2. Results and Discussion
Previously, Meng
et al. reported the purification of natural polyynes and their acetylated products from the root bark of
Oplopanax horridus (Devil’s Club) and evaluated them as anticancer reagents. Primary structure-activity analysis suggested that the terminal ethylenic bonds of polyacetylenes (
Figure 1) would influence their activity while the protection of single hydroxyl group might not influence the inhibitions very much [
25,
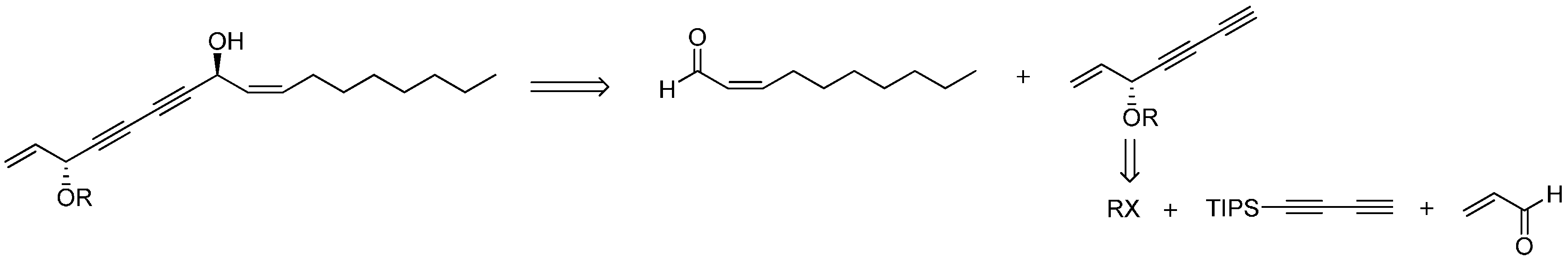
26]. Inspired by these results, we designed the synthetic route of falcarindiol analogues by constructing the first chiral center at the C-8 with an alkyne addition to aldehyde. The hydroxyl group (C-8), which was formed in this step, could be easily protected by an acetyl or benzoyl group, and then the intermediate polyynes with a single chiral center could be delivered into the second alkyne addition step. As depicted in our retrosynthetic analysis of falcarindiol analogues (
Figure 2), the main building blocks would be a protected diyne and suitable aldehydes.
Figure 2.
The retrosynthesis for falcarindiol.
Figure 2.
The retrosynthesis for falcarindiol.
The synthetic sequence started from the preparation of the diyne. The intermediate buta-1,3-diyn-1-yltriisopropylsilane (
4) was synthesized according to the method described by Trost
et al. with our modification, as shown in
Scheme 1 [
19]. 2-Methylbut-3-yn-2-ol (
1) was treated with Br
2 and aq. KOH to afford 4-bromo-2-methylbut-3-yn-2-ol (
2) in 90% yield. Then 4-bromo-2-methylbut-3-yn-2-ol (
2) underwent the Cadiot–Chodkiewicz cross-coupling reaction with triisopropylsilylacetylene in the presence of CuCl and amines to give 2-methyl-6-(triisopropylsilyl)hexa-3,5-diyn-2-ol (
3). After the removal of the 2-hydroxypropyl-group under basic condition, compound
4 was successfully synthesized.
Table 1 shows our screening of the reaction conditions for this step. As shown in
Table 1, the solvent used played a very important role in this deprotection step. Toluene was found to be the best solvent with the highest yield and less toxic (entry 5). In contrast to the literature [
27], the cleavage of 2-methyl-6-(triisopropylsilyl)hexa-3,5-diyn-2-ol (
3) to form the buta-1,3-diynyltriisopropylsilane was not significantly affected by the addition of K
3PO
4 (entry 4 and 5).
Scheme 1.
The total synthesis route of chiral falcarindiol analogues. (i) KOH, Br2, 0 °C, 0.5 h; then 25 °C, 0.5 h, 90%; (ii) BuNH2, CuCl, NH2OH·HCl, triisopropylsilylacetylene, 2 h, 0 °C, 89%; (iii) KOH, toluene, reflux, 4 h, 84%; (iv) R-BINOL, Cy2NH, ZnEt2, Et2O, 16 h, room temperature; Ti(OiPr)4, 2 h, room temperature; aldehyde, 4 h, room temperature, 80%–99%; (v) DMAP, CH2Cl2, 0 °C, Et3N, anhydride, 0 °C, 2 h; AcOH, TBAF, 0 °C,40 min, 72%–89%; (vi) R-BINOL, Cy2NH, ZnEt2, Et2O, room temperature, 16 h; Ti(OiPr)4, room temperature, 2 h; aldehyde, room temperature, 4 h, 17%–97%.
Scheme 1.
The total synthesis route of chiral falcarindiol analogues. (i) KOH, Br2, 0 °C, 0.5 h; then 25 °C, 0.5 h, 90%; (ii) BuNH2, CuCl, NH2OH·HCl, triisopropylsilylacetylene, 2 h, 0 °C, 89%; (iii) KOH, toluene, reflux, 4 h, 84%; (iv) R-BINOL, Cy2NH, ZnEt2, Et2O, 16 h, room temperature; Ti(OiPr)4, 2 h, room temperature; aldehyde, 4 h, room temperature, 80%–99%; (v) DMAP, CH2Cl2, 0 °C, Et3N, anhydride, 0 °C, 2 h; AcOH, TBAF, 0 °C,40 min, 72%–89%; (vi) R-BINOL, Cy2NH, ZnEt2, Et2O, room temperature, 16 h; Ti(OiPr)4, room temperature, 2 h; aldehyde, room temperature, 4 h, 17%–97%.
Table 1.
Screening of the conditions for the 2-hydroxypropyl cleavage.
Table 1.
Screening of the conditions for the 2-hydroxypropyl cleavage.
| Entry | Solvent | Base/eq. | Additive/eq. | T/°C | T/h | Yield% |
|---|
| 1 | benzene | KOH/2.2 | NULL | 81 | 4 | 79 |
| 2 | THF | KOH/1.0 | NULL | 25 | 6 | 9 |
| 3 | THF | KOH/2.2 | K3PO4/1.6 | 66 | 0.7 | NULL |
| 4 | toluene | KOH/1.0 | K3PO4/1.0 | 110 | 1.5 | 70 |
| 5 | toluene | KOH/2.2 | NULL | 110 | 4 | 84 |
With compound
4 in hand, we moved on to the construction of the 1-substituted-5-(triisopropylsilyl)pent-2,4-diynols containing a single chiral center (
Table 2). Compound
4 was separately reacted with acraldehyde, (
E)-pent-2-enal, cinnamaldehyde and decyl aldehyde in the presence of (
R)-BINOL/ZnEt
2/Ti(O
iPr)
4/Cy
2NH to give compounds
5a to
5d in 80%–99% yield (Entry 1 to 4). These compounds were converted to compounds
6 by firstly protecting the hydroxyl group of compounds
5 with an ester moiety and then removing the triisopropylsilyl protecting group. The compounds
5 were treated with DMAP, Et
3N and appropriate anhydride at 0 °C for 2 h in CH
2Cl
2, then the TIPS groups were removed with 1.1 equiv. of acetic acid and 1.2 equiv. of tetrabutylammonium fluoride (TBAF) to give compounds
6 in 72%–89% yields over two steps. The enantioselectivity of the BINOL/ZnEt
2/Ti(O
iPr)
4/Cy
2NH catalyzed addition of the diyne
4 to the aldehydes was evaluated by measuring the enantiomeric purity of compounds
6 which gave 81%–89% ee. The best enantioselectivity was achieved in the synthesis of
6a, which bears a vinyl group. According to our previously work and literature [
22,
24], using
R-BINOL as chiral source in this catalytic system will lead to the production of alkynol with an
S configuration. The NMR spectra and HPLC plots of compounds
6 are provided in the
Supplementary Materials.
Table 2.
Synthesis of the intermediate diynols 5 and the corresponding esters 6.
Then we subjected the diynes
6 to the asymmetric addition to aldehydes again by using the same catalyst system ((
R)-BINOL/ZnEt
2/Ti(O
iPr)
4/Cy
2NH) to synthesize a series of falcarindiol analogues. The results are summarized in
Table 3. Compound
6a was reacted with (
Z)-dec-2-enal to give the 3-OH protected falcarindiol
7a in 17% yield (Entry 1). Compound
6d reacted with acraldehyde to give 3-hydroxyheptadeca-1-en-4,6-diyn-8-yl benzoate
7b (Entry 2), which was a imitate of falcarindiol, in moderate yield (61%) and high selectivity (dr > 99:1). Similarly,
7c was obtained from the reaction of
6d with cinnamaldehyde in a high yield of 97% while the diastereoselectivity decreased to 6:1 on the basis of the HPLC analysis (Entry 3). The reaction of
6c with decyl aldehyde gave
7d with less than 10% yield, even if the reaction time was prolonged for 25 h.
7d is not stable and undergoes rapid degradation after the purification. As we applied
R-BINOL in the catalytic system twice, the configuration of the final product in our experiment, the
7a for instance, is assigned to be (3
S,8
S,
Z)-8-hydroxyheptadeca-1,9-dien-4,6-diyn-3-yl benzoat. This assignment is also in accordance with the optical rotation data of
7a [
4]. Besides, we realized that the sequence of alkynylation step of aldehydes could influence the purification and the stability of the final products. For example, the
7b or
7c, which is produced by alkynylation with decyl aldehyde in the first addition step, has higher yield than those alkynylated with acraldehyde (
7a) or cinnamaldehyde (
7d) first. The NMR spectra of compounds
7 were also provided in the
Supplementary Materials.
Table 3.
Synthesis of falcarindiol analogues a.
3. Experimental Section
3.1. General Methods
All commercial chemicals and solvents were used without further purification. Reactions were monitored by thin layer chromatography (TLC) on silica gel plates (F254) prepared in the laboratory and flash chromatography was performed on silica gel (300–400 mesh). The 1H- and 13C-NMR spectra were measured on Bruker 400 MHz spectrometer (Bruker, Fällanden, Switzerland) with CDCl3 as the solvent. Chemical shifts (ppm) were determined using tetramethylsilane (TMS) as the internal standard and the coupling constants (J) are given in hertz. Melting points were determined on a Hanon MP300 melting point measuring device. High-resolution mass spectra were obtained on a Finnigan LCQDECA spectrometer. The optical rotation was measured on a PERKIN ELEMER 341 Polarimeter. The HPLC analysis was using corresponding commercial chiral column (Daicel Chiralcel OD-3 column 250 × 4.6 mm, Daicel Chiralpak AD-H column 250 × 4.6 mm, or Daicel Chiralpak AS-H column 250 × 4.6 mm) as stated in the experimental procedures with UV detection at 254 nm.
3.2. Preparation of Buta-1,3-diyn-1-yltriisopropylsilane 4
Buta-1,3-diyn-1-yltriisopropylsilane (
4) was synthesized according to the literature with modifications [
12]. Bromine (5 mL, 97.6 mmol, 0.9 eq.) was slowly added to an aq. KOH solution (30 g, 535.7 mmol, 5.0 eq., 200 mL H
2O) at 0 °C and the stirring was maintained for 30 min. Then 2-methylbut-3-yn-2-ol (10 mL, 108.2 mmol, 1 eq.) was added drop wise to the mixture, which was stirred for 30 min. The reaction mixture was extracted with ether (60 mL × 3) after 40 min standing at room temperature. The organic layers were combined, dried over Na
2SO
4, and then evaporated under vacuum. Product
2 was isolated as yellow oil by chromatography (SiO
2, petroleum ether/AcOEt, 15:1,
v/v, yield 90%).
CuCl (364.4 mg, 3.7 mmol, 0.02 eq.) was added to a solution of butylamine (154 mL) in water (513 mL). The solution immediately turned blue, and the color changed to orange and then turned to brown by the addition of 0.5 g hydroxylamine hydrochloride. After triisopropylsilylacetylene (40.3 g, 220.8 mmol, 1.2 eq.) was added, the reaction was immediately cooled in an ice bath and the color turned orange. Then the bromoalkyne 2 (30.0 g, 184.0 mmol, 1.0 eq.) was added in slowly, and the reaction was stirred vigorously at 0 °C. Approximately every 2 min 0.5 g H2N(OH)·HCl was added, while the solution developed a brown color. The reaction was stirred for additional 2 h and monitored by TLC. The aqueous mixture was extracted with Et2O (3 × 50 mL); the combined organic phases were washed with brine (100 mL), and then dried over MgSO4. The crude product 3 was purified by column chromatography (petroleum ether:EtOAc = 10:1, yield = 89%) to afford a yellow oil.
A toluene (390 mL) solution of the TIPS protected dynol 3 (2.097 g, 7.9 mmol, 1 eq.) was heated at reflux in the presence of KOH (978.5 mg, 17.4 mmol, 2.2 eq.) for 4 h and then filtered through diatomaceous earth. After removal of the solvent under vacuum, the buta-1,3-diyn-1-yltriisopropylsilane 4 was isolated as a pale yellow oil by column chromatography (SiO2, petroleum ether only, yield 89%).
3.3. Preparation of 7-(Triisopropylsilyl)hepta-1-en-4,6-diyn-3-ol 5a
Under the protection of nitrogen, diethyl zinc (2.4 mL, 2 M in toluene, 4.8 mmol, 3.0 eq.) was slowly added to an ether (20 mL) solution of dicyclohexylamine (15.4 μL, 14.0 mg, 0.08 mmol, 0.05 eq.), R-BINOL (183.0 mg, 0.6 mmol, 0.4 eq.) and buta-1,3-diyn-1-yltriisopropylsilane 4 (1.0 g, 4.8 mmol, 3.0 eq.). The mixture was stirred at room temperature for 16 h, and then Ti(OiPr)4 (474 μL, 1.6 mmol, 1.0 eq.) was slowly added. After additional 2 h, acraldehyde (107 μL, 1.6 mmol, 1.0 eq.) was added and the resulting solution was stirred for 5 h. Then, the reaction was quenched with saturated ammonium chloride (6 mL). The resulting mixture was extracted with methylene chloride (5 mL × 3), dried over sodium sulfate and concentrated under vacuum. The residue was passed through a short silica gel column (petroleum ether/AcOEt, 30:1, v/v) to give 5a as light yellow oil in 91% yield. 1H-NMR (400 MHz, CDCl3) δ 6.02–5.94 (m, 1H), 5.56–5.46 (m, 1H), 5.32–5.26 (m, 1H), 4.97 (m, 1H), 1.93 (d, J = 6.4 Hz, 1H), 1.10 (s, 21H).
3.4. Preparation of (E)-1-(Triisopropylsilyl)nona-6-en-1,3-diyn-5-ol 5b
Under the protection of nitrogen, buta-1,3-diyn-1-yltriisopropylsilane 4 (742 μL, 3.0 mmol, 1.5 eq.), dicyclohexylamine (19.2 μL, 0.1 mmol, 0.05 eq.) and diethyl zinc (1.5 mL, 2 M in toluene, 3.0 mmol, 1.5 eq.) were sequentially added to an ether (20 mL) solution of R-BINOL (228.8 mg, 0.8 mmol, 0.4 eq.) in a 50 mL flask. The mixture was stirred at room temperature for 16 h, and then Ti(OiPr)4 (592 μL, 2.0 mmol, 1.0 eq.) was slowly added in. After additional 2 h, (E)-2-pentenal (196 μL, 2.0 mmol, 1.0 eq.) was added and the reaction mixture was stirred for 4 h. Then the reaction was quenched with the addition of saturated ammonium chloride (8 mL). The resulting mixture was extracted with methylene chloride (5 mL × 3), dried over sodium sulfate and concentrated under vacuum. The residue was passed through a short silica gel column (petroleum ether/AcOEt, 20:1, v/v) to give 5b as light yellow oil in 87% yield. 1H-NMR (400 MHz, CDCl3) δ 5.94 (m, 1H), 5.58 (m, 1H), 4.89 (m, 1H), 2.16–2.05 (m, 2H), 1.87 (d, J = 5.6 Hz, 1H), 1.08 (s, 21H), 1.02 (t, J =7.6 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 139.7, 136.6, 126.9, 88.8, 85.5, 75.6, 71.2, 63.5, 25.0, 18.5, 13.0, 11.2. HRMS: calcd for [M + Na+, C18H30NaOSi]+ 313.1964, Found 313.1941.
3.5. Preparation of (E)-1-Phenyl-7-(triisopropylsilyl)hepta-1-en-4,6-diyn-3-ol 5c
Under the protection of nitrogen, buta-1,3-diyn-1-yltriisopropylsilane 4 (1.4 g, 6.6 mmol, 2.0 eq.), dicyclohexylamine (66 μL, 0.3 mmol, 0.1 eq.) and diethyl zinc (8 mL, 2 M in toluene, 16 mmol, 5.0 eq.) were sequentially added to the ether (40 mL) solution of R-BINOL (188.8 mg, 0.7 mmol, 0.2 eq.) in a 100 mL flask. The mixture was stirred at room temperature for 24 h, and then Ti(OiPr)4 (488 μL, 1.6 mmol, 0.5 eq.) was slowly added in. After additional 2 h, cinnamaldehyde (415 μL, 3.3 mmol, 1.0 eq.) was added and the resulting solution was stirred for 12 h. Then the reaction was quenched with the addition of saturated ammonium chloride (15 mL). The resulting mixture was extracted with methylene chloride (10 mL × 3), dried over sodium sulfate and concentrated under vacuum. The residue was passed through a short silica gel column (petroleum ether/AcOEt, 40:1–20:1, v/v) to give 5c as light yellow oil in 87% yield. 1H-NMR (400 MHz, CDCl3) δ 7.45–7.37 (m, 2H), 7.37–7.30 (m, 2H), 7.28 (m, 1H), 6.76 (d, J = 16.0Hz, 1H), 6.27 (dd, J = 16.0, 7.2 Hz, 1H), 5.11 (m, 1H), 2.09 (d, J =6.4 Hz, 1H), 1.09 (s, 21H).
3.6. Preparation of 1-(Triisopropylsilyl)tetradeca-1,3-diyn-5-ol 5d
Under the protection of nitrogen, buta-1,3-diyn-1-yltriisopropylsilane 4 (2.6 g, 12.5 mmol, 3.0 eq.), dicyclohexylamine (40 μL, 0.2 mmol, 0.05 eq.) and diethyl zinc (6.3 mL, 12.5 mmol, 2 M in toluene, 3.0 eq.) were sequentially added to an ether (50 mL) solution of R-BINOL (477.6 mg, 1.7 mmol, 0.4 eq.) in a 100 mL flask. The mixture was stirred at room temperature for 16 h, and then Ti(OiPr)4 (1.2 mL, 4.2 mmol, 1.0 eq.) was slowly added in. The reaction was reacted for additional 2 h, after which decyl aldehyde (785 μL, 4.2 mmol, 1.0 eq.) was added and the mixture was stirred for 4 h. Then the reaction was quenched with saturated ammonium chloride (15 mL). The resulting mixture was extracted with methylene chloride (10 mL × 3), dried over sodium sulfate and concentrated under vacuum. The product was isolated by passing the residue through a short silica gel column (petroleum ether/AcOEt, 20:1, v/v) to give 5d as light yellow oil in 99% yield. 1H-NMR (400 MHz, CDCl3) δ 4.42 (m, 1H), 1.84 (d, J =4.8 Hz, 1H), 1.77–1.66 (m, 2H), 1.49–1.39 (m, 2H), 1.28 (m, 12H), 1.12–1.00 (m, 21H), 0.88 (t, J = 6.8 Hz, 3H).
3.7. Preparation of Hepta-1-en-4,6-diyn-3-yl benzoate 6a
DMAP (18.2 mg, 0.15 mmol, 0.1 eq.), benzoic anhydride (363.3 mg, 1.6 mmol, 1.1 eq.), and Et3N (220 μL, 1.6 mmol, 1.1 eq.) were sequentially added to a CH2Cl2 solution (8.0 mL) of 5a (416.5 mg, 1.5 mmol, 1 eq.) in a 25 mL flask at 0 °C. The reaction mixture was stirred for 2 h, and then quenched by saturated aq. NaHCO3 (15 mL). The resulting mixture was extracted with ether (10 mL × 3), and the organic layers were combined and dried with over sodium sulfate. After being concentrated under vacuum, the residue was dissolved in THF (8 mL) and stirred with acetic acid (90 μL, 1.6 mmol, 1.1 eq.) and TBAF (1.8 mL, 1.8 mmol, 1.2 eq. in THF) at 0 °C for 40 min. Then the reaction was quenched with 2 mL water and extracted with ether (10 mL × 3). The organic layers were combined and dried over sodium sulfate. Further purification was achieved by column chromatography (petroleum ether/AcOEt, 20:1, v/v) to give 6a as light yellow oil in 72% yield. ee = 89%, HPLC analysis: Chiralcel OD-3 column, F = 0.9 mL/min, hexane/iPrOH = 99.8/0.2, temp = 30 °C, λ = 254 nm, t1 = 20.6 min, t2 = 22.6 min. 1H-NMR (400 MHz, CDCl3) δ 8.16–8.01 (m, 2H), 7.69–7.54 (m, 1H), 7.45 (dd, J = 10.8, 4.7 Hz, 2H), 6.16 (dd, J = 5.6, 0.8 Hz, 1H), 6.00 (ddd, J = 16.8, 10.4, 5.6 Hz, 1H), 5.65 (d, J = 16.8Hz, 1H), 5.42 (d, J = 10.4 Hz, 1H), 2.24 (d, J = 0.8 Hz, 1H). 13C-NMR (100 MHz, CDCl3) δ 165.0, 133.5, 131.9, 129.9, 129.3, 128.5, 120.0, 71.4, 71.2, 69.3, 67.2, 64.7. HRMS: calcd for [M + Na+, C14H10NaO2]+ 233.0578, found 233.0547.
3.8. Preparation of (E)-Nona-6-en-1,3-diyn-5-yl benzoate 6b
DMAP (21.8 mg, 0.17 mmol, 0.1 eq.), benzoic anhydride (434.0 mg, 1.9 mmol, 1.1 eq.), and Et3N (262.9 μL, 1.9 mmol, 1.1 eq.) were sequentially added to a CH2Cl2 solution (9.0 mL) of 5b (506.7 mg, 1.7 mmol, 1 eq.) in a 25 mL flask at 0 °C. The reaction mixture was stirred for 2 h, and then quenched with saturated aq. NaHCO3 (15 mL). The resulting mixture was extracted with ether (10 mL × 3), and the organic layers were combined and dried over sodium sulfate. After being concentrated under vacuum, the residue was dissolved in THF (10 mL) and stirred with acetic acid (107 μL, 1.9 mmol, 1.1 eq.) and TBAF (2.1 mL, 2.1 mmol, 1.2 eq. in THF) at 0 °C for 40 min. Then the reaction was quenched with 2 mL water and extracted with ether (20 mL × 3). The organic layers were combined and dried over sodium sulfate. Further purification was achieved by column chromatography (petroleum ether/AcOEt, 40:1–20:1, v/v) to give 6b as light yellow oil in 86% yield. ee = 81%, HPLC analysis: Chiralcel AS-H column, F = 0.4 mL/min, hexane = 100%, temp = 30 °C, λ = 254 nm, t1 = 26.2 min, t2 = 34.4 min. 1H-NMR (400 MHz, CDCl3) δ 8.06 (m, 2H), 7.57 (m, 1H), 7.44 (m, 2H), 6.19–6.06 (m, 2H), 5.63 (ddt, J = 15.2, 6.4, 1.6 Hz, 1H), 2.24 (d, J = 0.8 Hz, 1H), 2.20–2.08 (m, 2H), 1.04 (t, J = 7.2 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 165.2, 139.3, 133.3, 129.9, 129.6, 128.4, 122.9, 72.3, 70.7, 69.1, 67.3, 64.8, 25.1, 12.8.
3.9. Preparation of (E)-1-Phenylhepta-1-en-4,6-diyn-3-yl acetate 6c
DMAP (3 mg, 0.024 mmol, 0.1 eq.), acetic anhydride (25 μL, 0.27 mmol, 1.1 eq.), and Et3N (37 μL, 0.27 mmol, 1.1 eq.) were sequentially added to a CH2Cl2 solution (1.4 mL) of 5c (82.6 mg, 0.24 mmol, 1 eq.) in a 5 mL flask at 0 °C. The reaction mixture was stirred for 2 h, and then quenched by saturated aq. NaHCO3 (2 mL). The resulting mixture was extracted with ether (3 mL × 3) and the organic layers were combined and dried over sodium sulfate. After being concentrated under vacuum, the residue was dissolved in THF (1 mL) and stirred with acetic acid (15 μL, 0.27 mmol, 1.1 eq.) and TBAF (300 μL, 3 mmol, 1.2 eq. in THF) at 0 °C for 1 h. Then the reaction was quenched by 2 mL water and extracted with ether. The organic layers were combined and dried over sodium sulfate. Further purification was achieved by column chromatography (petroleum ether/AcOEt, 50:1–30:1, v/v) to give 6c as light yellow oil in 72% yield. ee = 81%. HPLC analysis: Chiralpak AD-H column, F = 0.8 mL/min, hexane/iPrOH = 95.0/5.0, temp = 30 °C, λ = 254 nm, t1 = 9.1 min, t2 = 9.7 min. 1H-NMR (400 MHz, CDCl3) δ 7.46–7.38 (m, 2H), 7.38–7.28 (m, 3H), 6.84 (d, J = 15.8 Hz, 1H), 6.20 (dd, J = 15.8, 6.8 Hz, 1H), 6.07 (m, 1H), 2.26 (d, J = 0.8 Hz, 1H), 2.13 (s, 3H)
3.10. Preparation of Tetradeca-1,3-diyn-5-yl benzoate 6d
DMAP (53 mg, 0.4 mmol, 0.1 eq.), benzoic anhydride (1.1 g, 5.1 mmol, 1.2 eq.), and Et3N (639 μL, 4.7 mmol, 1.1 eq.) were sequentially added to a CH2Cl2 solution (25.0 mL) of 5d (1.538 g, 4.2 mmol, 1 eq.) in a 50 mL flask at 0 °C. The reaction mixture was stirred for 2 h, and then quenched with saturated aq. NaHCO3 (20 mL). The resulting mixture was extracted with ether (20 mL × 3) and the organic layers were combined and dried over sodium sulfate. After being concentrated under vacuum, the residue was dissolved in THF (25 mL) and stirred with acetic acid (260 μL, 4.7 mmol, 1.1 eq.) and TBAF (4.7 mL, 4.7 mmol, 1.2 eq. in THF) at 0 °C for 40 min. Then the reaction was quenched with 8 mL water and extracted with ether. The organic layers were combined and dried over sodium sulfate. Further purification was achieved by column chromatography (petroleum ether/AcOEt, 80:1–20:1, v/v) to give 6d as light yellow oil in 82% yield. ee = 82%, HPLC analysis: Chiralpak AS-H column, F = 0.8 mL/min, hexane 100%, temp = 30 °C, λ = 254, t1 = 8.6 min, t2 = 9.6 min. 1H-NMR (400 MHz, CDCl3) δ 8.09–8.01 (m, 2H), 7.58 (t, J = 7.4 Hz, 1H), 7.45 (t, J = 7.7 Hz, 2H), 5.63 (t, J = 6.6 Hz, 1H), 2.19 (s, 1H), 1.93 (m, 2H), 1.57–1.46 (m, 2H), 1.35–1.23 (m, 12H), 0.88 (t, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 165.4, 133.3, 129.8, 129.6, 128.4, 73.8 69.6, 68.6, 67.4, 64.5, 34.6, 31.9, 29.5, 29.40, 29.3, 29.1, 25.0, 22.7, 14.1. HRMS: calcd for [C21H26NaO2]+ 333.1830, Found 333.1836.
3.11. Preparation of (Z)-8-Hydroxyheptadeca-1,9-dien-4,6-diyn-3-yl benzoate 7a
Under the protection of nitrogen, hepta-1-en-4,6-diyn-3-yl benzoate 6a (1.1 mmol, 3.0 eq.), dicyclohexylamine (3.4 μL, 0.017 mmol, 0.05 eq.) and diethyl zinc (0.5 mL, 2 M in toluene, 1.1 mmol, 3.0 eq.) were sequentially added to an ether (5 mL) solution of R-BINOL (40.0 mg, 0.14 mmol, 0.4 eq.) in a well-dried 10mL flask. The mixture was stirred at room temperature for 16 h, and then a Ti(OiPr)4 solution (103.6 μL, 0.35 mmol, 1.0 eq.) was slowly added in. After additional 2 h, (Z)-dec-2-enal (54.0 μL, 0.35 mmol, 1.0 eq.) was added and the mixture was stirred for 4 h at room temperature. The reaction was quenched with saturated ammonium chloride (5 mL). The resulting mixture was extracted with methylene chloride (5 mL × 3), dried over sodium sulfate and concentrated under vacuum. Final product was obtained by purification with column chromatograph (petroleum ether/AcOEt, 10:1, v/v) to give 7a as light yellow oil in 17% yield. = 84.1 (c = 0.9, in CH2Cl2). dr = 14:1. 1H-NMR (400 MHz, CDCl3) δ 8.07 (m, 2H), 7.58 (m, 1H), 7.45 (m, 2H), 6.18 (d, J = 5.6 Hz, 1H), 6.00 (m, 1H), 5.95–5.84 (m, 1H), 5.64 (d, J = 17.2 Hz, 1H), 5.56 (dd, J = 17.2, 6.0 Hz, 1H), 5.41 (d, J = 10.0 Hz, 1H), 4.89 (t, J = 6.0 Hz, 1H), 2.05 (m, 2H), 1.93 (m, 1H), 1.37 (m, 2H), 1.29 (m, 8H), 0.88 (t, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 165.1, 135.4, 133.4, 132.0, 129.9, 129.4, 128.5, 127.5, 119.8, 79.5, 74.9, 71.0, 69.6, 65.0, 63.3, 32.0, 31.8, 29.1, 29.1, 28.8, 22.7, 14.1. HRMS [C24H28NaO3]+ calcd for 387.1936, Found 387.1923.
3.12. Preparation of 3-Hydroxyheptadeca-1-en-4,6-diyn-8-yl benzoate 7b
Under the protection of nitrogen, tetradeca-1,3-diyn-5-yl benzoate 6d (192 μL, 0.6 mmol, 3.0 eq.), dicyclohexylamine (2.0 μL, 0.01 mmol, 0.05 eq.) and diethyl zinc (0.3 mL, 2 M in toluene, 0.6 mmol, 3.0 eq.) were sequentially added to an ether (4 mL) solution of R-BINOL (22.9 mg, 0.08 mmol, 0.4 eq.) in a well-dried 10 mL flask. The mixture was stirred at room temperature for 16 h, and then a Ti(OiPr)4 solution (59 μL, 0.2 mmol, 1.0 eq.) was slowly added in. After additional 2 h, acraldehyde (13.4 μL, 0.2 mmol, 1.0 eq.) was added and the mixture was stirred for 4 h at room temperature. The reaction was quenched with saturated ammonium chloride (5 mL). The resulting mixture was extracted with methylene chloride (5 mL × 3), dried over sodium sulfate and concentrated under vacuum. The final product was obtained by purification with column chromatograph (petroleum ether/AcOEt, 10:1, V/V) which gave 7b as light yellow oil in 61% yield. dr > 99:1. 1H-NMR (400 MHz, CDCl3) δ 8.05 (m, 2H), 7.63–7.53 (m, 1H), 7.45 (m, 2H), 5.93 (m, 1H), 5.65 (t, J = 6.4 Hz, 1H), 5.47 (d, J = 17.2 Hz, 1H), 5.25 (d, J = 10.0 Hz, 1H), 4.93 (t, J = 5.6 Hz, 1H), 2.16 (d, J = 6.0 Hz, 1H), 1.98–1.85 (m, 2H), 1.57–1.46 (m, 2H), 1.35–1.24 (m, 12H), 0.88 (t, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 165.4, 135.7, 133.3, 129.8, 129.6, 128.4, 117.4, 78.1, 77.5, 70.2, 69.3, 64.7, 63.5, 34.6, 31.9, 29.5, 29.4, 29.3, 29.1, 25.0, 22.7, 14.1. HRMS [C24H30NaO3]+ calcd for 389.2093, Found 389.2091.
3.13. Preparation of (E)-3-Hydroxy-1-phenylheptadeca-1-en-4,6-diyn-8-yl benzoate 7c
Under the protection of nitrogen, tetradeca-1,3-diyn-5-yl benzoate 6d (192 μL, 0.6 mmol, 3.0 eq.), dicyclohexylamine (2.0 μL, 0.01 mmol, 0.05 eq.) and diethyl zinc (0.3 mL, 2 M in toluene, 0.6 mmol, 3.0 eq.) were sequentially added to an ether (4 mL) solution of R-BINOL (22.9 mg, 0.08 mmol, 0.4 eq.) in a well-dried 10 mL flask. The mixture was stirred at room temperature for 16 h, and Ti(OiPr)4 (59 μL, 0.2 mmol, 1.0 eq.) was slowly added in. After additional 2 h, cinnamaldehyde (25.2 μL, 0.2 mmol, 1.0 eq.) was added and the mixture was stirred for 4 h at room temperature. The reaction was quenched with saturated ammonium chloride (5 mL). The resulting mixture was extracted with methylene chloride (5 mL × 3), dried over sodium sulfate and concentrated under vacuum. The final product was obtained by purification with column chromatograph (petroleum ether/AcOEt, 10:1, v/v) which gave 7c as light yellow oil in 97% yield. dr = 6:1. HPLC analysis: Chiralcel OD-3 column, F = 0.8 mL/min, hexane/iPrOH = 94:6, T = 30 °C, λ = 254 nm, t1 = 51.8 min, t2 = 53.2 min, t3 = 58.3 min, t4 = 62.2 min. 1H-NMR (400 MHz, CDCl3) δ 8.05 (dt, J = 6.8, 0.8 Hz, 2H), 7.63–7.53 (m, 1H), 7.45 (m, 2H), 7.42–7.36 (m, 2H, 7.36–7.30 (m, 2H), 7.30–7.26 (m, 1H), 6.76 (d, J = 16.0 Hz, 1H), 6.25 (dd, J = 16.0, 6.0 Hz, 1H), 5.66 (t, J = 6.4 Hz, 1H), 5.11 (t, J = 6.0Hz, 1H), 2.09–2.01 (m, 1H), 1.93 (m, 2H), 1.52 (m, 2H), 1.40–1.22 (m, 12H), 0.88 (t, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 165.4, 135.8, 133.3, 132.8, 129.8, 129.6, 128.6, 128.4, 128.3, 126.9, 126.7, 78.1, 77.8, 70.4, 69.3, 64.7, 63.3, 34.6, 31.9, 29.5, 29.4, 29.3, 29.1, 25.0, 22.7, 14.1. HRMS [C30H34NaO3]+ calcd for 465.2406, Found 465.2391.
3.14. Preparation of (E)-8-hydroxy-1-phenylheptadeca-1-en-4,6-diyn-3-yl acetate 7d
Under the protection of nitrogen, (E)-1-phenylhepta-1-en-4,6-diyn-3-yl acetate 6c (203.3 mg, 0.9 mmol, 4.0 eq.), dicyclohexylamine (4.4 μL, 0.022 mmol, 0.1 eq.) and diethyl zinc (0.91 mL, 1 M in hexane, 0.91 mmol, 4.0 eq.) were sequentially added to an ether (5 mL) solution of R-BINOL (26.3 mg, 0.1 mmol, 0.4 eq.) in a well-dried 10 mL flask at 0 °C. The mixture was stirred at 0 °C for 16 h, and then Ti(OiPr)4 (68 μL, 0.35 mmol, 1.5 eq.) was slowly added in. After additional 3 h, decanal (42 μL, 0.22 mmol, 1.0 eq.) was added and the mixture was stirred for 24 h. The reaction was quenched with saturated ammonium chloride (5 mL). The resulting mixture was extracted with methylene chloride (5 mL × 3), dried over sodium sulfate and concentrated under vacuum. The final product 7d was obtained by purification with column chromotograph in less 10% yield with rapid degradation. 1H-NMR (400 MHz, CDCl3) δ 7.37 (m, 5H), 6.41 (dd, J =15.8, 6.4Hz, 1H), 6.32 (d, J = 6.4 Hz, 1H), 5.78 (d, J = 15.8 Hz, 1H), 4.47 (t, J = 6.4 Hz, 1H), 2.13 (s, 3H), 1.89 (s, 1H), 1.78–1.70 (m, 2H), 1.45 (m, 2H), 1.29 (m, 12H), 0.90 (t, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 169.7, 144.0, 137.6, 128.8, 128.6, 127.3, 110.7, 83.8, 76.1, 75.0, 74.9, 69.3, 63.0, 37.5, 31.9, 29.5, 29.5, 29.3, 29.2, 25.0, 22.7, 21.1, 14.1.






{kind=link}
{kind=link}
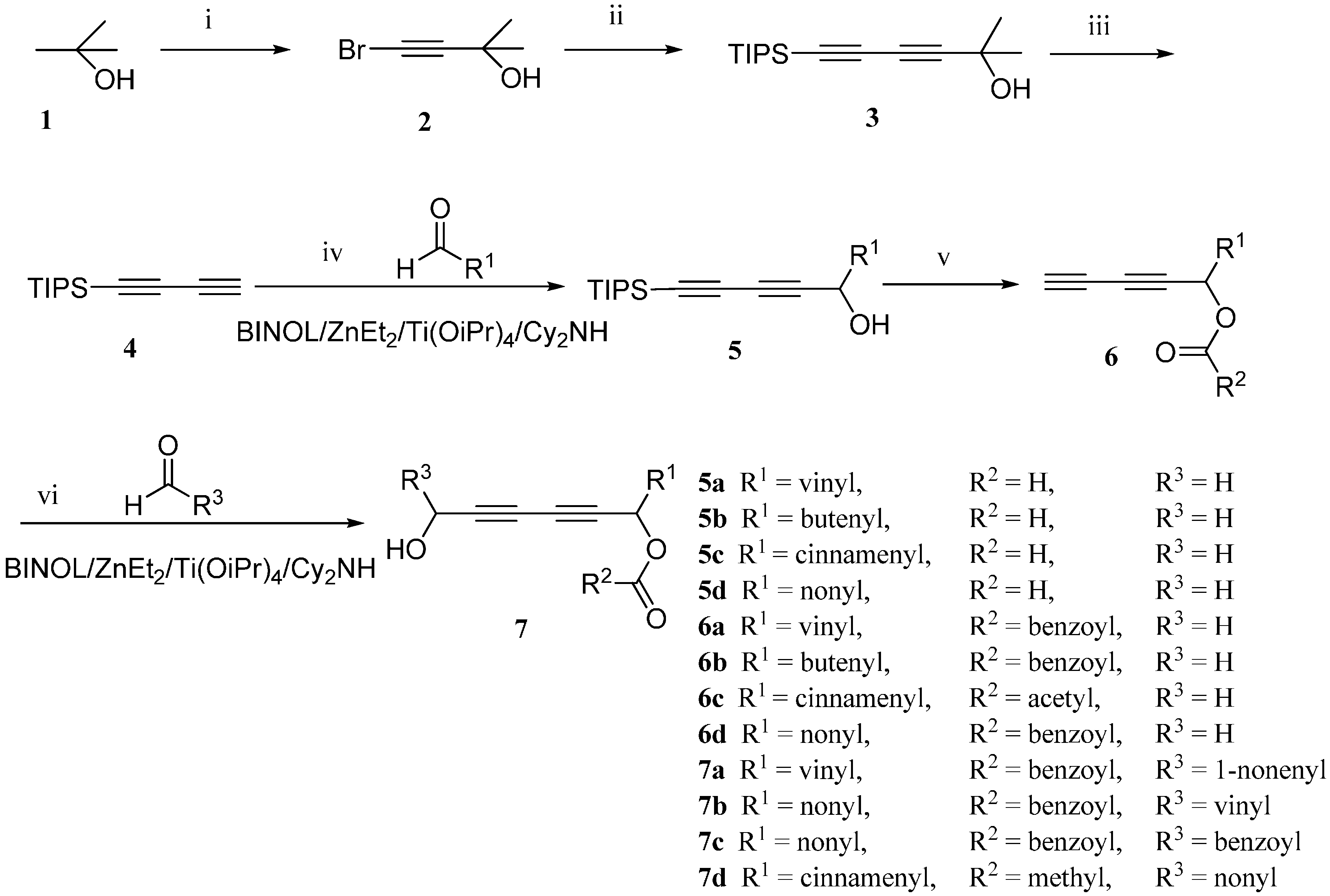{kind=link}











