
N-Monosubstituted Methoxy-oligo(ethylene glycol) Carbamate Ester Prodrugs of Resveratrol



Abstract
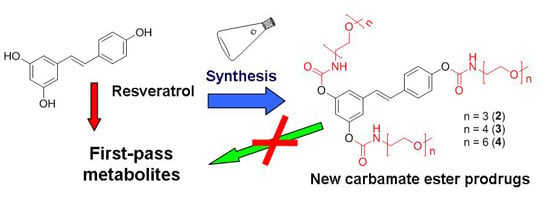
:
1. Introduction

2. Results and Discussion
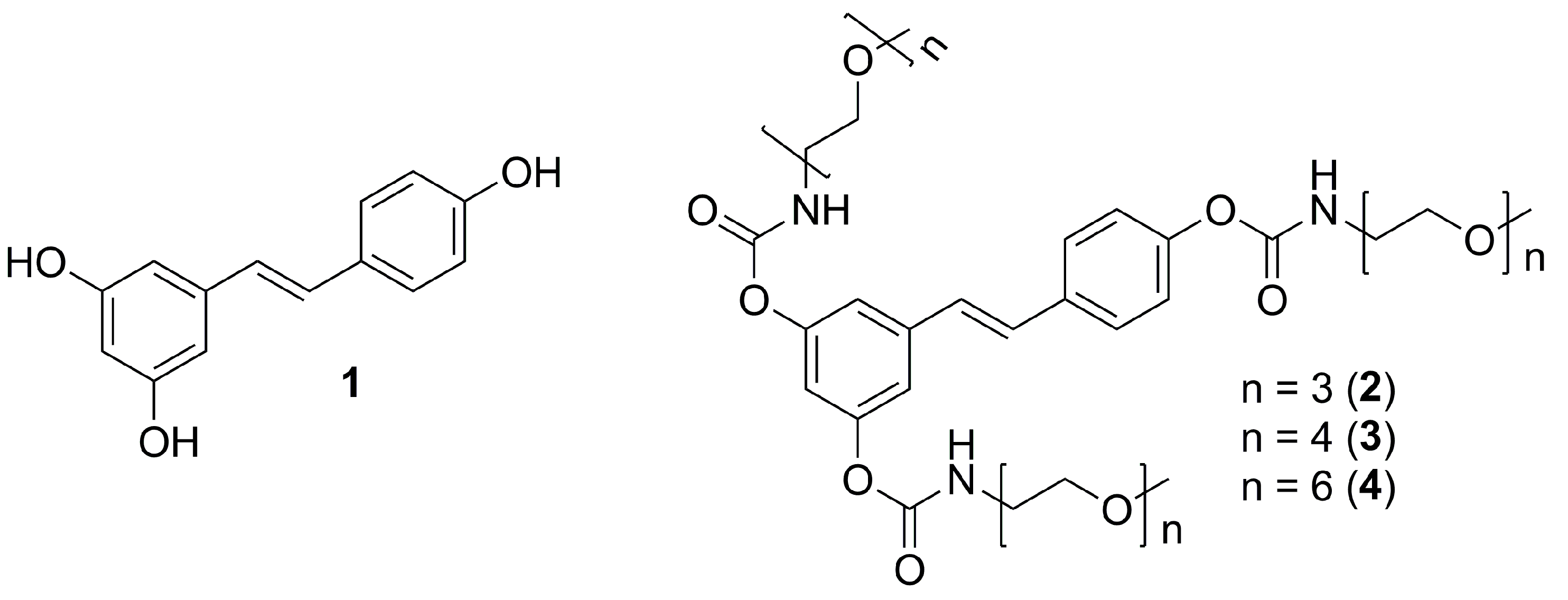
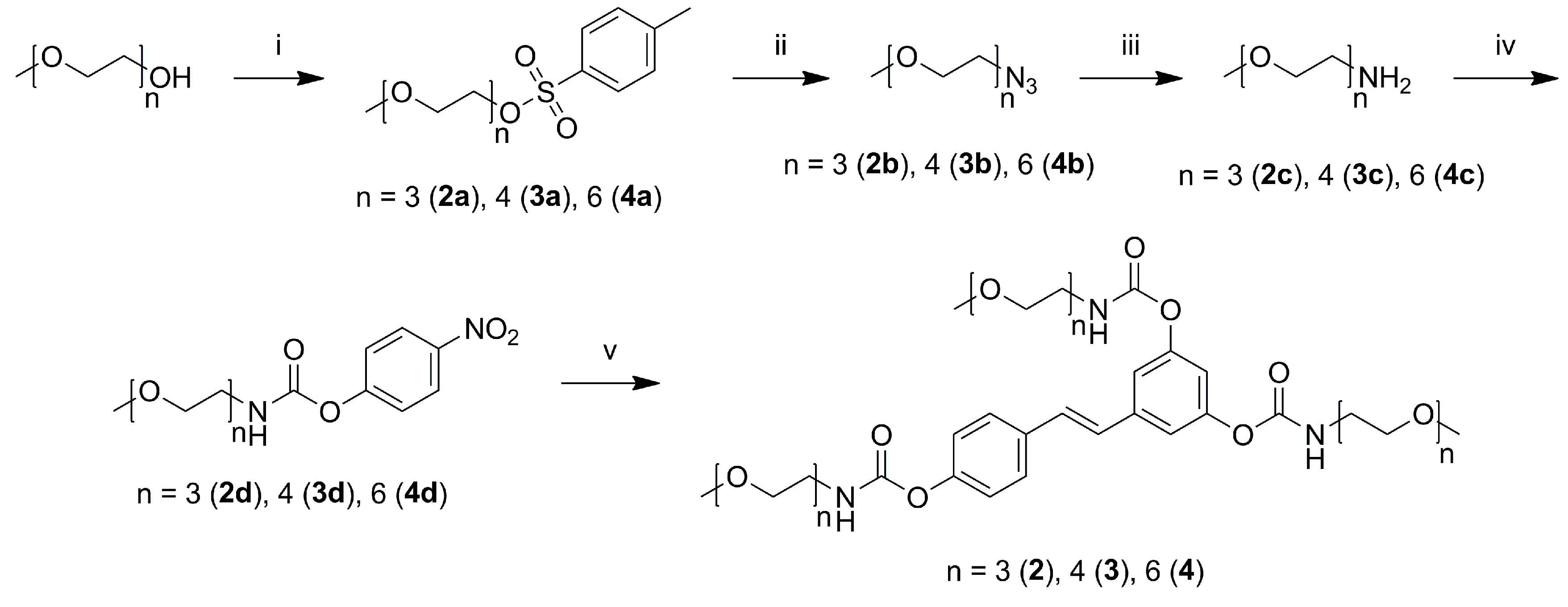
2.1. Synthesis

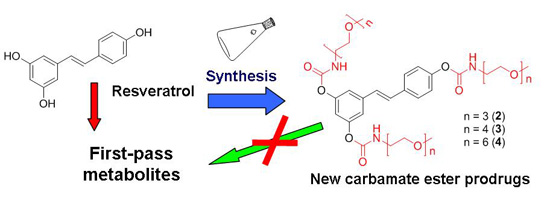
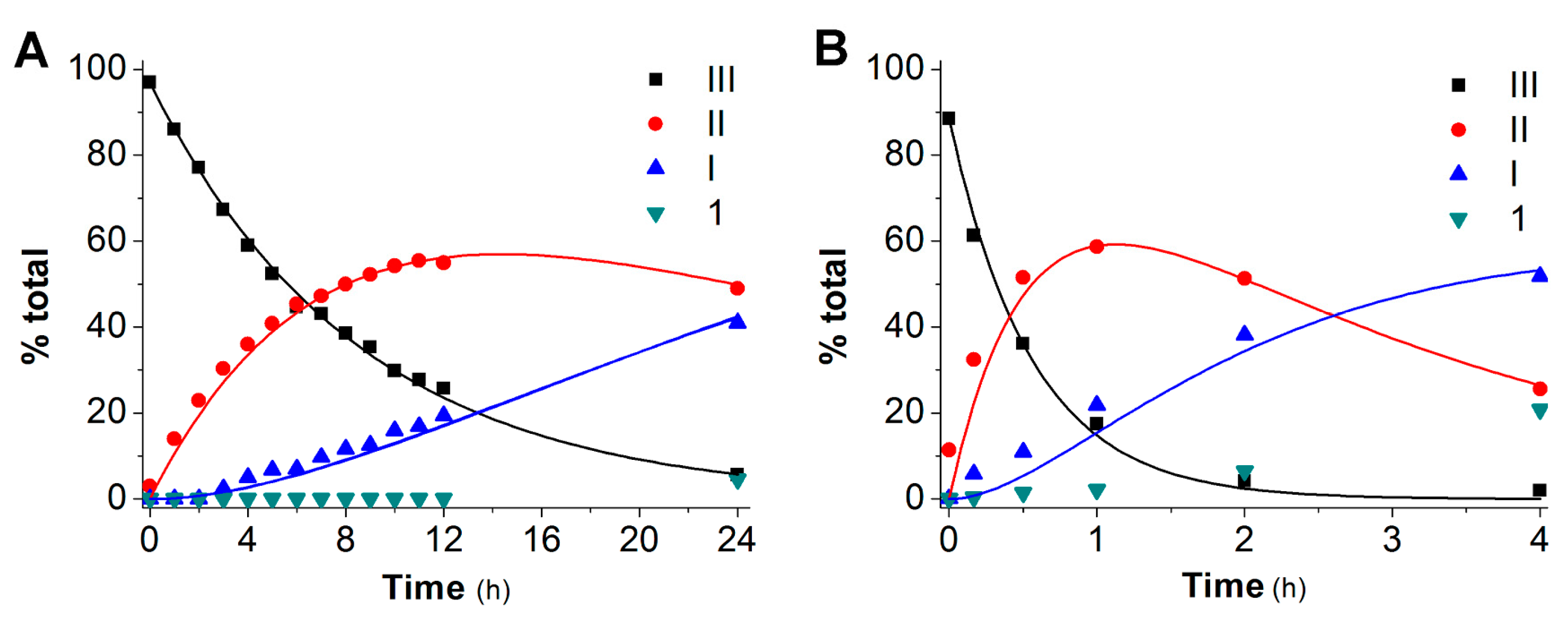
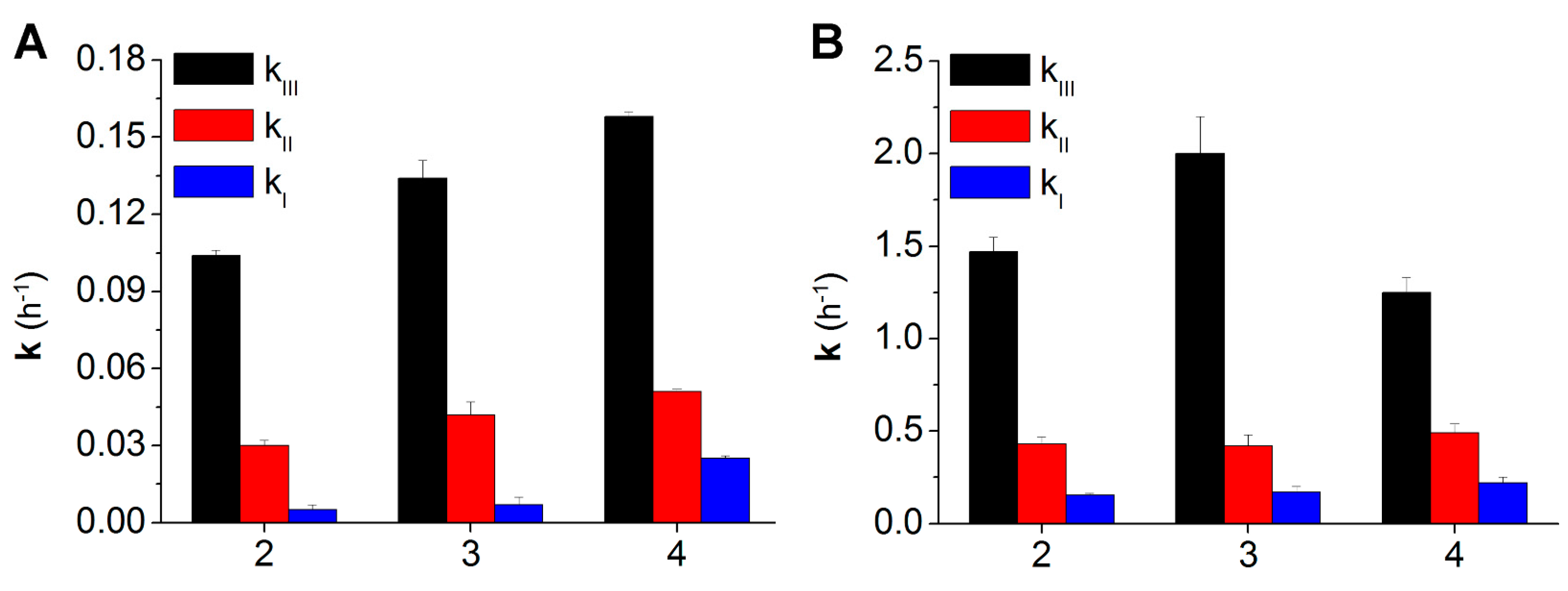
2.2. Hydrolysis Studies



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
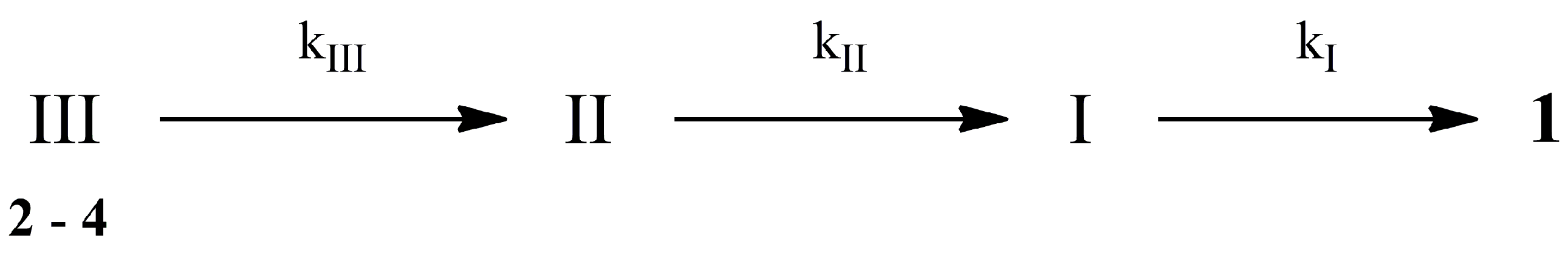{kind=link}
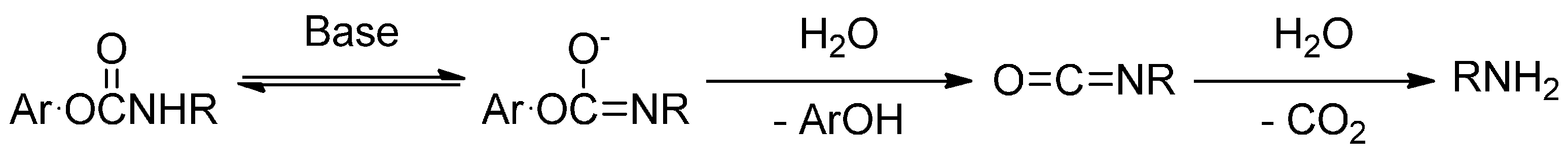{kind=link}
| Derivative | PBS 0.1 M, at pH 6.8, 37 °C | Blood | ||||||
|---|---|---|---|---|---|---|---|---|
| t1/2 (h) | kIII (h−1) | kII (h−1) | kI (h−1) | t1/2 (h) | kIII (h−1) | kII (h−1) | kI (h−1) | |
| 2 | 6.0 | 0.104 ± 0.002 | 0.030 ± 0.002 | 0.005 ± 0.002 | 0.5 | 1.47 ± 0.08 | 0.43 ± 0.04 | 0.155 ± 0.009 |
| 3 | 3.5 | 0.134 ± 0.007 | 0.042 ± 0.005 | 0.007 ± 0.003 | 0.3 | 2.0 ± 0.2 | 0.42 ± 0.06 | 0.17 ± 0.03 |
| 4 | 4.5 | 0.158 ± 0.002 | 0.051 ± 0.001 | 0.025 ± 0.001 | 0.5 | 1.25 ± 0.08 | 0.49 ± 0.05 | 0.22 ± 0.03 |


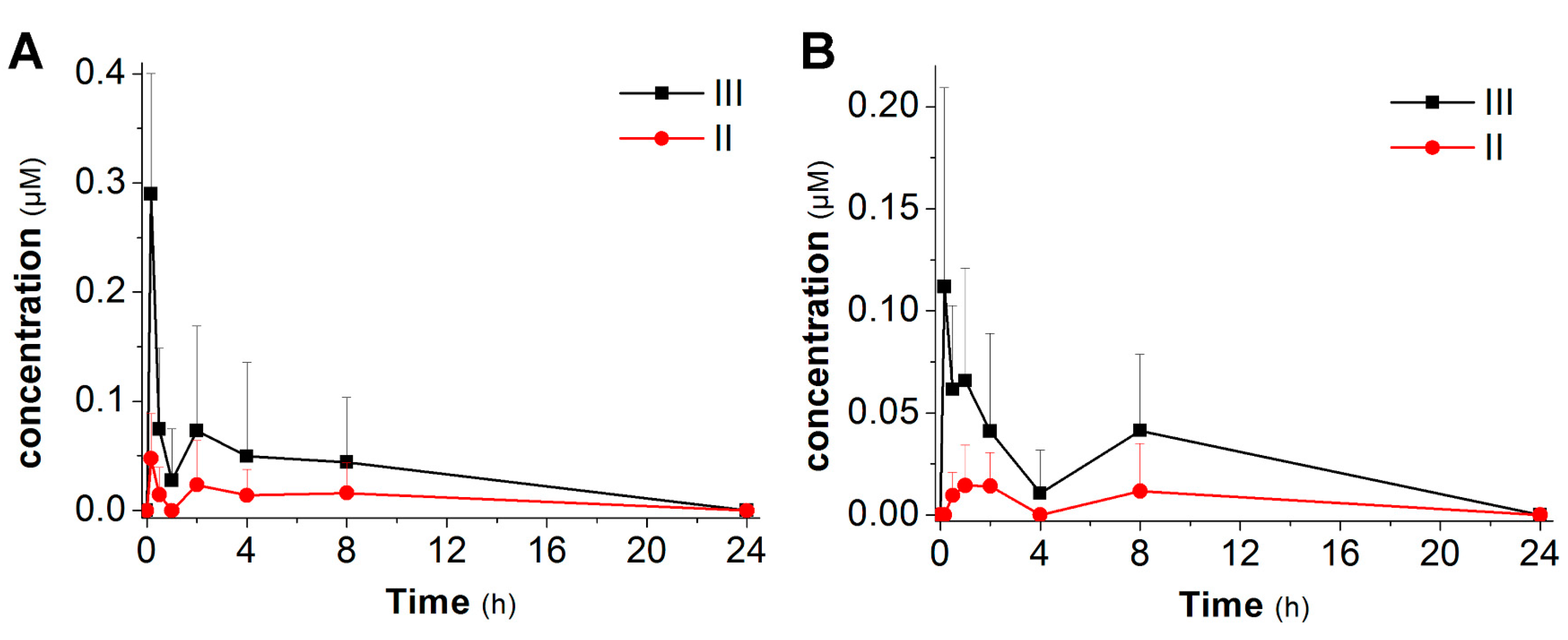
2.3. Pharmacokinetic Studies

3. Experimental Section
3.1. Materials and Instrumentation
3.2. Hydrolysis Assays
3.2.1. Hydrolysis under Physiological-Like Conditions
3.2.2. Stability in Rodent Whole Blood
3.2.3. Blood Sample Treatment and Analysis
3.3. Pharmacokinetic Studies
3.4. Synthesis
3.4.1. General Procedure for the Preparation of Methoxy-oligo(ethylene glycol)-p-toluenesulfonates (2a–4a)
3.4.2. General Procedure for the Preparation of Methoxy-oligo(ethylene glycol)-azides (2b–4b)
3.4.3. General Procedure for the Preparation of Methoxy-oligo(ethylene glycol)-amines (2c–4c)
3.4.4. General Procedure for the Preparation of Activated 4-Nitrophenyl Methoxy-oligo(ethylene glycol) Urethanes (2d–4d)
3.4.5. General Procedure for the Preparation of 3,4′,5-N-Monosubstituted-methoxy-oligo(ethylene glycol) Resveratrol Carbamate Esters (2–4)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Beyerle, J.; Frei, E.; Stiborova, M.; Habermann, N.; Ulrich, C.M. Biotransformation of xenobiotics in the human colon and rectum and its association with colorectal cancer. Drug Metab. Rev. 2015, 47, 199–221. [Google Scholar] [CrossRef] [PubMed]
- Bock, K.W. Homeostatic control of xeno- and endobiotics in the drug-metabolizing enzyme system. Biochem. Pharmacol. 2014, 90, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gundert-Remy, U.; Bernauer, U.; Blomeke, B.; Doring, B.; Fabian, E.; Goebel, C.; Hessel, S.; Jackh, C.; Lampen, A.; Oesch, F.; et al. Extrahepatic metabolism at the body’s internal-external interfaces. Drug Metab. Rev. 2014, 46, 291–324. [Google Scholar] [CrossRef] [PubMed]
- Hrycay, E.G.; Bandiera, S.M. The monooxygenase, peroxidase, and peroxygenase properties of cytochrome P450. Arch. Biochem. Biophys. 2012, 522, 71–89. [Google Scholar] [CrossRef] [PubMed]
- Hrycay, E.G.; Bandiera, S.M. Monooxygenase, peroxidase and peroxygenase properties and reaction mechanisms of cytochrome p450 enzymes. Adv. Exp. Med. Biol. 2015, 851, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Ako, R.; Wu, B. Crystal structures of human sulfotransferases: Insights into the mechanisms of action and substrate selectivity. Exp. Opin. Drug Metab. Toxicol. 2012, 8, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Runge-Morris, M.; Kocarek, T.A.; Falany, C.N. Regulation of the cytosolic sulfotransferases by nuclear receptors. Drug Metab. Rev. 2013, 45, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Oda, S.; Fukami, T.; Yokoi, T.; Nakajima, M. A comprehensive review of UDP-glucuronosyltransferase and esterases for drug development. Drug Metab. Pharmacokinet. 2015, 30, 30–51. [Google Scholar] [CrossRef] [PubMed]
- Ouzzine, M.; Gulberti, S.; Ramalanjaona, N.; Magdalou, J.; Fournel-Gigleux, S. The UDP-glucuronosyltransferases of the bloodbrain barrier: Their role in drug metabolism and detoxication. Front. Cell Neurosci. 2014, 8, 349. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; Obaidat, A.; Hagenbuch, B. OATPs, OATs and OCTs: The organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br. J. Pharmacol. 2012, 165, 1260–1287. [Google Scholar] [CrossRef] [PubMed]
- Homolya, L.; Varadi, A.; Sarkadi, B. Multidrug resistance-associated proteins: Export pumps for conjugates with glutathione, glucuronate or sulfate. Biofactors 2003, 17, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Wilkens, S. Structure and mechanism of ABC transporters. F1000Prime Rep. 2015, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F.; Wang, L.L.; Di, Y.M.; Xue, C.C.; Duan, W.; Li, C.G.; Li, Y. Substrates and inhibitors of human multidrug resistance associated proteins and the implications in drug development. Curr. Med. Chem. 2008, 15, 1981–2039. [Google Scholar] [CrossRef] [PubMed]
- Cottart, C.H.; Nivet-Antoine, V.; Laguillier-Morizot, C.; Beaudeux, J.L. Resveratrol bioavailability and toxicity in humans. Mol. Nutr. Food Res. 2010, 54, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Lancon, A.; Hanet, N.; Jannin, B.; Delmas, D.; Heydel, J.M.; Lizard, G.; Chagnon, M.C.; Artur, Y.; Latruffe, N. Resveratrol in human hepatoma HepG2 cells: Metabolism and inducibility of detoxifying enzymes. Drug Metab. Dispos. 2007, 35, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Lou, B.S.; Wu, P.S.; Hou, C.W.; Cheng, F.Y.; Chen, J.K. Simultaneous quantification of trans-resveratrol and its sulfate and glucuronide metabolites in rat tissues by stable isotope-dilution UPLC-MS/MS analysis. J. Pharm. Biomed. Anal. 2014, 94, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Maier-Salamon, A.; Bohmdorfer, M.; Riha, J.; Thalhammer, T.; Szekeres, T.; Jaeger, W. Interplay between metabolism and transport of resveratrol. Ann. N.Y. Acad. Sci. 2013, 1290, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Murakami, I.; Chaleckis, R.; Pluskal, T.; Ito, K.; Hori, K.; Ebe, M.; Yanagida, M.; Kondoh, H. Metabolism of skin-absorbed resveratrol into its glucuronized form in mouse skin. PLoS ONE 2014, 9, e115359. [Google Scholar] [CrossRef] [PubMed]
- Walle, T. Bioavailability of resveratrol. Ann. N.Y. Acad. Sci. 2011, 1215, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, E.; Soldo, T.; Erbersdobler, H.; Somoza, V. Bioactivity and metabolism of trans-resveratrol orally administered to Wistar rats. Mol. Nutr. Food Res. 2005, 49, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, E.; Somoza, V. Metabolism and bioavailability of trans-resveratrol. Mol. Nutr. Food Res. 2005, 49, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Juan, M.E.; Gonzalez-Pons, E.; Planas, J.M. Multidrug resistance proteins restrain the intestinal absorption of trans-resveratrol in rats. J. Nutr. 2010, 140, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Planas, J.M.; Alfaras, I.; Colom, H.; Juan, M.E. The bioavailability and distribution of trans-resveratrol are constrained by ABC transporters. Arch. Biochem. Biophys. 2012, 527, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Riha, J.; Brenner, S.; Bohmdorfer, M.; Giessrigl, B.; Pignitter, M.; Schueller, K.; Thalhammer, T.; Stieger, B.; Somoza, V.; Szekeres, T.; et al. Resveratrol and its major sulfated conjugates are substrates of organic anion transporting polypeptides (OATPs): Impact on growth of ZR-75-1 breast cancer cells. Mol. Nutr. Food Res. 2014, 58, 1830–1842. [Google Scholar] [CrossRef] [PubMed]
- Novelle, M.G.; Wahl, D.; Dieguez, C.; Bernier, M.; de Cabo, R. Resveratrol supplementation: Where are we now and where should we go? Ageing Res. Rev. 2015, 21, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Pezzuto, J.M. The pharmacology of resveratrol in animals and humans. Biochim. Biophys. Acta 2015, 1852, 1071–1113. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Xia, J.; Gao, J.; Inagaki, Y.; Tang, W.; Kokudo, N. Anti-tumor effects and cellular mechanisms of resveratrol. Drug Discov. Ther. 2015, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.S.; Canto, C. The molecular targets of resveratrol. Biochim. Biophys. Acta 2015, 1852, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Latruffe, N.; Lancon, A.; Frazzi, R.; Aires, V.; Delmas, D.; Michaille, J.J.; Djouadi, F.; Bastin, J.; Cherkaoui-Malki, M. Exploring new ways of regulation by resveratrol involving miRNAs, with emphasis on inflammation. Ann. N.Y. Acad. Sci. 2015, 1348, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Britton, R.G.; Kovoor, C.; Brown, K. Direct molecular targets of resveratrol: Identifying key interactions to unlock complex mechanisms. Ann. N.Y. Acad. Sci. 2015, 1348, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Bowers, J.L.; Tyulmenkov, V.V.; Jernigan, S.C.; Klinge, C.M. Resveratrol acts as a mixed agonist/antagonist for estrogen receptors alpha and beta. Endocrinology 2000, 141, 3657–3667. [Google Scholar] [PubMed]
- Chakraborty, S.; Levenson, A.S.; Biswas, P.K. Structural insights into Resveratrol’s antagonist and partial agonist actions on estrogen receptor alpha. BMC Struct. Biol. 2013, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Gehm, B.D.; McAndrews, J.M.; Chien, P.Y.; Jameson, J.L. Resveratrol, a polyphenolic compound found in grapes and wine, is an agonist for the estrogen receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 14138–14143. [Google Scholar] [CrossRef] [PubMed]
- Lopes Costa, A.; Le Bachelier, C.; Mathieu, L.; Rotig, A.; Boneh, A.; de Lonlay, P.; Tarnopolsky, M.A.; Thorburn, D.R.; Bastin, J.; Djouadi, F. Beneficial effects of resveratrol on respiratory chain defects in patients’ fibroblasts involve estrogen receptor and estrogen-related receptor alpha signaling. Hum. Mol. Genet. 2014, 23, 2106–2119. [Google Scholar] [CrossRef] [PubMed]
- Nwachukwu, J.C.; Srinivasan, S.; Bruno, N.E.; Parent, A.A.; Hughes, T.S.; Pollock, J.A.; Gjyshi, O.; Cavett, V.; Nowak, J.; Garcia-Ordonez, R.D.; et al. Resveratrol modulates the inflammatory response via an estrogen receptor-signal integration network. Elife 2014, 3, e02057. [Google Scholar] [CrossRef] [PubMed]
- Robb, E.L.; Stuart, J.A. The stilbenes resveratrol, pterostilbene and piceid affect growth and stress resistance in mammalian cells via a mechanism requiring estrogen receptor beta and the induction of Mn-superoxide dismutase. Phytochemistry 2014, 98, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Ruotolo, R.; Calani, L.; Fietta, E.; Brighenti, F.; Crozier, A.; Meda, C.; Maggi, A.; Ottonello, S.; Del Rio, D. Anti-estrogenic activity of a human resveratrol metabolite. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.C.; Connell, B.J.; Saleh, T.M. Resveratrol induced neuroprotection is mediated via both estrogen receptor subtypes, ERα and ERβ. Neurosci. Lett. 2013, 548, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Cottart, C.H.; Nivet-Antoine, V.; Beaudeux, J.L. Review of recent data on the metabolism, biological effects, and toxicity of resveratrol in humans. Mol. Nutr. Food Res. 2014, 58, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Cottart, C.H.; Nivet-Antoine, V.; Beaudeux, J.L. Is resveratrol an imposter? Mol. Nutr. Food Res. 2015, 59, 7. [Google Scholar] [CrossRef] [PubMed]
- Hausenblas, H.A.; Schoulda, J.A.; Smoliga, J.M. Resveratrol treatment as an adjunct to pharmacological management in type 2 diabetes mellitus—Systematic review and meta-analysis. Mol. Nutr. Food Res. 2015, 59, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Nunez-Sanchez, M.A.; Gonzalez-Sarrias, A.; Romo-Vaquero, M.; Garcia-Villalba, R.; Selma, M.V.; Tomas-Barberan, F.A.; Garcia-Conesa, M.T.; Espin, J.C. Dietary phenolics against colorectal cancer—From promising preclinical results to poor translation into clinical trials: Pitfalls and future needs. Mol. Nutr. Food Res. 2015, 59. [Google Scholar] [CrossRef] [PubMed]
- Smoliga, J.M.; Baur, J.A.; Hausenblas, H.A. Resveratrol and health—A comprehensive review of human clinical trials. Mol. Nutr. Food Res. 2011, 55, 1129–1141. [Google Scholar] [CrossRef] [PubMed]
- Visioli, F. The resveratrol fiasco. Pharmacol. Res. 2014, 90, 87. [Google Scholar] [CrossRef] [PubMed]
- Biasutto, L.; Mattarei, A.; Sassi, N.; Azzolini, M.; Romio, M.; Paradisi, C.; Zoratti, M. Improving the efficacy of plant polyphenols. Anticancer Agents Med. Chem. 2014, 14, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Biasutto, L.; Zoratti, M. Prodrugs of quercetin and resveratrol: A strategy under development. Curr. Drug Metab. 2014, 15, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Mattarei, A.; Carraro, M.; Azzolini, M.; Paradisi, C.; Zoratti, M.; Biasutto, L. New Water-Soluble Carbamate Ester Derivatives of Resveratrol. Molecules 2014, 19, 15900–15917. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.S.; Aher, N.; Patil, R.; Khandare, J. Poly(ethylene glycol)-Prodrug Conjugates: Concept, Design, and Applications. J. Drug Deliv. 2012, 2012, 17. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, R.B.; Choe, Y.H.; McGuire, J.; Conover, C.D. Effective drug delivery by PEGylated drug conjugates. Adv. Drug Deliv. Rev. 2003, 55, 217–250. [Google Scholar] [CrossRef]
- Kolate, A.; Baradia, D.; Patil, S.; Vhora, I.; Kore, G.; Misra, A. PEG—A versatile conjugating ligand for drugs and drug delivery systems. J. Control. Release 2014, 192, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Pasut, G.; Veronese, F.M. PEGylation for improving the effectiveness of therapeutic biomolecules. Drugs Today (Barc) 2009, 45, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Pasut, G.; Veronese, F.M. State of the art in PEGylation: The great versatility achieved after forty years of research. J. Control. Release 2012, 161, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Veronese, F.M.; Pasut, G. PEGylation, successful approach to drug delivery. Drug Discov. Today 2005, 10, 1451–1458. [Google Scholar] [CrossRef]
- Ginn, C.; Khalili, H.; Lever, R.; Brocchini, S. PEGylation and its impact on the design of new protein-based medicines. Future Med. Chem. 2014, 6, 1829–1846. [Google Scholar] [CrossRef] [PubMed]
- Mero, A.; Clementi, C.; Veronese, F.M.; Pasut, G. Covalent conjugation of poly(ethylene glycol) to proteins and peptides: Strategies and methods. Methods Mol. Biol. 2011, 751, 95–129. [Google Scholar] [PubMed]
- Milla, P.; Dosio, F.; Cattel, L. PEGylation of proteins and liposomes: A powerful and flexible strategy to improve the drug delivery. Curr. Drug Metab. 2012, 13, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Pasut, G. Pegylation of biological molecules and potential benefits: Pharmacological properties of certolizumab pegol. BioDrugs 2014, 28 (Suppl 1), 15–23. [Google Scholar] [CrossRef] [PubMed]
- Vllasaliu, D.; Fowler, R.; Stolnik, S. PEGylated nanomedicines: Recent progress and remaining concerns. Exp. Opin. Drug Deliv. 2014, 11, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Zundorf, I.; Dingermann, T. PEGylation—A well-proven strategy for the improvement of recombinant drugs. Pharmazie 2014, 69, 323–326. [Google Scholar] [PubMed]
- Siddalingappa, B.; Benson, H.A.E.; Brown, D.H.; Batty, K.T.; Chen, Y. Stabilization of Resveratrol in Blood Circulation by Conjugation to mPEG and mPEG-PLA Polymers: Investigation of Conjugate Linker and Polymer Composition on Stability, Metabolism, Antioxidant Activity and Pharmacokinetic Profile. PLoS ONE 2015, 10, e0118824. [Google Scholar] [CrossRef] [PubMed]
- Mattarei, A.; Azzolini, M.; Carraro, M.; Sassi, N.; Zoratti, M.; Paradisi, C.; Biasutto, L. Acetal derivatives as prodrugs of resveratrol. Mol. Pharm. 2013, 10, 2781–2792. [Google Scholar] [CrossRef] [PubMed]
- Kozerski, G.E.; Gallavan, R.H.; Ziemelis, M.J. Investigation of trialkoxysilane hydrolysis kinetics using liquid chromatography with inductively coupled plasma atomic emission spectrometric detection and non-linear regression modeling. Anal. Chim. Acta 2003, 489, 103–114. [Google Scholar] [CrossRef]
- Adams, P.; Baron, F.A. Esters of Carbamic Acid. Chem. Rev. 1965, 65, 567–602. [Google Scholar] [CrossRef]
- Dittert, L.W.; Higuchi, T. Rates of hydrolysis of carbamate and carbonate esters in alkaline solution. J. Pharm. Sci. 1963, 52, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Biasutto, L.; Marotta, E.; Garbisa, S.; Zoratti, M.; Paradisi, C. Determination of quercetin and resveratrol in whole blood—Implications for bioavailability studies. Molecules 2010, 15, 6570–6579. [Google Scholar] [CrossRef] [PubMed]
- Biasutto, L.; Marotta, E.; Bradaschia, A.; Fallica, M.; Mattarei, A.; Garbisa, S.; Zoratti, M.; Paradisi, C. Soluble polyphenols: Synthesis and bioavailability of 3,4′,5-tri(α-d-glucose-3-O-succinyl) resveratrol. Bioorg. Med. Chem. Lett. 2009, 19, 6721–6724. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 2–4 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mattarei, A.; Azzolini, M.; Zoratti, M.; Biasutto, L.; Paradisi, C. N-Monosubstituted Methoxy-oligo(ethylene glycol) Carbamate Ester Prodrugs of Resveratrol. Molecules 2015, 20, 16085-16102. https://doi.org/10.3390/molecules200916085
Mattarei A, Azzolini M, Zoratti M, Biasutto L, Paradisi C. N-Monosubstituted Methoxy-oligo(ethylene glycol) Carbamate Ester Prodrugs of Resveratrol. Molecules. 2015; 20(9):16085-16102. https://doi.org/10.3390/molecules200916085
Chicago/Turabian StyleMattarei, Andrea, Michele Azzolini, Mario Zoratti, Lucia Biasutto, and Cristina Paradisi. 2015. "N-Monosubstituted Methoxy-oligo(ethylene glycol) Carbamate Ester Prodrugs of Resveratrol" Molecules 20, no. 9: 16085-16102. https://doi.org/10.3390/molecules200916085
APA StyleMattarei, A., Azzolini, M., Zoratti, M., Biasutto, L., & Paradisi, C. (2015). N-Monosubstituted Methoxy-oligo(ethylene glycol) Carbamate Ester Prodrugs of Resveratrol. Molecules, 20(9), 16085-16102. https://doi.org/10.3390/molecules200916085