
Simultaneous Fitting of Absorption Spectra and Their Second Derivatives for an Improved Analysis of Protein Infrared Spectra

Abstract
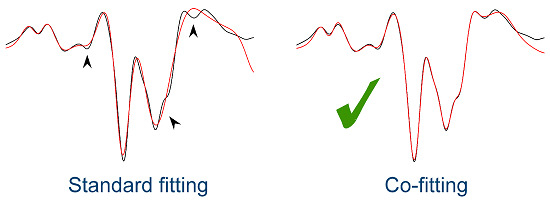
:
1. Introduction
2. Results and Discussion
2.1. Method Benchmarking and Quality Assessment


{kind=link}
{kind=link}
{kind=link}
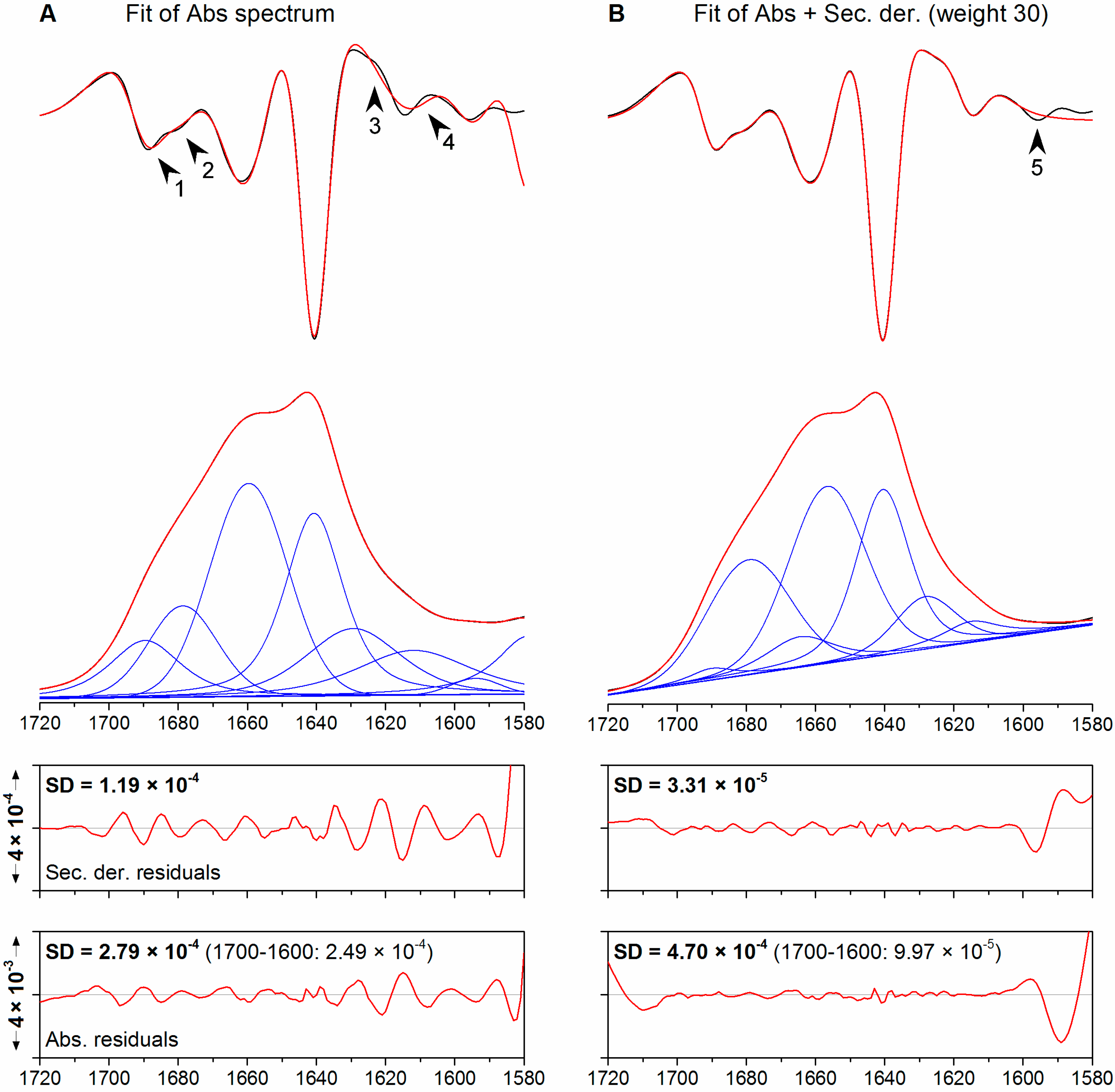{kind=link}
{kind=link}
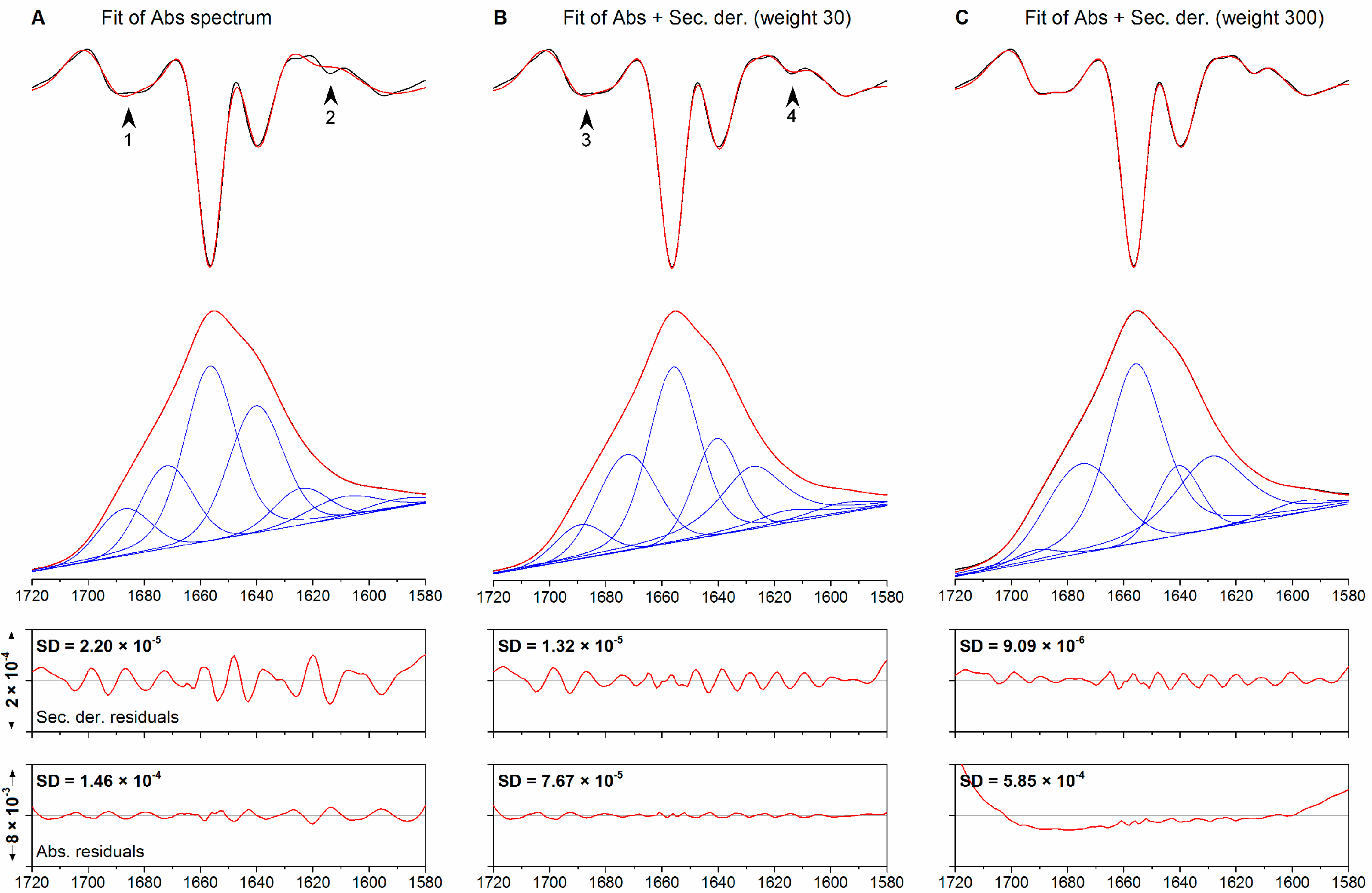{kind=link}
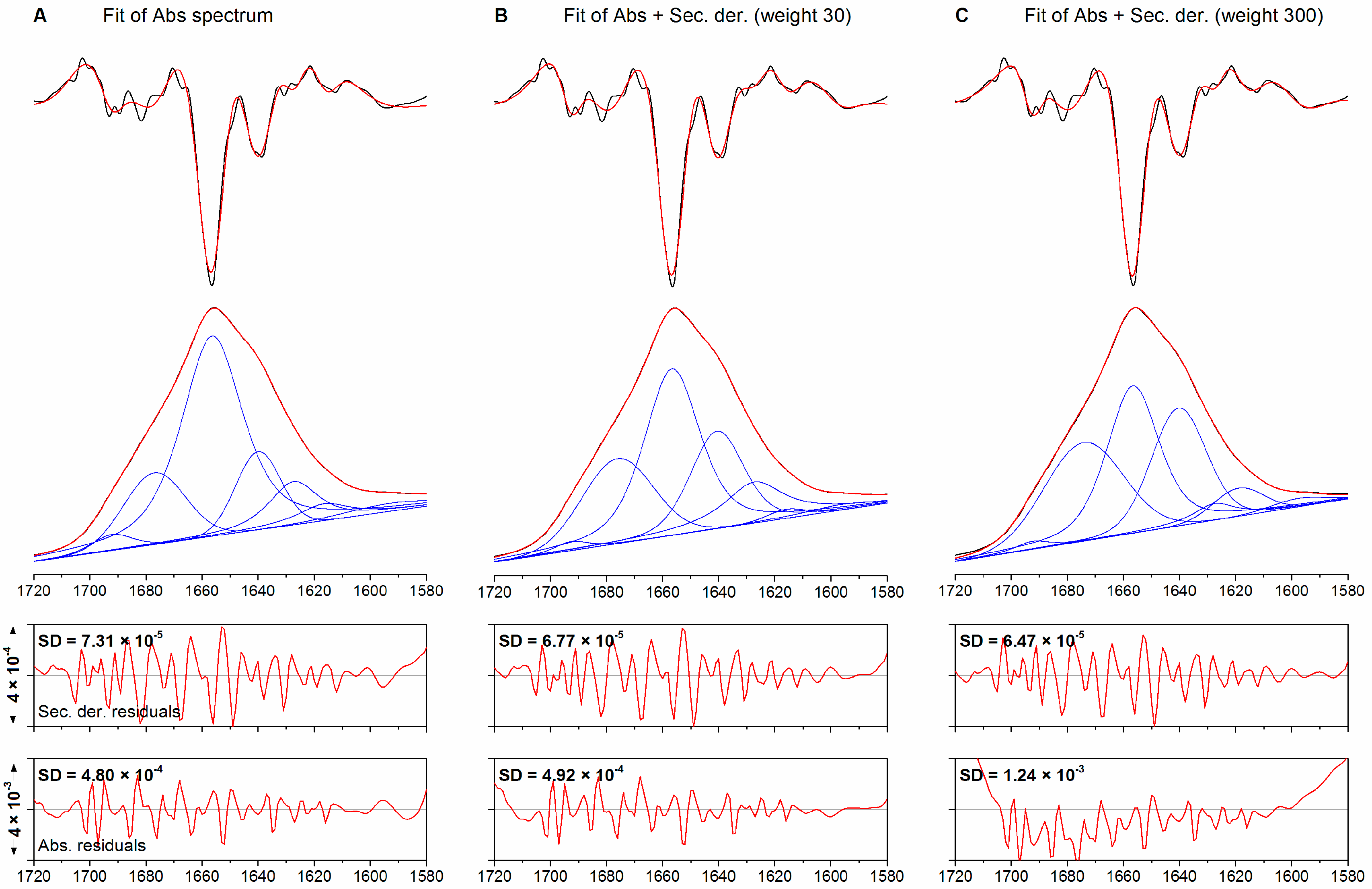{kind=link}
| Band | Initial Position/cm−1 | Final Position/cm−1 | Width (fg) | Intensity | Area/% |
|---|---|---|---|---|---|
| Parameters for Synthetic Spectrum | |||||
| 1 | 1690 | 20 (0.7) | 0.008 | 9.3 | |
| 2 | 1675 | 20 (0.7) | 0.006 | 7.1 | |
| 3 | 1652 | 35 (1.0) | 0.020 | 37.5 | |
| 4 | 1635 | 20 (0.7) | 0.020 | 23.6 | |
| 5 | 1625 | 19 (0.7) | 0.015 | 16.8 | |
| 6 | 1600 | 20 (0.7) | 0.005 | 5.7 | |
| 7 | 1585 | 20 (0.7) | 0.005 | 4.3 | |
| 8 | 1550 | 45 (1.0) | 0.025 | ||
| 9 | 1515 | 15 (0.7) | 0.002 | ||
| Fitting to Abs spectrum only | |||||
| 1 | See above | 1690.5 | 19.9 (0.63) | 0.008 | 8.4 |
| 2 | 1672.7 | 23.2 (1.00) | 0.011 | 11.5 | |
| 3 | 1653.9 | 22.9 (1.00) | 0.016 | 16.9 | |
| 4 | 1635.1 | 22.6 (0.83) | 0.030 | 32.7 | |
| 5 | 1624.3 | 20.0 (0.55) | 0.012 | 13.4 | |
| 6 | 1599.1 | 19.6 (1.00) | 0.006 | 4.9 | |
| 7 | 1579.0 | 23.7 (1.00) | 0.011 | 12.3 | |
| Fitting to Abs + Der, 30 | |||||
| 1 | See above | 1690.1 | 18.9 (0.81) | 0.007 | 8.1 |
| 2 | 1675.0 | 19.9 (0.75) | 0.006 | 7.4 | |
| 3 | 1652.0 | 33.5 (1.00) | 0.019 | 33.5 | |
| 4 | 1635.0 | 19.5 (0.74) | 0.021 | 24.4 | |
| 5 | 1625.0 | 17.0 (0.85) | 0.013 | 12.7 | |
| 6 | 1600.5 | 11.3 (0.61) | 0.001 | 0.6 | |
| 7 | 1579.0 | 35.1 (1.00) | 0.007 | 13.3 | |
| Fitting to Abs + Der, 3000 | |||||
| 1 | See above | 1690.1 | 20.1 (0.68) | 0.008 | 9.44 |
| 2 | 1674.8 | 22.0 (0.63) | 0.008 | 10.17 | |
| 3 | 1652.6 | 31.6 (1.00) | 0.019 | 31.51 | |
| 4 | 1635.2 | 20.3 (0.68) | 0.023 | 27.67 | |
| 5 | 1625.1 | 17.3 (0.88) | 0.014 | 12.93 | |
| 6 | 1600.5 | 12.9 (0.13) | 0.001 | 0.93 | |
| 7 | 1579.0 | 41.6 (1.00) | 0.003 | 7.34 | |
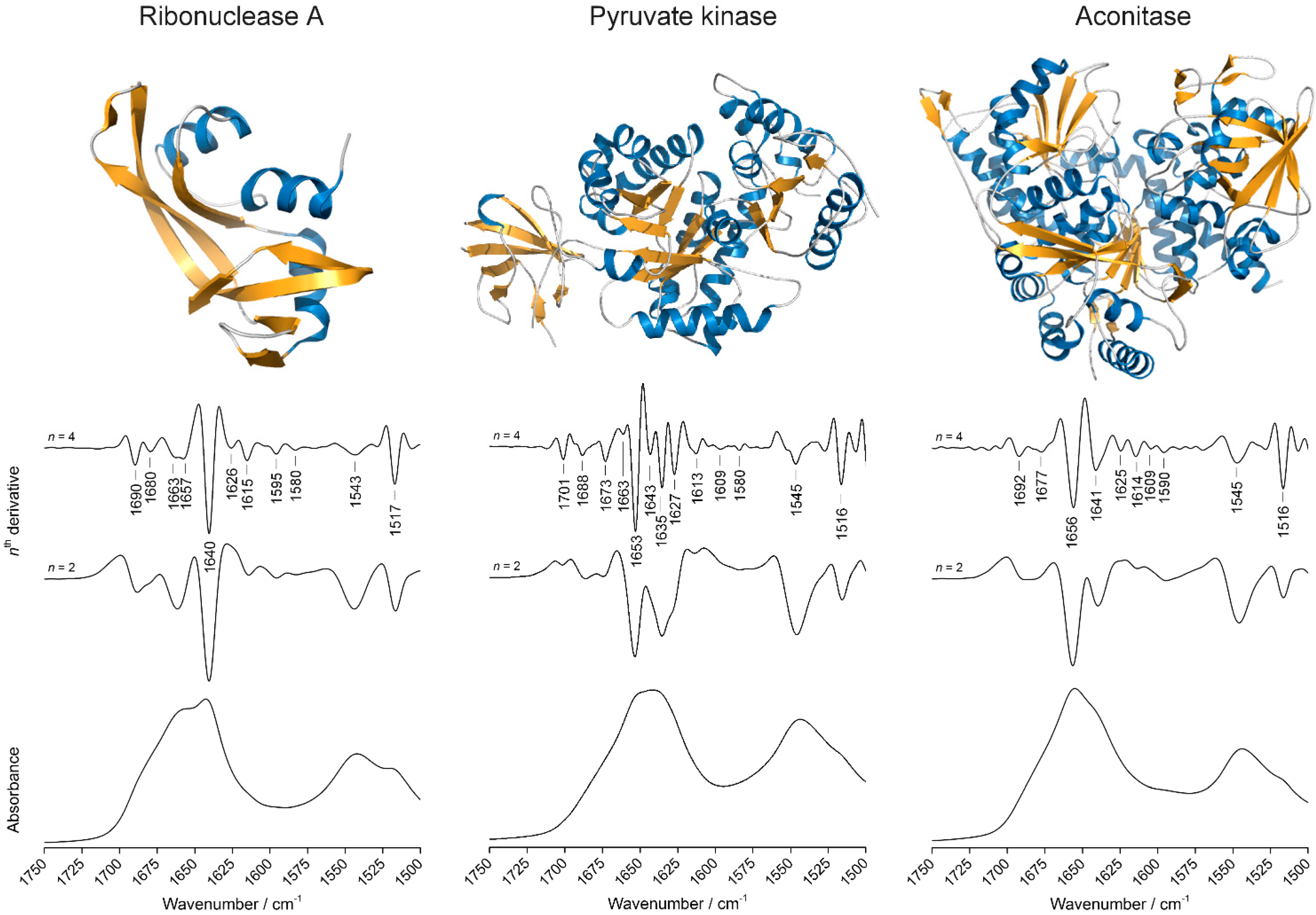
2.2. Overview of the Protein Infrared Spectra

2.3. Introduction to Co-Fitting of Protein Spectra at the Example of Ribonuclease A

| Band | Initial Position | Final Position | Width (fg) | Intensity | Area/% |
|---|---|---|---|---|---|
| Fitting of Abs spectrum alone | |||||
| 1 | 1690 | 1689.6 | 24.2 (0.37) | 0.074 | 10.5 |
| 2 | 1680 | 1678.6 | 23.5 (0.90) | 0.118 | 13.2 |
| 3 | 1663 | 1659.6 | 26.5 (1.00) | 0.274 | 33.3 |
| 4 | 1657 | 1656.2 | 23.5 (0.59) | 0.001 | 0.1 |
| 5 | 1640 | 1640.7 | 20.1 (0.41) | 0.235 | 27.5 |
| 6 | 1626 | 1629.1 | 32.5 (0.57) | 0.086 | 15.4 |
| 7 | 1615 | 1611.9 | 39.5 (0.02) | 0.058 | |
| 8 | 1595 | 1594.0 | 16.6 (0.42) | 0.021 | |
| 9 | 1580 | 1579.0 | 21.8 (1.00) | 0.074 | |
| Co-fitting (weight: 30) | |||||
| 1 | Same | 1690.2 | 15.4 (0.17) | 0.015 | 1.8 |
| 2 | as | 1679.3 | 28.3 (1.00) | 0.148 | 23.0 |
| 3 | above | 1664.6 | 23.1 (0.43) | 0.039 | 6.3 |
| 4 | 1656.7 | 26.3 (0.87) | 0.227 | 34.8 | |
| 5 | 1640.5 | 18.2 (0.57) | 0.213 | 25.4 | |
| 6 | 1628.2 | 20.8 (0.72) | 0.067 | 8.7 | |
| 7 | 1615.0 | 20.7 (0.65) | 0.027 | ||
| 8 | 1590.3 | 37.2 (0.00) | 0.000 | ||
| 9 | 1580.3 | 44.3 (0.00) | 0.000 | ||
2.4. Effect of Different Weights for the Second Derivative in Co-Fitting
2.4.1. Pyruvate Kinase
| Band | Initial Position | Final Position | Width (fg) | Intensity | Area/% |
|---|---|---|---|---|---|
| Fitting of Abs spectrum alone | |||||
| 1 | 1701 | 1701.8 | 9.2 (0.78) | 0.015 | 0.2 |
| 2 | 1688 | 1688.2 | 20.9 (1.00) | 0.103 | 3.9 |
| 3 | 1673 | 1670.6 | 29.1 (0.86) | 0.298 | 16.8 |
| 4 | 1663 | 1659.6 | 33.4 (1.00) | 0.235 | 14.4 |
| 5 | 1653 | 1654.0 | 17.9 (0.61) | 0.295 | 11.1 |
| 6 | 1643 | 1641.1 | 29.1 (0.76) | 0.376 | 22.2 |
| 7 | 1635 | 1635.9 | 27.6 (0.92) | 0.311 | 16.2 |
| 8 | 1627 | 1627.2 | 31.1 (0.70) | 0.260 | 14.9 |
| 9 | 1613 | 1613.4 | 28.6 (0.69) | 0.062 | |
| 10 | 1609 | 1609.8 | 31.3 (1.00) | 0.057 | |
| 11 | 1580 | 1579.0 | 27.2 (0.00) | 0.096 | |
| Co-fitting (weight: 30) | |||||
| 1 | Same | 1701.3 | 7.6 (0.94) | 0.007 | 0.1 |
| 2 | as | 1685.6 | 22.3 (1.00) | 0.141 | 6.3 |
| 3 | above | 1673.1 | 24.3 (0.54) | 0.157 | 9.2 |
| 4 | 1658.2 | 29.6 (0.73) | 0.545 | 36.2 | |
| 5 | 1652.8 | 15.6 (0.63) | 0.280 | 10.2 | |
| 6 | 1643.7 | 15.7 (0.83) | 0.301 | 10.2 | |
| 7 | 1635.5 | 17.2 (0.81) | 0.484 | 18.2 | |
| 8 | 1627.0 | 16.3 (0.74) | 0.262 | 9.6 | |
| 9 | 1617.7 | 21.2 (0.77) | 0.210 | ||
| 10 | 1613.1 | 29.7 (0.49) | 0.019 | ||
| 11 | 1579.0 | 15.6 (0.00) | 0.056 | ||
| Co-fitting (weight: 300) | |||||
| 1 | Same | 1701.1 | 12.7 (0.52) | 0.019 | 0.5 |
| 2 | as | 1688.5 | 18.7 (0.98) | 0.084 | 3.0 |
| 3 | above | 1672.8 | 27.5 (0.84) | 0.323 | 17.8 |
| 4 | 1660.1 | 22.5 (0.50) | 0.340 | 17.5 | |
| 5 | 1652.7 | 15.3 (0.70) | 0.371 | 12.0 | |
| 6 | 1644.1 | 18.7 (0.40) | 0.398 | 17.6 | |
| 7 | 1636.4 | 18.4 (0.73) | 0.472 | 18.2 | |
| 8 | 1626.9 | 18.6 (0.70) | 0.339 | 13.4 | |
| 9 | 1616.0 | 18.4 (0.68) | 0.107 | ||
| 10 | 1612.1 | 26.1 (0.69) | 0.056 | ||
| 11 | 1581.5 | 18.8 (0.91) | 0.075 | ||

2.4.2. Aconitase
| Band | Initial Position | Final Position | Width (fg) | Intensity | Area/% |
|---|---|---|---|---|---|
| Fitting of Abs spectrum alone | |||||
| 1 | 1692 | 1687.1 | 22.0 (1.00) | 0.049 | 9.3 |
| 2 | 1677 | 1672.1 | 22.0 (1.00) | 0.086 | 16.5 |
| 3 | 1656 | 1656.6 | 21.7 (0.70) | 0.182 | 38.9 |
| 4 | 1641 | 1640.3 | 22.8 (0.86) | 0.132 | 27.9 |
| 5 | 1625 | 1624.2 | 22.3 (1.00) | 0.038 | 7.4 |
| 6 | 1614 | 1615.4 | 28.9 (0.89) | 0.011 | |
| 7 | 1609 | 1609.1 | 29.5 (1.00) | 0.021 | |
| 8 | 1590 | 1590.0 | 26.0 (1.00) | 0.009 | |
| Co-fitting (weight: 30) | |||||
| 1 | Same | 1689.1 | 20.8 (0.96) | 0.035 | 6.3 |
| 2 | as | 1672.6 | 24.7 (1.00) | 0.100 | 20.7 |
| 3 | above | 1655.8 | 21.3 (0.75) | 0.182 | 36.4 |
| 4 | 1640.5 | 19.0 (1.00) | 0.100 | 16.0 | |
| 5 | 1627.8 | 27.8 (0.22) | 0.065 | 20.5 | |
| 6 | 1615.7 | 28.9 (0.00) | 0.013 | ||
| 7 | 1609.6 | 28.5 (0.48) | 0.021 | ||
| 8 | 1596.2 | 27.5 (0.38) | 0.010 | ||
| Co-fitting (weight: 300) | |||||
| 1 | Same | 1691.5 | 18.3 (0.61) | 0.012 | 2.1 |
| 2 | as | 1675.0 | 30.0 (1.00) | 0.093 | 22.0 |
| 3 | above | 1655.8 | 23.9 (0.52) | 0.187 | 42.6 |
| 4 | 1640.6 | 18.2 (1.00) | 0.073 | 10.4 | |
| 5 | 1628.7 | 30.5 (0.42) | 0.076 | 22.9 | |
| 6 | 1613.8 | 15.0 (0.47) | 0.035 | ||
| 7 | 1609.0 | 27.6 (0.27) | 0.003 | ||
| 8 | 1597.7 | 25.4 (1.00) | 0.012 | ||

2.5. Fitting in the Presence of Water Vapor Bands

3. Experimental Section
3.1. Materials
3.2. Preparation of Protein Samples
3.3. Infrared Spectroscopy
3.4. Spectral Analysis and Curve-Fitting
3.5. Secondary Structure Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| SD | standard deviation |
| SDAbs | standard deviation of the difference between two absorption spectra |
| SDDer | standard deviation of the difference between two second derivative spectra |
References
- Yang, H.; Yang, S.; Kong, J.; Dong, A.; Yu, S. Obtaining information about protein secondary structures in aqueous solution using Fourier transform infrared spectroscopy. Nat. Protoc. 2015, 10, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta Bioenerg. 2007, 1767, 1073–1101. [Google Scholar] [CrossRef] [PubMed]
- Barth, A.; Zscherp, C. What vibrations tell about proteins. Q. Rev. Biophys. 2002, 35, 369–430. [Google Scholar] [CrossRef] [PubMed]
- Mäntele, W. The analysis of protein conformation by infrared spectroscopy: An introduction of the editor to a scientific dispute. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 138, 964–966. [Google Scholar] [CrossRef] [PubMed]
- Manning, M.C. Use of infrared spectroscopy to monitor protein structure and stability. Expert Rev. Proteomics 2005, 2, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Gilmanshin, R.; Williams, S.; Callender, R.H.; Woodruff, W.H.; Dyer, R.B. Fast events in protein folding: Relaxation dynamics of secondary and tertiary structure in native apomyoglobin. Proc. Natl. Acad. Sci. USA 1997, 94, 3709–3713. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.; Causgrove, T.P.; Gilmanshin, R.; Fang, K.S.; Callender, R.H.; Woodruff, W.H.; Dyer, R.B. Fast Events in Protein Folding: Helix Melting and Formation in a Small Peptide. Biochemistry 1996, 35, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Heimburg, T.; Schünemann, J.; Weber, K.; Geisler, N. FTIR-Spectroscopy of Multistranded Coiled Coil Proteins. Biochemistry 1999, 38, 12727–12734. [Google Scholar] [CrossRef] [PubMed]
- Karjalainen, E.L.; Barth, A. Vibrational Coupling between Helices Influences the Amide I Infrared Absorption of Proteins: Application to Bacteriorhodopsin and Rhodopsin. J. Phys. Chem. B 2012, 116, 4448–4456. [Google Scholar] [CrossRef] [PubMed]
- Karjalainen, E.L.; Ravi, H.K.; Barth, A. Simulation of the Amide I Absorption of Stacked β-Sheets. J. Phys. Chem. B 2011, 115, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Ham, S.; Cho, M. Inter-peptide interaction and delocalization of amide I vibrational excitons in myoglobin and flavodoxin. J. Chem. Phys. 2002, 117, 6821. [Google Scholar] [CrossRef]
- Strasfeld, D.B.; Ling, Y.L.; Gupta, R.; Raleigh, D.P.; Zanni, M.T. Strategies for Extracting Structural Information from 2D–IR Spectroscopy of Amyloid: Application to Islet Amyloid Polypeptide. J. Phys. Chem. B 2009, 113, 15679–15691. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer-Stenner, R.; Measey, T.J. Simulation of infrared, Raman and VCD amide I band profiles of self-assembled peptides. Spectroscopy 2010, 24, 25–36. [Google Scholar] [CrossRef]
- Chehín, R.; Iloro, I.; Marcos, M.J.; Villar, E.; Shnyrov, V.L.; Arrondo, J.L. Thermal and pH-induced conformational changes of a β-sheet protein monitored by infrared spectroscopy. Biochemistry 1999, 38, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Herman, P.; Staiano, M.; Marabotti, A.; Varriale, A.; Scirè, A.; Tanfani, F.; Vecer, J.; Rossi, M.; D’Auria, S. D-Trehalose/D-maltose-binding protein from the hyperthermophilic archaeon Thermococcus litoralis: The binding of trehalose and maltose results in different protein conformational states. Proteins Struct. Funct. Bioinform. 2006, 63, 754–767. [Google Scholar] [CrossRef] [PubMed]
- Abbott, G.W.; Ramesh, B.; Srai, S.K. Interaction between Soluble and Membrane-Embedded Potassium Channel Peptides Monitored by Fourier Transform Infrared Spectroscopy. PLoS ONE 2012, 7, e49070. [Google Scholar] [CrossRef] [PubMed]
- Adato, R.; Altug, H. In situ ultra-sensitive infrared absorption spectroscopy of biomolecule interactions in real time with plasmonic nanoantennas. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Barth, A.; Zscherp, C. Substrate binding and enzyme function investigated by infrared spectroscopy. FEBS Lett. 2000, 477, 151–156. [Google Scholar] [CrossRef]
- Baldassarre, M.; Scirè, A.; Tanfani, F. Turning pyridoxal-5'-phosphate-dependent enzymes into thermostable binding proteins: D-Serine dehydratase from baker’s yeast as a case study. Biochimie 2012, 94, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Dodd, J.G.; DeNoyer, L.K. Handbook of Vibrational Spectroscopy; Chalmers, J.M., Griffiths, P.R., Eds.; John Wiley & Sons Ltd.: Chichester, UK, 2002. [Google Scholar]
- Arrondo, J.L.; Muga, A.; Castresana, J.; Goñi, F.M. Quantitative studies of the structure of proteins in solution by Fourier-transform infrared spectroscopy. Prog. Biophys. Mol. Biol. 1993, 59, 23–56. [Google Scholar] [CrossRef]
- Byler, D.M.; Wilson, R.M.; Randall, C.S.; Sokoloski, T.D. Second derivative infrared spectroscopy as a non-destructive tool to assess the purity and structural integrity of proteins. Pharm. Res. 1995, 12, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Susi, H.; Byler, D.M. Protein structure by Fourier transform infrared spectroscopy: Second derivative spectra. Biochem. Biophys. Res. Commun. 1983, 115, 391–397. [Google Scholar] [CrossRef]
- Kauppinen, J.K.; Moffat, D.J.; Mantsch, H.H.; Cameron, D.G. Fourier self-deconvolution: A method for resolving intrinsically overlapped bands. Appl. Spectrosc. 1981, 35, 271–276. [Google Scholar] [CrossRef]
- Lórenz-Fonfría, V.A.; Padrós, E. Curve-fitting of Fourier manipulated spectra comprising apodization, smoothing, derivation and deconvolution. Spec. Acta Part A 2004, 60, 2703–2710. [Google Scholar] [CrossRef] [PubMed]
- Holler, F.; Burns, D.H.; Callis, J.B. Direct Use of Second Derivatives in Curve-Fitting Procedures. Appl. Spectrosc. 1989, 43, 877–882. [Google Scholar] [CrossRef]
- Gasda, P.J.; Ogliore, R.C. Modeling the Raman spectrum of graphitic material in rock samples with fluorescence backgrounds: Accuracy of fitting and uncertainty estimation. Appl. Spectrosc. 2014, 68, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Dong, A.; Huang, P.; Caughey, W.S. Protein secondary structures in water from second-derivative amide I infrared spectra. Biochemistry 1990, 29, 3303–3308. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Li, H.; Ou, Y.; Tao, W.; Dong, A.; Kong, J.; Ji, C.; Yu, S. The Secondary Structure of Calcineurin Regulatory Region and Conformational Change Induced by Calcium/Calmodulin Binding. J. Biol. Chem. 2008, 283, 11407–11413. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.R. Basic Aspects of the Technique and Applications of Infrared Spectroscopy of Peptides and Proteins. In Infrared Analysis of Peptids and Proteins Principles and Applications; ACS Symposium Series; Singh, B.R., Ed.; ACS: Washington, DC, USA, 1999; Volume 750, pp. 2–37. [Google Scholar]
- Mikhailyuk, I.K.; Lokstein, H.; Razjivin, A.P. A method of spectral subband decomposition by simultaneous fitting the initial spectrum and a set of its derivatives. J. Biochem. Biophys. Methods 2005, 63, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Kumar, S.; Montigny, C.; le Maire, M.; Barth, A. Quality assessment of recombinant proteins by infrared spectroscopy. Characterisation of a protein aggregation related band of the Ca2+-ATPase. Analyst 2014, 139, 4231–4240. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, A.; Bott, R.; Sjölin, L. The refined crystal structure of ribonuclease A at 2.0 Å resolution. J. Biol. Chem. 1982, 257, 1325–1332. [Google Scholar] [PubMed]
- Larsen, T.M.; Laughlin, L.T.; Holden, H.M.; Rayment, I.; Reed, G.H. Structure of Rabbit Muscle Pyruvate Kinase Complexed with Mn2+, K+, and Pyruvate. Biochemistry 1994, 33, 6301–6309. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, S.J.; Lauble, H.; Prasad, G.S.; Stout, C.D. The mechanism of aconitase: 1.8 Å resolution crystal structure of the S642A: Citrate complex. Protein Sci. 1999, 8, 2655–2662. [Google Scholar] [CrossRef] [PubMed]
- Krimm, S.; Bandekar, J. Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. Adv. Protein Chem. 1986, 38, 181–364. [Google Scholar] [PubMed]
- Olinger, J.M.; Hill, D.M.; Jakobsen, R.J.; Brody, R.S. Fourier transform infrared studies of ribonuclease in H2O and 2H2O solutions. Biochim. Biophys. Acta 1986, 896, 89–98. [Google Scholar] [CrossRef]
- Barth, A. The infrared absorption of amino acid side chains. Prog. Biophys. Mol. Biol. 2000, 74, 141–173. [Google Scholar] [CrossRef]
- Yu, S.; Lee, L.L.-Y.; Ching Lee, J. Effects of metabolites on the structural dynamics of rabbit muscle pyruvate kinase. Biophys. Chem. 2003, 103, 1–11. [Google Scholar] [CrossRef]
- Surewicz, W.K.; Mantsch, H.H.; Chapman, D. Determination of protein secondary structure by transform infrared spectroscopy: A critical assessment. Biochemistry 1993, 32, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Mantsch, H.H. The Use and Misuse of FTIR Spectroscopy in the Determination of Protein Structure. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 95–120. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldassarre, M.; Li, C.; Eremina, N.; Goormaghtigh, E.; Barth, A. Simultaneous Fitting of Absorption Spectra and Their Second Derivatives for an Improved Analysis of Protein Infrared Spectra. Molecules 2015, 20, 12599-12622. https://doi.org/10.3390/molecules200712599
Baldassarre M, Li C, Eremina N, Goormaghtigh E, Barth A. Simultaneous Fitting of Absorption Spectra and Their Second Derivatives for an Improved Analysis of Protein Infrared Spectra. Molecules. 2015; 20(7):12599-12622. https://doi.org/10.3390/molecules200712599
Chicago/Turabian StyleBaldassarre, Maurizio, Chenge Li, Nadejda Eremina, Erik Goormaghtigh, and Andreas Barth. 2015. "Simultaneous Fitting of Absorption Spectra and Their Second Derivatives for an Improved Analysis of Protein Infrared Spectra" Molecules 20, no. 7: 12599-12622. https://doi.org/10.3390/molecules200712599
APA StyleBaldassarre, M., Li, C., Eremina, N., Goormaghtigh, E., & Barth, A. (2015). Simultaneous Fitting of Absorption Spectra and Their Second Derivatives for an Improved Analysis of Protein Infrared Spectra. Molecules, 20(7), 12599-12622. https://doi.org/10.3390/molecules200712599