. Heparin-binding sites in granulocyte-macrophage colony-stimulating factor. Localization and regulation by histidine ionization. J. Biol. Chem. 2005, 280, 31949–31956. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.A.; Higashiyama, S.; Wood, K.; Pollitt, N.S.; Damm, D.; McEnroe, G.; Garrick, B.; Ashton, N.; Lau, K.; Hancock, N.; et al. Characterization of sequences within heparin-binding egf-like growth factor that mediate interaction with heparin. J. Biol. Chem. 1994, 269, 2541–2549. [Google Scholar] [PubMed]
- Chakravarty, L.; Rogers, L.; Quach, T.; Breckenridge, S.; Kolattukudy, P.E. Lysine 58 and histidine 66 at the c-terminal alpha-helix of monocyte chemoattractant protein-1 are essential for glycosaminoglycan binding. J. Biol. Chem. 1998, 273, 29641–29647. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.A.; Piper, M.; Fukuhara, N.; Strochlic, L.; Cho, G.; Howitt, J.A.; Ahmed, Y.; Powell, A.K.; Turnbull, J.E.; Holt, C.E.; et al. A molecular mechanism for the heparan sulfate dependence of slit-robo signaling. J. Biol. Chem. 2006, 281, 39693–39698. [Google Scholar] [CrossRef] [PubMed]
- Brickman, Y.G.; Ford, M.D.; Small, D.H.; Bartlett, P.F.; Nurcombe, V. Heparan sulfates mediate the binding of basic fibroblast growth factor to a specific receptor on neural precursor cells. J. Biol. Chem. 1995, 270, 24941–24948. [Google Scholar] [CrossRef] [PubMed]
- Ori, A.; Free, P.; Courty, J.; Wilkinson, M.C.; Fernig, D.G. Identification of heparin-binding sites in proteins by selective labeling. Mol. Cell. Proteomics 2009, 8, 2256–2265. [Google Scholar] [CrossRef] [PubMed]
- Stahl, S.; Gaetzner, S.; Mueller, T.D.; Felbor, U. Endostatin phenylalanines 31 and 34 define a receptor binding site. Genes Cells 2005, 10, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Levy-Adam, F.; Abboud-Jarrous, G.; Guerrini, M.; Beccati, D.; Vlodavsky, I.; Ilan, N. Identification and characterization of heparin/heparan sulfate binding domains of the endoglycosidase heparanase. J. Biol. Chem. 2005, 280, 20457–20466. [Google Scholar] [CrossRef] [PubMed]
- Holloway, D.E.; Chavali, G.B.; Hares, M.C.; Subramanian, V.; Acharya, K.R. Structure of murine angiogenin: Features of the substrate- and cell-binding regions and prospects for inhibitor-binding studies. Acta Crystallogr. Sect. D Biol. Crystallogr. 2005, 61, 1568–1578. [Google Scholar] [CrossRef]
- Moroianu, J.; Riordan, J.F. Nuclear translocation of angiogenin in proliferating endothelial cells is essential to its angiogenic activity. Proc. Natl. Acad. Sci. USA 1994, 91, 1677–1681. [Google Scholar] [CrossRef] [PubMed]
- Novotny, W.F.; Maffi, T.; Mehta, R.L.; Milner, P.G. Identification of novel heparin-releasable proteins, as well as the cytokines midkine and pleiotrophin, in human postheparin plasma. Arterioscler. Thromb. 1993, 13, 1798–1805. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Talukder, A.H.; Ishihara, M.; Hara, S.; Yoshida, K.; Muramatsu, T.; Kaneda, N. Limited proteolysis by chymotrypsin of midkine and inhibition by heparin binding. Biochem. Biophys. Res. Commun. 1996, 228, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Deepa, S.S.; Yamada, S.; Zako, M.; Goldberger, O.; Sugahara, K. Chondroitin sulfate chains on syndecan-1 and syndecan-4 from normal murine mammary gland epithelial cells are structurally and functionally distinct and cooperate with heparan sulfate chains to bind growth factors. A novel function to control binding of midkine, pleiotrophin, and basic fibroblast growth factor. J. Biol. Chem. 2004, 279, 37368–37376. [Google Scholar] [CrossRef] [PubMed]
- Stabile, H.; Mitola, S.; Moroni, E.; Belleri, M.; Nicoli, S.; Coltrini, D.; Peri, F.; Pessi, A.; Orsatti, L.; Talamo, F.; et al. Bone morphogenic protein antagonist drm/gremlin is a novel proangiogenic factor. Blood 2007, 109, 1834–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, T.A.; Sandhu, S.K.; Alam, S.M.; de Bono, J.S. Hgf/c-met targeted therapeutics: Novel strategies for cancer medicine. Curr. Drug Targets 2011, 12, 2045–2058. [Google Scholar] [CrossRef] [PubMed]
- Rubin, J.S.; Day, R.M.; Breckenridge, D.; Atabey, N.; Taylor, W.G.; Stahl, S.J.; Wingfield, P.T.; Kaufman, J.D.; Schwall, R.; Bottaro, D.P. Dissociation of heparan sulfate and receptor binding domains of hepatocyte growth factor reveals that heparan sulfate-c-met interaction facilitates signaling. J. Biol. Chem. 2001, 276, 32977–32983. [Google Scholar] [CrossRef] [PubMed]
- Ashikari, S.; Habuchi, H.; Kimata, K. Characterization of heparan sulfate oligosaccharides that bind to hepatocyte growth factor. J. Biol. Chem. 1995, 270, 29586–29593. [Google Scholar] [CrossRef] [PubMed]
- Salbach, P.B.; Bruckmann, M.; Turovets, O.; Kreuzer, J.; Kubler, W.; Walter-Sack, I. Heparin-mediated selective release of hepatocyte growth factor in humans. Br. J. Clin. Pharmacol. 2000, 50, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Zioncheck, T.F.; Richardson, L.; Liu, J.; Chang, L.; King, K.L.; Bennett, G.L.; Fugedi, P.; Chamow, S.M.; Schwall, R.H.; Stack, R.J. Sulfated oligosaccharides promote hepatocyte growth factor association and govern its mitogenic activity. J. Biol. Chem. 1995, 270, 16871–16878. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Ishii, T.; Hara, H.; Sugiura, N.; Kimata, K.; Akamatsu, N. Hepatocyte growth factor immobilized onto culture substrates through heparin and matrigel enhances DNA synthesis in primary rat hepatocytes. Exp. Cell Res. 1994, 211, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Urbinati, C.; Ravelli, C.; Tanghetti, E.; Belleri, M.; Giacopuzzi, E.; Monti, E.; Presta, M.; Rusnati, M. Substrate-immobilized hiv-1 tat drives vegfr2/alpha(v)beta(3)-integrin complex formation and polarization in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, e25–e34. [Google Scholar] [CrossRef] [PubMed]
- Bugatti, A.; Chiodelli, P.; Rosenbluh, J.; Loyter, A.; Rusnati, M. Bsa conjugates bearing multiple copies of the basic domain of HIV-1 tat: Prototype for the development of multitarget inhibitors of extracellular tat. Antivir. Res. 2010, 87, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Urbinati, C.; Caputo, A.; Possati, L.; Lortat-Jacob, H.; Giacca, M.; Ribatti, D.; Presta, M. Pentosan polysulfate as an inhibitor of extracellular hiv-1 tat. J. Biol. Chem. 2001, 276, 22420–22425. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Tulipano, G.; Spillmann, D.; Tanghetti, E.; Oreste, P.; Zoppetti, G.; Giacca, M.; Presta, M. Multiple interactions of HIV-I tat protein with size-defined heparin oligosaccharides. J. Biol. Chem. 1999, 274, 28198–28205. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.C.; Samaniego, F.; Nair, B.C.; Buonaguro, L.; Ensoli, B. HIV-1 tat protein exits from cells via a leaderless secretory pathway and binds to extracellular matrix-associated heparan sulfate proteoglycans through its basic region. Aids 1997, 11, 1421–1431. [Google Scholar] [CrossRef] [PubMed]
- Mitola, S.; Soldi, R.; Zanon, I.; Barra, L.; Gutierrez, M.I.; Berkhout, B.; Giacca, M.; Bussolino, F. Identification of specific molecular structures of human immunodeficiency virus type 1 tat relevant for its biological effects on vascular endothelial cells. J. Virol. 2000, 74, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Benelli, R.; Presta, M.; Rusnati, M.; Ziche, M.; Rubartelli, A.; Paglialunga, G.; Bussolino, F.; Noonan, D. HIV-tat protein is a heparin-binding angiogenic growth factor. Oncogene 1996, 12, 289–297. [Google Scholar] [PubMed]
- Garcia-Olivas, R.; Hoebeke, J.; Castel, S.; Reina, M.; Fager, G.; Lustig, F.; Vilaro, S. Differential binding of platelet-derived growth factor isoforms to glycosaminoglycans. Histochem. Cell Biol. 2003, 120, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Raines, E.W.; Ross, R. Compartmentalization of pdgf on extracellular binding sites dependent on exon-6-encoded sequences. J. Cell Biol. 1992, 116, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Rolny, C.; Spillmann, D.; Lindahl, U.; Claesson-Welsh, L. Heparin amplifies platelet-derived growth factor (pdgf)- bb-induced pdgf alpha -receptor but not pdgf beta-receptor tyrosine phosphorylation in heparan sulfate-deficient cells. Effects on signal transduction and biological responses. J. Biol. Chem. 2002, 277, 19315–19321. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, L.; van Meeteren, L.A. Transforming growth factor beta family members in regulation of vascular function: In the light of vascular conditional knockouts. Exp. Cell Res. 2013, 319, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- McCaffrey, T.A.; Falcone, D.J.; Vicente, D.; Du, B.; Consigli, S.; Borth, W. Protection of transforming growth factor-beta 1 activity by heparin and fucoidan. J. Cell. Physiol. 1994, 159, 51–59. [Google Scholar] [CrossRef]
- Vilchis-Landeros, M.M.; Montiel, J.L.; Mendoza, V.; Mendoza-Hernandez, G.; Lopez-Casillas, F. Recombinant soluble betaglycan is a potent and isoform-selective transforming growth factor-beta neutralizing agent. Biochem. J. 2001, 355, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Azfer, A.; Zhelyabovska, O.; Fatma, S.; Kolattukudy, P.E. Monocyte chemotactic protein (mcp)-1 promotes angiogenesis via a novel transcription factor, mcp-1-induced protein (mcpip). J. Biol. Chem. 2008, 283, 14542–14551. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.K.; Paavola, C.D.; Johnson, Z.; Gaudry, J.P.; Geretti, E.; Borlat, F.; Kungl, A.J.; Proudfoot, A.E.; Handel, T.M. Identification of the glycosaminoglycan binding site of the CC chemokine, MCP-1: Implications for structure and function in vivo. J. Biol. Chem. 2004, 279, 22294–22305. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Sweeney, M.D.; Saad, O.M.; Crown, S.E.; Hsu, A.R.; Handel, T.M.; Leary, J.A. Chemokine-glycosaminoglycan binding: Specificity for CCR2 ligand binding to highly sulfated oligosaccharides using FTICR mass spectrometry. J. Biol. Chem. 2005, 280, 32200–32208. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Palmer, A.C.; Fritchley, S.J.; Maley, Y.; Kirby, J.A. Multimerization of monocyte chemoattractant protein-1 is not required for glycosaminoglycan-dependent transendothelial chemotaxis. Biochem. J. 2001, 358, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, H.; Watanabe, N.; Hirose, M.; Sun, X.; Atarashi, K.; Kimura, T.; Shikata, K.; Matsuda, M.; Ogawa, D.; Heljasvaara, R.; et al. Collagen xviii, a basement membrane heparan sulfate proteoglycan, interacts with l-selectin and monocyte chemoattractant protein-1. J. Biol. Chem. 2003, 278, 13069–13076. [Google Scholar] [CrossRef] [PubMed]
- Lortat-Jacob, H.; Baltzer, F.; Grimaud, J.A. Heparin decreases the blood clearance of interferon-gamma and increases its activity by limiting the processing of its carboxyl-terminal sequence. J. Biol. Chem. 1996, 271, 16139–16143. [Google Scholar] [CrossRef] [PubMed]
- Douglas, M.S.; Rix, D.A.; Dark, J.H.; Talbot, D.; Kirby, J.A. Examination of the mechanism by which heparin antagonizes activation of a model endothelium by interferon-gamma (ifn-gamma). Clin. Exp. Immunol. 1997, 107, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Caccuri, F.; Giagulli, C.; Bugatti, A.; Benetti, A.; Alessandri, G.; Ribatti, D.; Marsico, S.; Apostoli, P.; Slevin, M.A.; Rusnati, M.; et al. HIV-1 matrix protein p17 promotes angiogenesis via chemokine receptors cxcr1 and cxcr2. Proc. Natl. Acad. Sci. USA 2012, 109, 14580–14585. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, M.A.; Baronio, M.; Poiesi, C. HIV-1 p17 matrix protein interacts with heparan sulfate side chain of CD44v3, syndecan-2, and syndecan-4 proteoglycans expressed on human activated CD4+ t cells affecting tumor necrosis factor alpha and interleukin 2 production. J. Biol. Chem. 2011, 286, 19541–19548. [Google Scholar] [CrossRef] [PubMed]
- Poiesi, C.; De Francesco, M.A.; Baronio, M.; Manca, N. HIV-1 p17 binds heparan sulfate proteoglycans to activated cd4(+) t cells. Virus Res. 2008, 132, 25–32. [Google Scholar] [CrossRef]
- Valdembri, D.; Serini, G.; Vacca, A.; Ribatti, D.; Bussolino, F. In vivo activation of jak2/stat-3 pathway during angiogenesis induced by gm-csf. FASEB J. 2002, 16, 225–227. [Google Scholar] [PubMed]
- Wettreich, A.; Sebollela, A.; Carvalho, M.A.; Azevedo, S.P.; Borojevic, R.; Ferreira, S.T.; Coelho-Sampaio, T. Acidic ph modulates the interaction between human granulocyte-macrophage colony-stimulating factor and glycosaminoglycans. J. Biol. Chem. 1999, 274, 31468–31475. [Google Scholar] [CrossRef] [PubMed]
- Lantz, M.; Thysell, H.; Nilsson, E.; Olsson, I. On the binding of tumor necrosis factor (tnf) to heparin and the release in vivo of the tnf-binding protein i by heparin. J. Clin. Investig. 1991, 88, 2026–2031. [Google Scholar] [CrossRef] [PubMed]
- Tufvesson, E.; Westergren-Thorsson, G. Tumour necrosis factor-alpha interacts with biglycan and decorin. FEBS Lett. 2002, 530, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Waters, J.P.; Pober, J.S.; Bradley, J.R. Tumour necrosis factor and cancer. J. Pathol. 2013, 230, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, M.; Starzec, A.; Vassy, R.; Perret, G.Y.; Crepin, M. Distinct heparin binding sites on vegf165 and its receptors revealed by their interaction with a non sulfated glycoaminoglycan (napac). Biochim. Biophys. Acta 2008, 1780, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Lee, S.T. The fourth immunoglobulin-like loop in the extracellular domain of flt-1, a vegf receptor, includes a major heparin-binding site. Biochem. Biophys. Res. Commun. 1999, 264, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, V.P.; Paredes-Gamero, E.J.; Monteiro, H.P.; Rocha, H.A.; Trindade, E.S.; Nader, H.B. Heparin-integrin interaction in endothelial cells: Downstream signaling and heparan sulfate expression. J. Cell. Physiol. 2012, 227, 2740–2749. [Google Scholar] [CrossRef] [PubMed]
- Taraboletti, G.; Rusnati, M.; Ragona, L.; Colombo, G. Targeting tumor angiogenesis with tsp-1-based compounds: Rational design of antiangiogenic mimetics of endogenous inhibitors. Oncotarget 2010, 1, 662–673. [Google Scholar] [PubMed]
- Ferrari do Outeiro-Bernstein, M.A.; Nunes, S.S.; Andrade, A.C.; Alves, T.R.; Legrand, C.; Morandi, V. A recombinant nh(2)-terminal heparin-binding domain of the adhesive glycoprotein, thrombospondin-1, promotes endothelial tube formation and cell survival: A possible role for syndecan-4 proteoglycan. Matrix Biol. 2002, 21, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.V.; Benslimane-Ahmim, Z.; Egot, M.; Lokajczyk, A.; Grelac, F.; Galy-Fauroux, I.; Juliano, L.; le-Bonniec, B.; Takiya, C.M.; Fischer, A.M.; et al. A motif within the n-terminal domain of tsp-1 specifically promotes the proangiogenic activity of endothelial colony-forming cells. Biochem. Pharmacol. 2012, 84, 1014–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clamp, A.; Blackhall, F.H.; Henrioud, A.; Jayson, G.C.; Javaherian, K.; Esko, J.; Gallagher, J.T.; Merry, C.L. The morphogenic properties of oligomeric endostatin are dependent on cell surface heparan sulfate. J. Biol. Chem. 2006, 281, 14813–14822. [Google Scholar] [CrossRef] [PubMed]
- Reis, R.C.; Schuppan, D.; Barreto, A.C.; Bauer, M.; Bork, J.P.; Hassler, G.; Coelho-Sampaio, T. Endostatin competes with bfgf for binding to heparin-like glycosaminoglycans. Biochem. Biophys. Res. Commun. 2005, 333, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Rosenkilde, M.M.; Schwartz, T.W. The chemokine system—A major regulator of angiogenesis in health and disease. APMIS 2004, 112, 481–495. [Google Scholar] [CrossRef]
- Goger, B.; Halden, Y.; Rek, A.; Mosl, R.; Pye, D.; Gallagher, J.; Kungl, A.J. Different affinities of glycosaminoglycan oligosaccharides for monomeric and dimeric interleukin-8: A model for chemokine regulation at inflammatory sites. Biochemistry 2002, 41, 1640–1646. [Google Scholar] [CrossRef] [PubMed]
- Hoogewerf, A.J.; Kuschert, G.S.; Proudfoot, A.E.; Borlat, F.; Clark-Lewis, I.; Power, C.A.; Wells, T.N. Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry 1997, 36, 13570–13578. [Google Scholar] [CrossRef] [PubMed]
- Whittall, C.; Kehoe, O.; King, S.; Rot, A.; Patterson, A.; Middleton, J. A chemokine self-presentation mechanism involving formation of endothelial surface microstructures. J. Immunol. 2013, 190, 1725–1736. [Google Scholar] [CrossRef] [PubMed]
- Halden, Y.; Rek, A.; Atzenhofer, W.; Szilak, L.; Wabnig, A.; Kungl, A.J. Interleukin-8 binds to syndecan-2 on human endothelial cells. Biochem. J. 2004, 377, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Presta, M.; Belleri, M.; Vecchi, A.; Hesselgesser, J.; Mantovani, A.; Horuk, R. Noncompetitive, chemokine-mediated inhibition of basic fibroblast growth factor-induced endothelial cell proliferation. J. Biol. Chem. 1998, 273, 7911–7919. [Google Scholar] [CrossRef] [PubMed]
- Mercurius, K.O.; Morla, A.O. Cell adhesion and signaling on the fibronectin 1st type iii repeat; requisite roles for cell surface proteoglycans and integrins. BMC Cell Biol. 2001, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Fredenburgh, J.C.; Leslie, B.A.; Stafford, A.R.; Lim, T.; Chan, H.H.; Weitz, J.I. Zn2+ mediates high affinity binding of heparin to the alphac domain of fibrinogen. J. Biol. Chem. 2013, 288, 29394–29402. [Google Scholar] [CrossRef] [PubMed]
- Ushiro, S.; Ono, M.; Izumi, H.; Kohno, K.; Taniguchi, N.; Higashiyama, S.; Kuwano, M. Heparin-binding epidermal growth factor-like growth factor: P91 activation induction of plasminogen activator/inhibitor, and tubular morphogenesis in human microvascular endothelial cells. Jpn. J. Cancer Res. Gann 1996, 87, 68–77. [Google Scholar] [CrossRef]
- Gorsi, B.; Liu, F.; Ma, X.; Chico, T.J.; Shrinivasan, A.; Kramer, K.L.; Bridges, E.; Monteiro, R.; Harris, A.L.; Patient, R.; et al. The heparan sulfate editing enzyme Sulf1 plays a novel role in zebrafish VegfA mediated arterial venous identity. Angiogenesis 2014, 17, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Yang, J.; Wang, D.; Cao, L.; Tan, W.; Qian, H.; Sun, B.; Qian, Q.; Yin, Z.; Wu, M.; et al. Hsulf-1 gene exhibits anticancer efficacy through negatively regulating vegfr-2 signaling in human cancers. PLoS ONE 2011, 6, e23274. [Google Scholar] [CrossRef]
- Neufeld, G.; Gospodarowicz, D. Protamine sulfate inhibits mitogenic activities of the extracellular matrix and fibroblast growth factor, but potentiates that of epidermal growth factor. J. Cell. Physiol. 1987, 132, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Dell’Era, P.; Urbinati, C.; Tanghetti, E.; Massardi, M.L.; Nagamine, Y.; Monti, E.; Presta, M. A distinct basic fibroblast growth factor (fgf-2)/fgf receptor interaction distinguishes urokinase-type plasminogen activator induction from mitogenicity in endothelial cells. Mol. Biol. Cell 1996, 7, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.; Folkman, J. Protamine is an inhibitor of angiogenesis. Nature 1982, 297, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.K.; Larsson, H.; Dixelius, J.; Johansson, I.; Lee, C.; Oellig, C.; Bjork, I.; Claesson-Welsh, L. A fragment of histidine-rich glycoprotein is a potent inhibitor of tumor vascularization. Cancer Res. 2004, 64, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Miao, R.Q.; Chen, V.; Chao, L.; Chao, J. Structural elements of kallistatin required for inhibition of angiogenesis. Am. J. Physiol. Cell Physiol. 2003, 284, C1604–C1613. [Google Scholar] [PubMed]
- Vanwildemeersch, M.; Olsson, A.K.; Gottfridsson, E.; Claesson-Welsh, L.; Lindahl, U.; Spillmann, D. The anti-angiogenic his/pro-rich fragment of histidine-rich glycoprotein binds to endothelial cell heparan sulfate in a Zn2+-dependent manner. J. Biol. Chem. 2006, 281, 10298–10304. [Google Scholar] [CrossRef] [PubMed]
- Luster, A.D.; Greenberg, S.M.; Leder, P. The ip-10 chemokine binds to a specific cell surface heparan sulfate site shared with platelet factor 4 and inhibits endothelial cell proliferation. J. Exp. Med. 1995, 182, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Maurer, A.M.; Han, Z.C.; Dhermy, D.; Briere, J. Inhibitory effect of platelet factor 4 on human erythroleukemic cells is dependent on cell surface heparan sulfate. J. Lab. Clin. Med. 1996, 127, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Chadderton, N.S.; Stringer, S.E. Interaction of platelet factor 4 with fibroblast growth factor 2 is stabilised by heparan sulphate. Int. J. Biochem. Cell Biol. 2003, 35, 1052–1055. [Google Scholar] [CrossRef] [PubMed]
- Gengrinovitch, S.; Greenberg, S.M.; Cohen, T.; Gitay-Goren, H.; Rockwell, P.; Maione, T.E.; Levi, B.Z.; Neufeld, G. Platelet factor-4 inhibits the mitogenic activity of vegf121 and vegf165 using several concurrent mechanisms. J. Biol. Chem. 1995, 270, 15059–15065. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, M.G.; Tsoi, C.K.; Jarvelainen, H.T.; Wight, T.N. Selective expression and processing of biglycan during migration of bovine aortic endothelial cells. The role of endogenous basic fibroblast growth factor. J. Biol. Chem. 1997, 272, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Korner, G.; Ishai-Michaeli, R.; Bashkin, P.; Bar-Shavit, R.; Fuks, Z. Extracellular matrix-resident growth factors and enzymes: Possible involvement in tumor metastasis and angiogenesis. Cancer Metastasis Rev. 1990, 9, 203–226. [Google Scholar] [CrossRef] [PubMed]
- Cummings, R.D. The repertoire of glycan determinants in the human glycome. Mol. BioSyst. 2009, 5, 1087–1104. [Google Scholar] [CrossRef] [PubMed]
- Rek, A.; Krenn, E.; Kungl, A.J. Therapeutically targeting protein-glycan interactions. Br. J. Pharmacol. 2009, 157, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Takahashi, K.; Campion, S.L.; Liu, Y.; Gustavsen, G.G.; Pena, L.A.; Zamora, P.O. Synthetic peptide F2A4-K-NS mimics fibroblast growth factor-2 in vitro and is angiogenic in vivo. Int. J. Mol. Med. 2006, 17, 833–839. [Google Scholar] [PubMed]
- Lee, T.Y.; Folkman, J.; Javaherian, K. Hspg-binding peptide corresponding to the exon 6a-encoded domain of vegf inhibits tumor growth by blocking angiogenesis in murine model. PLoS ONE 2010, 5, e9945. [Google Scholar] [CrossRef] [PubMed]
- Hamma-Kourbali, Y.; Bernard-Pierrot, I.; Heroult, M.; Dalle, S.; Caruelle, D.; Milhiet, P.E.; Fernig, D.G.; Delbe, J.; Courty, J. Inhibition of the mitogenic, angiogenic and tumorigenic activities of pleiotrophin by a synthetic peptide corresponding to its c-thrombospondin repeat-i domain. J. Cell. Physiol. 2008, 214, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Mader, J.S.; Smyth, D.; Marshall, J.; Hoskin, D.W. Bovine lactoferricin inhibits basic fibroblast growth factor- and vascular endothelial growth factor165-induced angiogenesis by competing for heparin-like binding sites on endothelial cells. Am. J. Pathol. 2006, 169, 1753–1766. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Favaloro, E.J. Recombinant platelet factor 4: A therapeutic, anti-neoplastic chimera? Semin. Thromb. Hemost. 2010, 36, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Parry, G.C.; Levin, E.G. Inhibition of tumor cell migration by LD22–4, an N-terminal fragment of 24-kDa FGF2, is mediated by neuropilin 1. Cancer Res. 2013, 73, 3316–3325. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Lai, H.; Zouaoui, R.; Duffner, J.; Zhou, H.; Jayaraman, L.P.; Zhao, G.; Ganguly, T.; Kishimoto, T.K.; Venkataraman, G. Bioactivity screening of partially desulfated low-molecular-weight heparins: A structure/activity relationship study. Glycobiology 2011, 21, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; McCallum, S.A.; Xiao, Z.; Zhang, F.; Linhardt, R.J. Binding affinities of vascular endothelial growth factor (vegf) for heparin-derived oligosaccharides. Biosci. Rep. 2011, 32, 71–81. [Google Scholar] [CrossRef]
- Kim, J.; Al-Hilal, T.A.; Chung, S.W.; Kim, S.Y.; Ryu, G.H.; Son, W.C.; Byun, Y. Antiangiogenic and anticancer effect of an orally active low molecular weight heparin conjugates and its application to lung cancer chemoprevention. J. Control. Release 2015, 199, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.W.; Bae, S.M.; Lee, M.; Al-Hilal, T.A.; Lee, C.K.; Kim, J.K.; Kim, I.S.; Kim, S.Y.; Byun, Y. Lht7, a chemically modified heparin, inhibits multiple stages of angiogenesis by blocking vegf, fgf2 and pdgf-b signaling pathways. Biomaterials 2014, 37C, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Pisano, C.; Aulicino, C.; Vesci, L.; Casu, B.; Naggi, A.; Torri, G.; Ribatti, D.; Belleri, M.; Rusnati, M.; Presta, M. Undersulfated, low-molecular-weight glycol-split heparin as an antiangiogenic vegf antagonist. Glycobiology 2005, 15, 1C–6C. [Google Scholar] [CrossRef] [PubMed]
- Basappa; Murugan, S.; Kavitha, C.V.; Purushothaman, A.; Nevin, K.G.; Sugahara, K.; Rangappa, K.S. A small oxazine compound as an anti-tumor agent: A novel pyranoside mimetic that binds to vegf, hb-egf, and tnf-alpha. Cancer Lett. 2010, 297, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.J.; Huang, N.K.; Chang, T.T.; Wang, D.L.; Lu, M.K. Study for anti-angiogenic activities of polysaccharides isolated from antrodia cinnamomea in endothelial cells. Life Sci. 2005, 76, 3029–3042. [Google Scholar] [CrossRef] [PubMed]
- Koyanagi, S.; Tanigawa, N.; Nakagawa, H.; Soeda, S.; Shimeno, H. Oversulfation of fucoidan enhances its anti-angiogenic and antitumor activities. Biochem. Pharmacol. 2003, 65, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Hamma-Kourbali, Y.; Vassy, R.; Starzec, A.; Le Meuth-Metzinger, V.; Oudar, O.; Bagheri-Yarmand, R.; Perret, G.; Crepin, M. Vascular endothelial growth factor 165 (vegf(165)) activities are inhibited by carboxymethyl benzylamide dextran that competes for heparin binding to vegf(165) and vegf(165).Kdr complexes. J. Biol. Chem. 2001, 276, 39748–39754. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, C.; Cochran, S.; Jimmink, S.; Ferro, V. Synthesis of a heparan sulfate mimetic library targeting fgf and vegf via click chemistry on a monosaccharide template. ChemMedChem 2012, 7, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Maynard, H.D.; Hubbell, J.A. Discovery of a sulfated tetrapeptide that binds to vascular endothelial growth factor. Acta Biomater. 2005, 1, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Lake, A.C.; Vassy, R.; Di Benedetto, M.; Lavigne, D.; Le Visage, C.; Perret, G.Y.; Letourneur, D. Low molecular weight fucoidan increases vegf165-induced endothelial cell migration by enhancing vegf165 binding to vegfr-2 and nrp1. J. Biol. Chem. 2006, 281, 37844–37852. [Google Scholar] [CrossRef] [PubMed]
- Rezzola, S.; Monte, M.D.; Belleri, M.; Bugatti, A.; Chiodelli, P.; Corsini, M.; Cammalleri, M.; Cancarini, A.; Morbidelli, L.; Oreste, P.; et al. Therapeutic potential of anti-angiogenic multi-target n,o-sulfated E. Coli k5 polysaccharide in diabetic retinopathy. Diabetes 2015. [Google Scholar] [CrossRef]
- Casu, B.; Guerrini, M.; Guglieri, S.; Naggi, A.; Perez, M.; Torri, G.; Cassinelli, G.; Ribatti, D.; Carminati, P.; Giannini, G.; et al. Undersulfated and glycol-split heparins endowed with antiangiogenic activity. J. Med. Chem. 2004, 47, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.G.; Mrabat, H.; Yu, L.; Hales, C.A.; Li, B.; Moore, C.N.; Zhang, F.; Linhardt, R.J. Anti-proliferative effects of o-acyl-low-molecular-weight heparin derivatives on bovine pulmonary artery smooth muscle cells. Glycoconj. J. 2011, 28, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Kasbauer, C.W.; Paper, D.H.; Franz, G. Sulfated beta-(1-->4)-galacto-oligosaccharides and their effect on angiogenesis. Carbohydr. Res. 2001, 330, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Foxall, C.; Wei, Z.; Schaefer, M.E.; Casabonne, M.; Fugedi, P.; Peto, C.; Castellot, J.J., Jr.; Brandley, B.K. Sulfated malto-oligosaccharides bind to basic fgf, inhibit endothelial cell proliferation, and disrupt endothelial cell tube formation. J. Cell. Physiol. 1996, 168, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Giraux, J.L.; Matou, S.; Bros, A.; Tapon-Bretaudiere, J.; Letourneur, D.; Fischer, A.M. Modulation of human endothelial cell proliferation and migration by fucoidan and heparin. Eur. J. Cell Biol. 1998, 77, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Zugmaier, G.; Lippman, M.E.; Wellstein, A. Inhibition by pentosan polysulfate (pps) of heparin-binding growth factors released from tumor cells and blockage by pps of tumor growth in animals. J. Natl. Cancer Inst. 1992, 84, 1716–1724. [Google Scholar] [CrossRef] [PubMed]
- Presta, M.; Oreste, P.; Zoppetti, G.; Belleri, M.; Tanghetti, E.; Leali, D.; Urbinati, C.; Bugatti, A.; Ronca, R.; Nicoli, S.; et al. Antiangiogenic activity of semisynthetic biotechnological heparins: Low-molecular-weight-sulfated escherichia coli k5 polysaccharide derivatives as fibroblast growth factor antagonists. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 71–76. [Google Scholar] [PubMed]
- Benelli, U.; Bocci, G.; Danesi, R.; Lepri, A.; Bernardini, N.; Bianchi, F.; Lupetti, M.; Dolfi, A.; Campagni, A.; Agen, C.; et al. The heparan sulfate suleparoide inhibits rat corneal angiogenesis and in vitro neovascularization. Exp. Eye Res. 1998, 67, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Sakairi, N.; Kuzuhara, H.; Okamoto, T.; Yajima, M. Synthesis and biological evaluation of 2-amino-2-deoxy- and 6-amino-6-deoxy-cyclomaltoheptaose polysulfates as synergists for angiogenesis inhibition. Bioorg. Med. Chem. 1996, 4, 2187–2192. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.; Burns, W.W., III; Paper, D.H. Selective inhibition of cell proliferation and DNA synthesis by the polysulphated carbohydrate l-carrageenan. Cancer Chemother. Pharmacol. 1995, 36, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Roy, S.; Cochran, E.; Zouaoui, R.; Chu, C.L.; Duffner, J.; Zhao, G.; Smith, S.; Galcheva-Gargova, Z.; Karlgren, J.; et al. M402, a novel heparan sulfate mimetic, targets multiple pathways implicated in tumor progression and metastasis. PLoS ONE 2011, 6, e21106. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Avci, F.Y.; Munoz, E.M.; McDowell, L.M.; Chen, M.; Pedersen, L.C.; Zhang, L.; Linhardt, R.J.; Liu, J. Enzymatic redesigning of biologically active heparan sulfate. J. Biol. Chem. 2005, 280, 42817–42825. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Geng, M.; Li, J.; Guan, H.; Ding, J. Studies of marine sulfated polymannuroguluronate on endothelial cell proliferation and endothelial immunity and related mechanisms. J. Pharmacol. Sci. 2003, 92, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ping Li, C.; Cochran, S.; Ferro, V. Application of the four-component ugi condensation for the preparation of sulfated glycoconjugate libraries. Bioorg. Med. Chem. Lett. 2004, 14, 2221–2226. [Google Scholar] [CrossRef] [PubMed]
- Cochran, S.; Li, C.P.; Bytheway, I. An experimental and molecular-modeling study of the binding of linked sulfated tetracyclitols to fgf-1 and fgf-2. ChemBioChem 2005, 6, 1882–1890. [Google Scholar] [CrossRef] [PubMed]
- Ashikari-Hada, S.; Habuchi, H.; Kariya, Y.; Itoh, N.; Reddi, A.H.; Kimata, K. Characterization of growth factor-binding structures in heparin/heparan sulfate using an octasaccharide library. J. Biol. Chem. 2004, 279, 12346–12354. [Google Scholar] [CrossRef] [PubMed]
- Urbinati, C.; Bugatti, A.; Oreste, P.; Zoppetti, G.; Waltenberger, J.; Mitola, S.; Ribatti, D.; Presta, M.; Rusnati, M. Chemically sulfated escherichia coli k5 polysaccharide derivatives as extracellular hiv-1 tat protein antagonists. FEBS Lett. 2004, 568, 171–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, K.; Gooderham, N.J.; Davies, D.S.; Edwards, R.J. Interaction of the transactivating protein hiv-1 tat with sulphated polysaccharides. Biochem. Pharmacol. 1999, 57, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.X.; Li, J.; Sun, Y.X.; Qi, X.; Wang, Q.J.; Xin, X.L.; Geng, M.Y. Sulfated polymannuroguluronate, a novel anti-aids drug candidate, inhibits hiv-1 tat-induced angiogenesis in kaposi’s sarcoma cells. Biochem. Pharmacol. 2007, 74, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, E.A.; Lortat-Jacob, H.; Priestley, G.V.; Nakamoto, B.; Papayannopoulou, T. Sulfated polysaccharides increase plasma levels of sdf-1 in monkeys and mice: Involvement in mobilization of stem/progenitor cells. Blood 2002, 99, 44–51. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, R.; Rerek, M.; Wood, E.J. Fucoidan modulates the effect of transforming growth factor (tgf)-beta1 on fibroblast proliferation and wound repopulation in in vitro models of dermal wound repair. Biol. Pharm. Bull. 2004, 27, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Lubineau, A.; Lortat-Jacob, H.; Gavard, O.; Sarrazin, S.; Bonnaffe, D. Synthesis of tailor-made glycoconjugate mimetics of heparan sulfate that bind ifn-gamma in the nanomolar range. Chemistry 2004, 10, 4265–4282. [Google Scholar] [CrossRef] [PubMed]
- Cassinelli, G.; Lanzi, C.; Tortoreto, M.; Cominetti, D.; Petrangolini, G.; Favini, E.; Zaffaroni, N.; Pisano, C.; Penco, S.; Vlodavsky, I.; et al. Antitumor efficacy of the heparanase inhibitor sst0001 alone and in combination with antiangiogenic agents in the treatment of human pediatric sarcoma models. Biochem. Pharmacol. 2013, 85, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Waltenberger, J.; Mayr, U.; Frank, H.; Hombach, V. Suramin is a potent inhibitor of vascular endothelial growth factor. A contribution to the molecular basis of its antiangiogenic action. J. Mol. Cell. Cardiol. 1996, 28, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- Takano, S.; Gately, S.; Neville, M.E.; Herblin, W.F.; Gross, J.L.; Engelhard, H.; Perricone, M.; Eidsvoog, K.; Brem, S. Suramin, an anticancer and angiosuppressive agent, inhibits endothelial cell binding of basic fibroblast growth factor, migration, proliferation, and induction of urokinase-type plasminogen activator. Cancer Res. 1994, 54, 2654–2660. [Google Scholar] [PubMed]
- Marchetti, D.; Reiland, J.; Erwin, B.; Roy, M. Inhibition of heparanase activity and heparanase-induced angiogenesis by suramin analogues. Int. J. Cancer 2003, 104, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Groen, H.J.; de Vries, E.G.; Wynendaele, W.; van der Graaf, W.T.; Fokkema, E.; Lechuga, M.J.; Poggesi, I.; Dirix, L.Y.; van Oosterom, A.T. Pnu-145156e, a novel angiogenesis inhibitor, in patients with solid tumors: A phase i and pharmacokinetic study. Clin. Cancer Res. 2001, 7, 3928–3933. [Google Scholar] [PubMed]
- Raman, K.; Karuturi, R.; Swarup, V.P.; Desai, U.R.; Kuberan, B. Discovery of novel sulfonated small molecules that inhibit vascular tube formation. Bioorg. Med. Chem. Lett. 2012, 22, 4467–4470. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Urbinati, C. Polysulfated/sulfonated compounds for the development of drugs at the crossroad of viral infection and oncogenesis. Curr. Pharm. Des. 2009, 15, 2946–2957. [Google Scholar] [CrossRef] [PubMed]
- Urbinati, C.; Chiodelli, P.; Rusnati, M. Polyanionic drugs and viral oncogenesis: A novel approach to control infection, tumor-associated inflammation and angiogenesis. Molecules 2008, 13, 2758–2785. [Google Scholar] [CrossRef] [PubMed]
- Correia-da-Silva, M.; Sousa, E.; Pinto, M.M. Emerging sulfated flavonoids and other polyphenols as drugs: Nature as an inspiration. Med. Res. Rev. 2014, 34, 223–279. [Google Scholar] [CrossRef] [PubMed]
- Bugatti, A.; Urbinati, C.; Ravelli, C.; De Clercq, E.; Liekens, S.; Rusnati, M. Heparin-mimicking sulfonic acid polymers as multitarget inhibitors of human immunodeficiency virus type 1 tat and gp120 proteins. Antimicrob. Agents Chemother. 2007, 51, 2337–2345. [Google Scholar] [CrossRef] [PubMed]
- Liekens, S.; Leali, D.; Neyts, J.; Esnouf, R.; Rusnati, M.; Dell’Era, P.; Maudgal, P.C.; de Clercq, E.; Presta, M. Modulation of fibroblast growth factor-2 receptor binding, signaling, and mitogenic activity by heparin-mimicking polysulfonated compounds. Mol. Pharmacol. 1999, 56, 204–213. [Google Scholar] [PubMed]
- Zhang, F.; Zhang, Z.; Lin, X.; Beenken, A.; Eliseenkova, A.V.; Mohammadi, M.; Linhardt, R.J. Compositional analysis of heparin/heparan sulfate interacting with fibroblast growth factor. Fibroblast growth factor receptor complexes. Biochemistry 2009, 48, 8379–8386. [Google Scholar] [CrossRef]
- Karoli, T.; Liu, L.; Fairweather, J.K.; Hammond, E.; Li, C.P.; Cochran, S.; Bergefall, K.; Trybala, E.; Addison, R.S.; Ferro, V. Synthesis, biological activity, and preliminary pharmacokinetic evaluation of analogues of a phosphosulfomannan angiogenesis inhibitor (pi-88). J. Med. Chem. 2005, 48, 8229–8236. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Xin, X.; Meng, L.; Tong, L.; Lin, L.; Geng, M.; Ding, J. The marine-derived oligosaccharide sulfate (mdos), a novel multiple tyrosine kinase inhibitor, combats tumor angiogenesis both in vitro and in vivo. PLoS ONE 2008, 3, e3774. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chuang, Y.J.; Swanson, R.; Li, J.; Seo, K.; Leung, L.; Lau, L.F.; Olson, S.T. Antiangiogenic antithrombin down-regulates the expression of the proangiogenic heparan sulfate proteoglycan, perlecan, in endothelial cells. Blood 2004, 103, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Aviezer, D.; Iozzo, R.V.; Noonan, D.M.; Yayon, A. Suppression of autocrine and paracrine functions of basic fibroblast growth factor by stable expression of perlecan antisense cdna. Mol. Cell. Biol. 1997, 17, 1938–1946. [Google Scholar] [PubMed]
- Van Wijk, X.M.; Thijssen, V.L.; Lawrence, R.; van den Broek, S.A.; Dona, M.; Naidu, N.; Oosterhof, A.; van de Westerlo, E.M.; Kusters, L.J.; Khaled, Y.; et al. Interfering with udp-glcnac metabolism and heparan sulfate expression using a sugar analogue reduces angiogenesis. ACS Chem. Biol. 2013, 8, 2331–2338. [Google Scholar] [CrossRef] [PubMed]
- Raman, K.; Ninomiya, M.; Nguyen, T.K.; Tsuzuki, Y.; Koketsu, M.; Kuberan, B. Novel glycosaminoglycan biosynthetic inhibitors affect tumor-associated angiogenesis. Biochem. Biophys. Res. Commun. 2011, 404, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Ashikari-Hada, S.; Habuchi, H.; Kariya, Y.; Kimata, K. Heparin regulates vascular endothelial growth factor165-dependent mitogenic activity, tube formation, and its receptor phosphorylation of human endothelial cells. Comparison of the effects of heparin and modified heparins. J. Biol. Chem. 2005, 280, 31508–31515. [Google Scholar] [CrossRef] [PubMed]
- Roghani, M.; Moscatelli, D. Basic fibroblast growth factor is internalized through both receptor-mediated and heparan sulfate-mediated mechanisms. J. Biol. Chem. 1992, 267, 22156–22162. [Google Scholar] [PubMed]
- Miao, H.Q.; Ishai-Michaeli, R.; Atzmon, R.; Peretz, T.; Vlodavsky, I. Sulfate moieties in the subendothelial extracellular matrix are involved in basic fibroblast growth factor sequestration, dimerization, and stimulation of cell proliferation. J. Biol. Chem. 1996, 271, 4879–4886. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ai, X.; Freeman, S.D.; Pownall, M.E.; Lu, Q.; Kessler, D.S.; Emerson, C.P., Jr. Qsulf1, a heparan sulfate 6-o-endosulfatase, inhibits fibroblast growth factor signaling in mesoderm induction and angiogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 4833–4838. [Google Scholar] [CrossRef] [PubMed]
- Uchimura, K.; Morimoto-Tomita, M.; Bistrup, A.; Li, J.; Lyon, M.; Gallagher, J.; Werb, Z.; Rosen, S.D. Hsulf-2, an extracellular endoglucosamine-6-sulfatase, selectively mobilizes heparin-bound growth factors and chemokines: Effects on vegf, fgf-1, and sdf-1. BMC Biochem. 2006, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.C.; Rahimi, N.; Forsten-Williams, K.; Nugent, M.A. Heparan sulfate proteoglycans function as receptors for fibroblast growth factor-2 activation of extracellular signal-regulated kinases 1 and 2. Circ. Res. 2004, 94, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Sasisekharan, R.; Moses, M.A.; Nugent, M.A.; Cooney, C.L.; Langer, R. Heparinase inhibits neovascularization. Proc. Natl. Acad. Sci. USA 1994, 91, 1524–1528. [Google Scholar] [CrossRef] [PubMed]
- Collins, B.E.; Paulson, J.C. Cell surface biology mediated by low affinity multivalent protein-glycan interactions. Curr. Opin. Chem. Biol. 2004, 8, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Ishwar, A.R.; Jeong, K.J.; Panitch, A.; Akkus, O. Raman spectroscopic investigation of peptide-glycosaminoglycan interactions. Appl. Spectrosc. 2009, 63, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Raj, P.A.; Marcus, E.; Rein, R. Conformational requirements of suramin to target angiogenic growth factors. Angiogenesis 1998, 2, 183–199. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, P.L.; Liu, J.; Linhardt, R.J. Chemoenzymatic synthesis of glycosaminoglycans: Re-creating, re-modeling and re-designing nature’s longest or most complex carbohydrate chains. Glycobiology 2013, 23, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Guedes, N.; Czechura, P.; Echeverria, B.; Ruiz, A.; Michelena, O.; Martin-Lomas, M.; Reichardt, N.C. Toward the solid-phase synthesis of heparan sulfate oligosaccharides: Evaluation of iduronic acid and idose building blocks. J. Org. Chem. 2013, 78, 6911–6934. [Google Scholar] [CrossRef] [PubMed]
- Freeman, C.; Liu, L.; Banwell, M.G.; Brown, K.J.; Bezos, A.; Ferro, V.; Parish, C.R. Use of sulfated linked cyclitols as heparan sulfate mimetics to probe the heparin/heparan sulfate binding specificity of proteins. J. Biol. Chem. 2005, 280, 8842–8849. [Google Scholar] [CrossRef] [PubMed]
- Corallini, A.; Betti, M.; Rusnati, M.; Campioni, D.; Ciomei, M.; Sola, F.; Calza, N.; Zauli, G.; Presta, M.; Barbanti-Brodano, G.; et al. Characterization of the effects of two polysulfonated distamycin a derivatives, pnu145156e and pnu153429, on hiv type 1 tat protein. AIDS Res. Hum. Retrovir. 1998, 14, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Raghuraman, A.; Mosier, P.D.; Desai, U.R. Finding a needle in a haystack: Development of a combinatorial virtual screening approach for identifying high specificity heparin/heparan sulfate sequence(s). J. Med. Chem. 2006, 49, 3553–3562. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Riverst, G.; Zhu, Y.; Jacobson, A.; Peyers, J.; Grundstrom, G.; Burch, P.; Hussein, S.; Marolewski, A.; Herlihy, W.; et al. Identification of inhibitors of heparin-growth factor interactions from combinatorial libraries of four-component condensation reactions. Bioorg. Med. Chem. 2001, 9, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Herbert, J.M.; Cottineau, M.; Driot, F.; Pereillo, J.M.; Maffrand, J.P. Activity of pentosan polysulphate and derived compounds on vascular endothelial cell proliferation and migration induced by acidic and basic fgf in vitro. Biochem. Pharmacol. 1988, 37, 4281–4288. [Google Scholar] [CrossRef] [PubMed]
- Schwartsmann, G.; Sprinz, E.; Kalakun, L.; Yamagushi, N.; Sander, E.; Grivicich, I.; Koya, R.; Mans, D.R. Phase ii study of pentosan polysulfate (pps) in patients with aids-related kaposi’s sarcoma. Tumori 1996, 82, 360–363. [Google Scholar] [PubMed]
- Maione, T.E.; Gray, G.S.; Petro, J.; Hunt, A.J.; Donner, A.L.; Bauer, S.I.; Carson, H.F.; Sharpe, R.J. Inhibition of angiogenesis by recombinant human platelet factor-4 and related peptides. Science 1990, 247, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Ogawa, K.; Katsube, T.; Shimao, K.; Konno, S.; Shimakawa, T.; Yoshimatsu, K.; Naritaka, Y.; Yagawa, H.; Hirose, K. Platelet factor 4 gene transfection into tumor cells inhibits angiogenesis, tumor growth and metastasis. Anticancer Res. 2005, 25, 847–851. [Google Scholar] [PubMed]
- Adulnirath, A.; Chung, S.W.; Park, J.; Hwang, S.R.; Kim, J.Y.; Yang, V.C.; Kim, S.Y.; Moon, H.T.; Byun, Y. Cyclic rgdyk-conjugated lmwh-taurocholate derivative as a targeting angiogenesis inhibitor. J. Control. Release 2012, 164, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Leali, D.; Belleri, M.; Urbinati, C.; Coltrini, D.; Oreste, P.; Zoppetti, G.; Ribatti, D.; Rusnati, M.; Presta, M. Fibroblast growth factor-2 antagonist activity and angiostatic capacity of sulfated escherichia coli k5 polysaccharide derivatives. J. Biol. Chem. 2001, 276, 37900–37908. [Google Scholar] [PubMed]
- Rusnati, M.; Vicenzi, E.; Donalisio, M.; Oreste, P.; Landolfo, S.; Lembo, D. Sulfated k5 escherichia coli polysaccharide derivatives: A novel class of candidate antiviral microbicides. Pharmacol. Ther. 2009, 123, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Borgenstrom, M.; Jalkanen, M.; Salmivirta, M. Sulfated derivatives of escherichia coli k5 polysaccharides as modulators of fibroblast growth factor signaling. J. Biol. Chem. 2003, 278, 49882–49889. [Google Scholar] [CrossRef] [PubMed]
- Maddineni, J.; Jeske, W.P.; Baltasar, F.; Cornelli, U.; Manoni, M.; Hoppensteadt, D.A.; Fareed, J. Modulatory effects of escherichia coli capsular-derived sulfaminoheparosans and heparins on tissue factor-mediated activation of platelets: Flow cytometric analysis. Clin. Appl. Thromb. Hemost. 2006, 12, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Oreste, P.; Zoppetti, G. Semi-synthetic heparinoids. In Handbook of Experimental Pharmacology; Springer: Berlin Heidelberg, Germany, 2012; pp. 403–422. [Google Scholar]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiodelli, P.; Bugatti, A.; Urbinati, C.; Rusnati, M. Heparin/Heparan Sulfate Proteoglycans Glycomic Interactome in Angiogenesis: Biological Implications and Therapeutical Use. Molecules 2015, 20, 6342-6388. https://doi.org/10.3390/molecules20046342
Chiodelli P, Bugatti A, Urbinati C, Rusnati M. Heparin/Heparan Sulfate Proteoglycans Glycomic Interactome in Angiogenesis: Biological Implications and Therapeutical Use. Molecules. 2015; 20(4):6342-6388. https://doi.org/10.3390/molecules20046342
Chicago/Turabian StyleChiodelli, Paola, Antonella Bugatti, Chiara Urbinati, and Marco Rusnati. 2015. "Heparin/Heparan Sulfate Proteoglycans Glycomic Interactome in Angiogenesis: Biological Implications and Therapeutical Use" Molecules 20, no. 4: 6342-6388. https://doi.org/10.3390/molecules20046342
APA StyleChiodelli, P., Bugatti, A., Urbinati, C., & Rusnati, M. (2015). Heparin/Heparan Sulfate Proteoglycans Glycomic Interactome in Angiogenesis: Biological Implications and Therapeutical Use. Molecules, 20(4), 6342-6388. https://doi.org/10.3390/molecules20046342