
Development of Orally Active Thrombin Inhibitors for the Treatment of Thrombotic Disorder Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

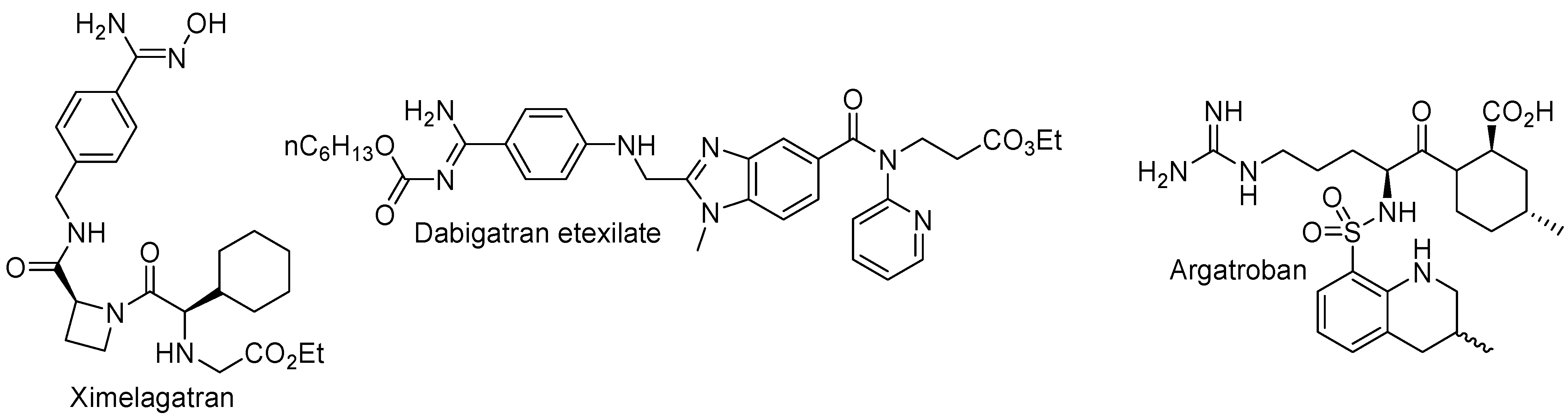
2. Orally Active Thrombin Inhibitors
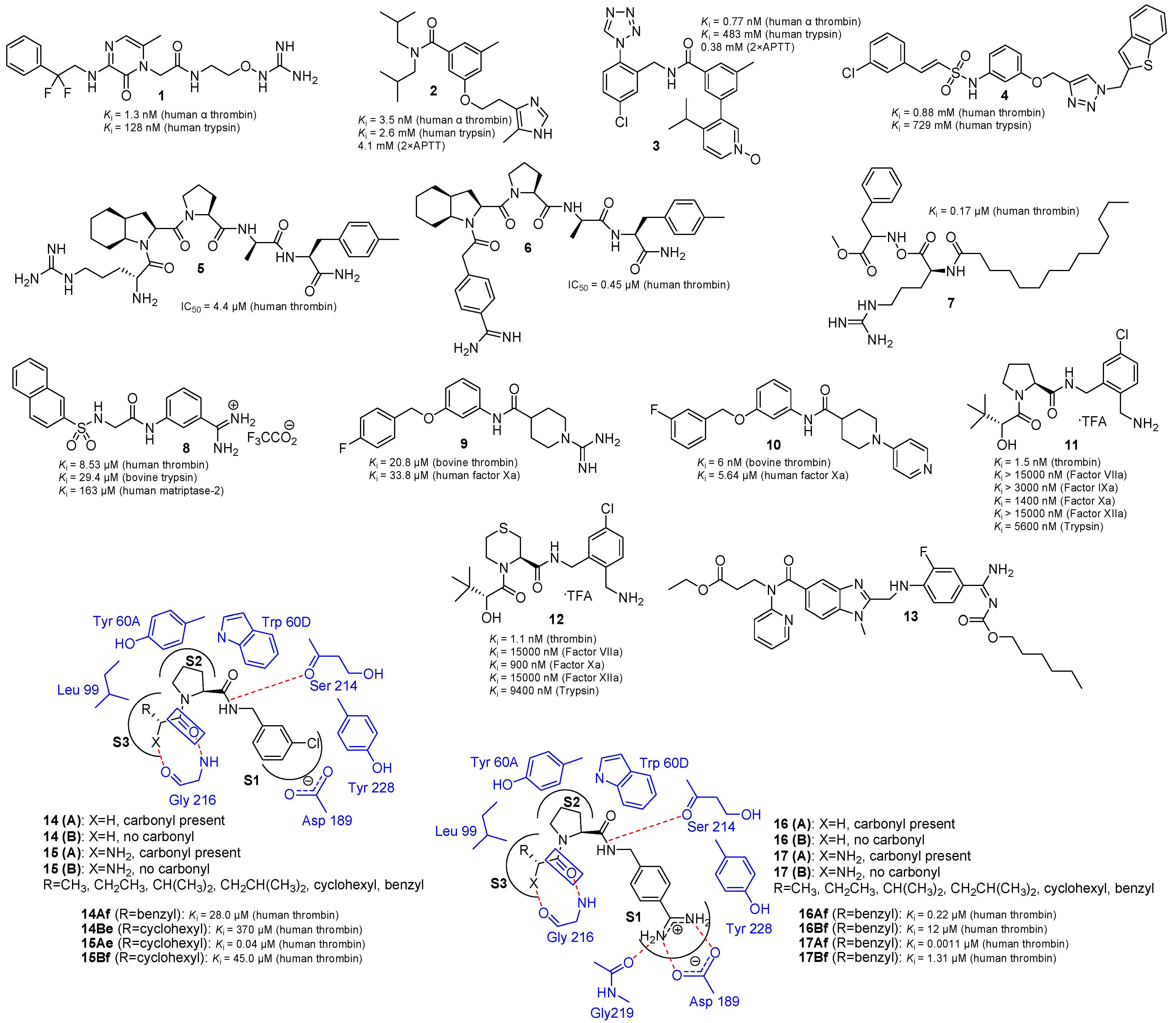
2.1. Selective and Orally Active Thrombin Inhibitors


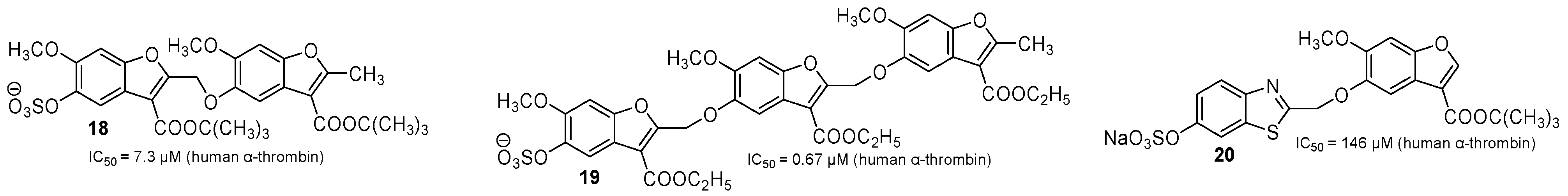
2.2. Allosteric Thrombin Inhibitors

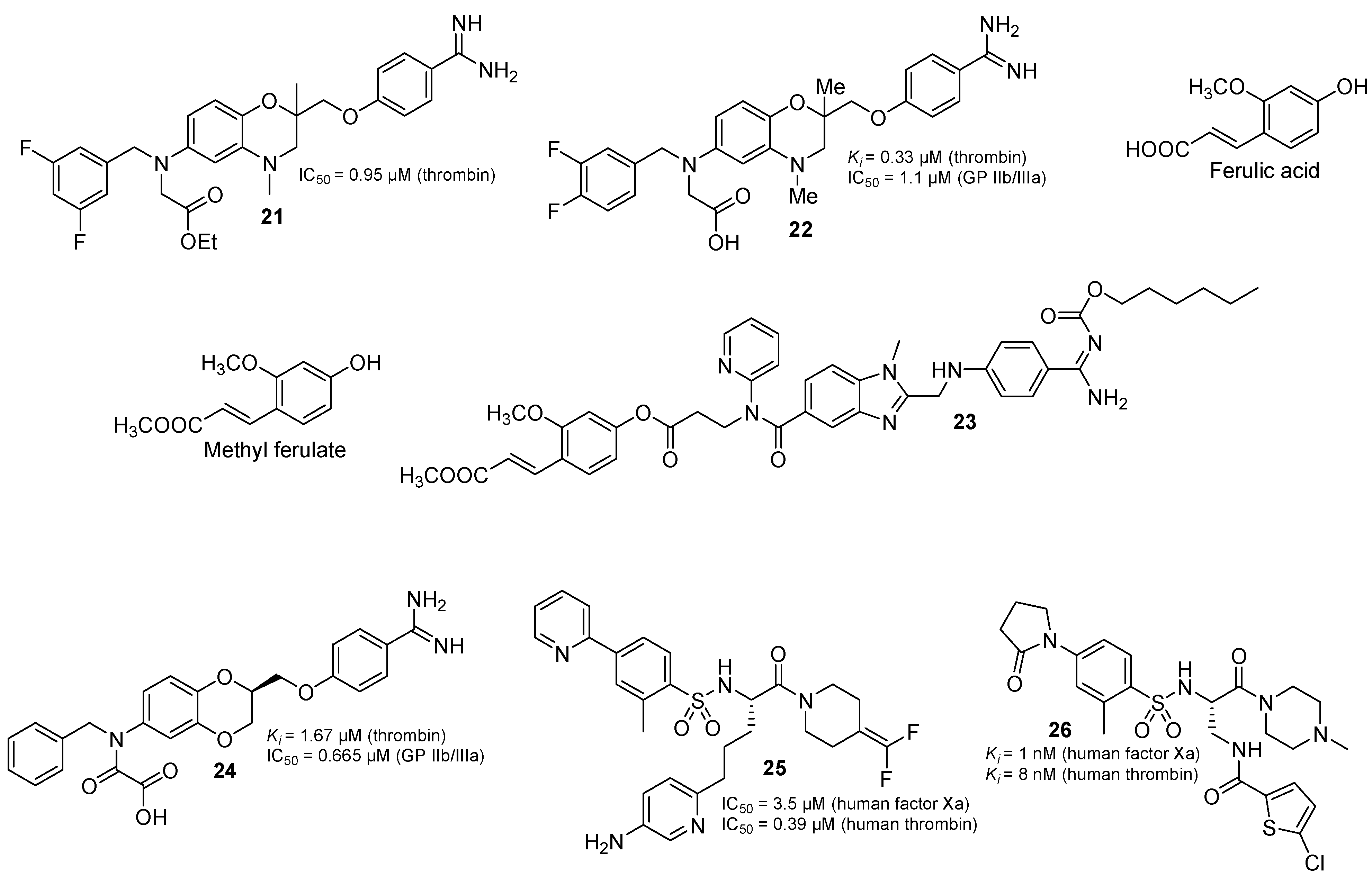
2.3. Thrombin Inhibitor in Multi-Target Drugs

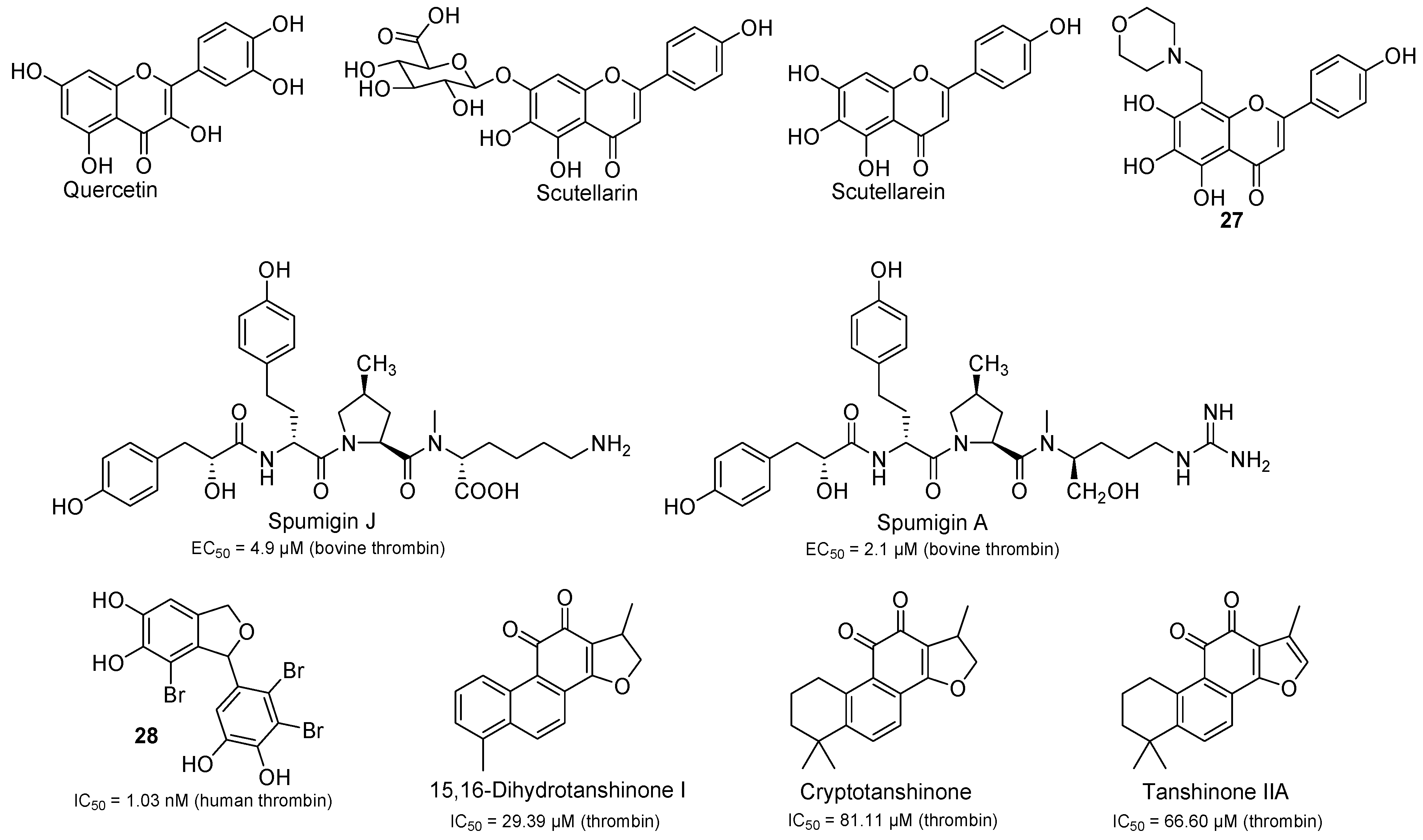
2.4. Natural Products with Thrombin Inhibitory Activity

3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Collins, B.; Hollidge, C. Antithrombotic drug market. Nat. Rev. Drug. Discov. 2003, 2, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Coburn, C.A. Small-molecule direct thrombin inhibitors: 1997–2000. Exp. Opin. Ther. Pat. 2001, 11, 721–738. [Google Scholar] [CrossRef]
- Tapparelli, C.; Metternich, R.; Ehrhardt, C.; Cook, N.S. Synthetic low-molecular weight thrombin inhibitors: Molecular design and pharmacological profile. Trends Pharmacol. Sci. 1993, 14, 366–376. [Google Scholar] [CrossRef]
- Hirsh, J.; Fuster, V. Guide to anticoagulant therapy. Part 2: Oral anticoagulants. American Heart Association. Circulation 1994, 89, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Veldman, A.; Hoffman, M.; Ehrenforth, S. New insights into the coagulation system and implications for new therapeutic options with recombinant factor VIIa. Curr. Med. Chem. 2003, 10, 797–811. [Google Scholar] [CrossRef] [PubMed]
- Spyropoulos, A.C. Brave new world: The current and future use of novel anticoagulants. Thromb. Res. 2008, 123, S29–S35. [Google Scholar] [CrossRef] [PubMed]
- Blizzard, T.A.; Singh, S.; Patil, B.; Chidurala, N.; Komanduri, V.; Debnath, S.; Belyakov, S.; Crespo, A.; Struck, A.; Kurtz, M.; et al. Heterocyclic core analogs of a direct thrombin inhibitor Timothy A. Bioorg. Med. Chem. Lett. 2014, 24, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Straub, A.; Roehrig, S.; Hillisch, A. Oral, direct thrombin and factor Xa inhibitors: The replacement for warfarin, leeches, and pig intestines? Angew. Chem. Int. Ed. 2011, 50, 4574–4590. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.Y.; Jin, Y.; Desai, U.R. An update on recent patents on thrombin inhibitors (2010–2013). Expert Opin. Ther. Pat. 2014, 24, 47–67. [Google Scholar] [CrossRef] [PubMed]
- Steinmetzer, T.; Stürzebecher, J. Progress in the development of synthetic thrombin inhibitors as new orally active anticoagulants. Curr. Med. Chem. 2004, 11, 2297–2321. [Google Scholar] [CrossRef] [PubMed]
- Das, J.; Kimball, S.D. Thrombin active site inhibitors. Bioorg. Med. Chem. 1995, 3, 999–1007. [Google Scholar] [CrossRef]
- Srivastava, S.; Goswami, L.N.; Dikshit, D.K. Progress in the design of low molecular weight thrombin inhibitors. Med. Res. Rev. 2005, 25, 66–92. [Google Scholar] [CrossRef] [PubMed]
- Schärer, K.; Morgenthaler, M.; Seiler, P.; Diederich, F.; Banner, D.W.; Tschopp, T.; Obst-Sander, U. Enantiomerically pure thrombin inhibitors for exploring the molecular-recognition features of the oxyanion hole. Helv. Chim. Acta 2004, 87, 2517–2538. [Google Scholar] [CrossRef]
- Bajusz, S.; Szell, E.; Bagdy, D.; Barabas, E.; Horvath, G.; Dioszegi, M.; Fittler, Z.; Szabo, G.; Juhasz, A.; Tomori, E.; et al. Highly active and selective anticoagulants: d-Phe-Pro-Arg-H, a free tripeptide aldehyde prone to spontaneous inactivation, and its stable N-methyl derivative, d-MePhe-Pro-Arg-H. J. Med. Chem. 1990, 33, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Obst, U.; Banner, D.W.; Weber, L.; Diederich, F. Molecular recognition at the thrombin active site: Structure-based design and synthesis of potent and selective thrombin inhibitors and the X-ray crystal structures of two thrombin-inhibitor complexes. Chem. Biol. 1997, 4, 287–295. [Google Scholar] [CrossRef]
- Lu, T.; Markotan, T.; Ballentine, S.K.; Giardino, E.C.; Spurlino, J.; Brown, K.; Maryanoff, B.E.; Tomczuk, B.E.; Damiano, B.P.; Shukla, U.; et al. Discovery and clinical evaluation of 1-{N-[2-(amidinoaminooxy)ethyl]amino}carbonylmethyl-6-methyl-3-[2,2-difluoro-2-phenylethylamino]pyrazinone (RWJ-671818), a thrombin inhibitor with an oxyguanidine P1 motif. J. Med. Chem. 2010, 53, 1843–1856. [Google Scholar] [CrossRef] [PubMed]
- Tomczuk, B.; Lu, T.; Soll, R.M.; Fedde, C.; Wang, A.; Murphy, L.; Crysler, C.; Dasgupta, M.; Eisennagel, S.; Spurlino, J.; et al. Oxyguanidines: Application to non-peptidic phenyl-based thrombin inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 1495–1498. [Google Scholar] [CrossRef]
- Lu, T.; Tomczuk, B.; Illig, C.R.; Bone, R.; Murphy, L.; Spurlino, J.; Salemme, F.R.; Soll, R.M. In vitro evaluation and crystallographic analysis of a new class of selective, non-amide-based thrombin inhibitors. Bioorg. Med. Chem. Lett. 1998, 8, 1595–1600. [Google Scholar] [CrossRef]
- Isaacs, R.C.A.; Newton, C.L.; Cutrona, K.J.; Mercer, S.P.; Payne, L.S.; Stauffer, K.J.; Williams, P.D.; Cook, J.J.; Krueger, J.A.; Dale Lewis, S.; et al. Design, synthesis and SAR of a series of 1,3,5-trisubstituted benzenes as thrombin inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 1536–1540. [Google Scholar] [CrossRef] [PubMed]
- Siles, R.; Kawasaki, Y.; Ross, P.; Freire, E. Synthesis and biochemical evaluation of triazole/tetrazole-containing sulfonamides against thrombin and related serine proteases. Bioorg. Med. Chem. Lett. 2011, 21, 5305–5309. [Google Scholar] [CrossRef] [PubMed]
- Nieman, M.T.; Warnock, M.; Hasan, A.A.K.; Mahdi, F.; Lucchesi, B.R.; Brown, N.J.; Murphey, L.J.; Schmaier, A.H. The preparation and characterization of novel peptide antagonists to thrombin and factor VIIa and activation of protease-activated receptor 1. J. Pharmacol. Exp. Ther. 2004, 311, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Girnys, E.A.; Porter, V.R.; Mosberg, H.I. Conformationally restricted analogs of the direct thrombin inhibitor FM 19. Bioorg. Med. Chem. 2011, 19, 7425–7434. [Google Scholar] [CrossRef] [PubMed]
- Poyarkov, A.A.; Poyarkova, S.A.; Smirnova, I.V.; Kukhar, V.P. Liporetro-d-peptides-A novel class of highly selective thrombin inhibitors. Thromb. Res. 2012, 129, e97–e105. [Google Scholar] [CrossRef] [PubMed]
- Poyarkov, A.; Rocabayera, X.; Poyarkova, S.; Kukhar, V. Influence of aromatic and aliphatic moieties on thrombin inhibitors potency. Open Biochem. J. 2008, 2, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Ilaš, J.; Tomašić, T.; Kikelj, D. Novel potent and selective thrombin inhibitors based on a central 1,4-benzoxazin-3(4H)-one scaffold. J. Med. Chem. 2008, 51, 2863–2867. [Google Scholar] [CrossRef] [PubMed]
- Di Fenza, A.; Heine, A.; Koert, U.; Klebe, G. Understanding binding selectivity toward trypsin and factor Xa: The role of aromatic interactions. Chem. Med. Chem. 2007, 2, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Dosa, S.; Stirnberg, M.; Lulsdorff, V.; Hausler, D.; Maurer, E.; Gutschow, M. Active site mapping of trypsin, thrombin and matriptase-2 by sulfamoyl benzamidines. Bioorg. Med. Chem. 2012, 20, 6489–6505. [Google Scholar] [CrossRef] [PubMed]
- De Candia, M.; Fiorella, F.; Lopopolo, G.; Carotti, A.; Rosaria Romano, M.; Diego Lograno, M.; Martel, S.; Carrupt, P.A.; Belviso, B.D.; Caliandro, R.; et al. Synthesis and biological evaluation of direct thrombin inhibitors bearing 4-(piperidin-1-yl)pyridine at the P1 position with potent anticoagulant activity. J. Med. Chem. 2013, 56, 8696–8711. [Google Scholar] [CrossRef] [PubMed]
- Morrissette, M.M.; Stauffer, K.J.; Williams, P.D.; Lyle, T.A.; Vacca, J.P.; Krueger, J.A.; Lewis, S.D.; Lucas, B.J.; Wong, B.K.; White, R.B.; et al. Low molecular weight thrombin inhibitors with excellent potency, metabolic stability, and oral bioavailability. Bioorg. Med. Chem. Lett. 2004, 14, 4161–4164. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.S. New oral Xa and II a inhibitors: Updates on clinical trial results. J. Thromb. Thromb. 2008, 25, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Lip, G.Y.; Larsen, T.B.; Skjøth, F.; Rasmussen, L.H. Indirect comparisons of new oral anticoagulant drugs for efficacy and safety when used for stroke prevention in atrial fibrillation. J. Am. Coll. Cardiol. 2012, 60, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Hauel, N.H.; Nar, H.; Priepke, H.; Ries, U.; Stassen, J.M.; Wienen, W. Structure based design of novel potent nonpeptide thrombin inhibitors. J. Med. Chem. 2002, 45, 1757–1766. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, M.W. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 2005, 3, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Li, M.L.; Ren, Y.J.; Dong, M.H.; Ren, W.X. Design, synthesis and structural exploration of novel fluorinated dabigatran derivatives as direct thrombin inhibitors. Eur. J. Med. Chem. 2015, 96, 122–138. [Google Scholar] [CrossRef] [PubMed]
- Said, A.M.; Hangauer, D.G. Binding cooperativity between a ligand carbonyl group and a hydrophobic side chain can be enhanced by additional H-bonds in a distance dependent manner: A case study with thrombin inhibitors. Eur. J. Med. Chem. 2015, 96, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Huntington, J.A. Molecular recognition mechanisms of thrombin. J. Thromb. Haemost. 2005, 3, 1861–1872. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Liu, L.W.; Esmon, C.T.; Johnson, A.E. The fifth and sixth growth factor-like domains of thrombomodulin bind to the anionbinding exosite of thrombin and alter its specificity. J. Biol. Chem. 1992, 267, 11023–11028. [Google Scholar] [PubMed]
- Li, W.; Johnson, D.J.; Adams, T.E.; Pozzi, N.; de Filippis, V.; Huntington, J.A. Thrombin inhibition by serpins disrupts exosite II. J. Biol. Chem. 2010, 285, 38621–38629. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.Y.; Thakkar, J.N.; Mohammed, B.M.; Martin, E.J.; Brophy, D.F.; Kishimoto, T.; Desai, U.R. Targeting the GPIbα binding site of thrombin to simultaneously induce dual anticoagulant and antiplatelet effects. J. Med. Chem. 2014, 57, 3030–3039. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.Y.; Desai, U.R. Substantial non-electrostatic forces are needed to induce allosteric disruption of thrombin’s active site through exosite 2. Biochem. Biophys. Res. Commun. 2014, 452, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Henry, B.L.; Abdel Aziz, M.; Zhou, Q.; Desai, U.R. Sulfated, low molecular weight lignins are potent inhibitors of plasmin, in addition to thrombin and factor Xa: Novel opportunity for controlling complex pathologies. Thromb. Haemost. 2010, 103, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Singh Sidhu, P.; Liang, A.; Mehta, A.Y.; Abdel Aziz, M.H.; Zhou, Q.; Desai, U.R. Rational design of potent, small, synthetic allosteric inhibitors of thrombin. J. Med. Chem. 2011, 54, 5522–5531. [Google Scholar] [CrossRef] [PubMed]
- Abdel Aziz, M.H.; Sidhu, P.S.; Liang, A.; Kim, J.Y.; Mosier, P.D.; Zhou, Q.; Farrell, D.H.; Desai, U.R. Designing allosteric regulators of thrombin. Monosulfated benzofuran dimers selectively interact with Arg173 of exosite 2 to induce inhibition. J. Med. Chem. 2012, 55, 6888–6897. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, P.S.; Abdel Aziz, M.H.; Sarkar, A.; Mehta, A.Y.; Zhou, Q.; Desai, U.R. Designing allosteric regulators of thrombin. Exosite 2 features multiple subsites that can be targeted by sulfated small molecules for inducing inhibition. J. Med. Chem. 2013, 56, 5059–5070. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, P.S.; Zhou, Q.; Desai, U.R. A simple, general approach of allosteric coagulation enzyme inhibition through monosulfated hydrophobic scaffolds. Bioorg. Med. Chem. Lett. 2014, 24, 5716–5720. [Google Scholar] [CrossRef] [PubMed]
- Muro, S.; Muzykantov, V.R. Targeting of antioxidant and anti-thrombotic drugs to endothelial cell adhesion molecules. Curr. Pharm. Des. 2005, 11, 2383–2401. [Google Scholar] [CrossRef] [PubMed]
- Ilić, M.; Kontogiorgis, C.; Hadjipavlou-Litina, D.; Ilaš, J.; Kikelj, D. Thrombin inhibitors with lipid peroxidation and lipoxygenase inhibitory activities. Bioorg. Med. Chem. Lett. 2011, 21, 4705–4709. [Google Scholar] [CrossRef] [PubMed]
- Ilić, M.; Kikelj, D.; Ilaš, J. Fluorinated dual antithrombotic compounds based on 1,4-benzoxazine scaffold. Eur. J. Med. Chem. 2012, 50, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Chan, Y.Y.; Kuo, P.C.; Chen, Y.F.; Chang, R.J.; Chen, I.S.; Wu, S.J.; Wu, T.S. The constituents of roots and stems of Illigera luzonensis and their anti-platelet aggregation effects. Int. J. Mol. Sci. 2014, 15, 13424–13436. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Z.; Diao, X.J.; Yang, W.H.; Li, F.; He, G.W.; Gong, G.Q.; Xu, Y.G. Design, synthesis and antithrombotic evaluation of novel dabigatran prodrugs containing methyl ferulate. Bioorg. Med. Chem. Lett. 2013, 23, 2089–2092. [Google Scholar] [CrossRef] [PubMed]
- Douketis, J.D. Combination warfarin-ASA therapy: Which patients should receive it, which patients should not, and why? Thromb. Res. 2011, 127, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Ilić, M.; Dunkel, P.; Ilaš, J.; Chabielska, E.; Zakrzeska, A.; Mátyus, P.; Kikelj, D. Towards dual antithrombotic compounds—Balancing thrombin inhibitory and fibrinogen GPIIb/IIIa binding inhibitory activities of 2,3-dihydro-1,4-benzodioxine derivatives through regio- and stereoisomerism. Eur. J. Med. Chem. 2013, 62, 329–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meneyrol, J.; Follmann, M.; Lassalle, G.; Wehner, V.; Barre, G.; Rousseaux, T.; Altenburger, J.M.; Petit, F.; Bocskei, Z.; Schreuder, H.; et al. 5-Chlorothiophene-2-carboxylic acid [(S)-2-[2-methyl-3-(2-oxopyrrolidin-1-yl)benzenesulfonylamino]-3-(4-methylpiperazin-1-yl)-3-oxopropyl]amide (SAR107375), a selective and potent orally active dual thrombin and Factor Xa inhibitor. J. Med. Chem. 2013, 56, 9441–9456. [Google Scholar] [CrossRef] [PubMed]
- Li, H.X.; Han, S.Y.; Wang, X.W.; Ma, X.; Zhang, K.; Wang, L.; Ma, Z.Z.; Tu, P.F. Effect of the carthamins yellow from Carthamus tinctorius L. on hemorheological disorders of blood stasis in rats. Food Chem. Toxicol. 2009, 47, 1797–1802. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lai, X.Y.; Ling, X.M.; Zhao, Y.Y.; Cui, J.R. Interactions between thrombin with flavonoids from Abelmoschus manihot (L.) Medicus by CZE. Chromatographia 2006, 64, 45–50. [Google Scholar] [CrossRef]
- Singh, B.; Kaur, P.; Gopichand; Singh, R.D.; Ahuja, P.S. Biology and chemistry of Ginkgo biloba. Fitoterapia 2008, 79, 401–418. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ma, H.Y.; Yang, N.Y.; Tang, Y.P.; Guo, J.M.; Tao, W.W.; Duan, J.A. A series of natural flavonoids as thrombin inhibitors: Structure-activity relationships. Thromb. Res. 2010, 126, e365–e378. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.H.; Li, N.G.; Tang, Y.P.; Li, W.; Yin, L.; Yang, J.P.; Tang, H.; Duan, J.A. Metabolism-based synthesis, biologic evaluation and SARs analysis of O-methylated analogs of quercetin as thrombin inhibitors. Eur. J. Med. Chem. 2012, 54, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Li, N.G.; Shen, M.Z.; Wang, Z.J.; Tang, Y.P.; Shi, Z.H.; Fu, Y.F.; Shi, Q.P.; Tang, H.; Duan, J.A. Design, synthesis and biological evaluation of glucose-containing scutellarein derivatives as neuroprotective agents based on metabolic mechanism of scutellarin in vivo. Bioorg. Med. Chem. Lett. 2013, 23, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.W.; Feng, T.M.; Shan, L.C.; Cai, B.Z.; Chu, W.F.; Niu, H.L.; Lu, Y.J.; Yang, B.F. Scutellarin-induced endothelium-independent relaxation in rat aorta. Phytother. Res. 2008, 22, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.H.; Li, N.G.; Tang, Y.P.; Zhang, L.; Tang, H.; Wang, Z.J.; Liu, L.; Song, S.L.; Guo, J.M.; Ding, A.W. Synthesis and bio-activity evaluation of scutellarein as a potent agent for the therapy of ischemic cerebrovascular disease. Int. J. Mol. Sci. 2011, 12, 8208–8216. [Google Scholar] [CrossRef] [PubMed]
- Li, N.G.; Song, S.L.; Shen, M.Z.; Tang, Y.P.; Shi, Z.H.; Tang, H.; Shi, Q.P.; Fu, Y.F.; Duan, J.A. Mannich bases of scutellarein as thrombin-inhibitors: Design, synthesis, biological activity and solubility. Bioorg. Med. Chem. 2012, 20, 6919–6923. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Li, X.; Li, J.; Guo, S.; Su, H.; Fan, X. Antithrombotic effects of bromophenol, analga-derived thrombin inhibitor. Chin. J. Oceanol. Limnol. 2010, 28, 96–98. [Google Scholar] [CrossRef]
- Hankey, G.J.; Eikelboom, J.W. Dabigatran etexilate: A new oral thrombin inhibitor. Circulation 2011, 123, 1436–1450. [Google Scholar] [CrossRef] [PubMed]
- Anas, A.R.J.; Kisugi, T.; Umezawa, T.; Matsuda, F.; Campitelli, M.R.; Quinn, R.J.; Okino, T. Thrombin inhibitors from the freshwater cyanobacterium anabaena compacta. J. Nat. Prod. 2012, 75, 1546–1552. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.Y.; Wang, Y.P. Pharmacological actions and therapeutic applications of Salvia miltiorrhiza depside salt and its active components. Acta Pharmacol. Sin. 2012, 33, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Song, H.P.; Li, P.; Zhou, P.; Dong, X.; Chen, J. Screening of direct thrombin inhibitors from Radix Salviae Miltiorrhizae by a peak fractionation approach. J. Pharm. Biomed. Anal. 2015, 109, 85–90. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, L.-W.; Dai, W.-C.; Li, N.-G. Development of Orally Active Thrombin Inhibitors for the Treatment of Thrombotic Disorder Diseases. Molecules 2015, 20, 11046-11062. https://doi.org/10.3390/molecules200611046
He L-W, Dai W-C, Li N-G. Development of Orally Active Thrombin Inhibitors for the Treatment of Thrombotic Disorder Diseases. Molecules. 2015; 20(6):11046-11062. https://doi.org/10.3390/molecules200611046
Chicago/Turabian StyleHe, Li-Wei, Wei-Chen Dai, and Nian-Guang Li. 2015. "Development of Orally Active Thrombin Inhibitors for the Treatment of Thrombotic Disorder Diseases" Molecules 20, no. 6: 11046-11062. https://doi.org/10.3390/molecules200611046
APA StyleHe, L. -W., Dai, W. -C., & Li, N. -G. (2015). Development of Orally Active Thrombin Inhibitors for the Treatment of Thrombotic Disorder Diseases. Molecules, 20(6), 11046-11062. https://doi.org/10.3390/molecules200611046