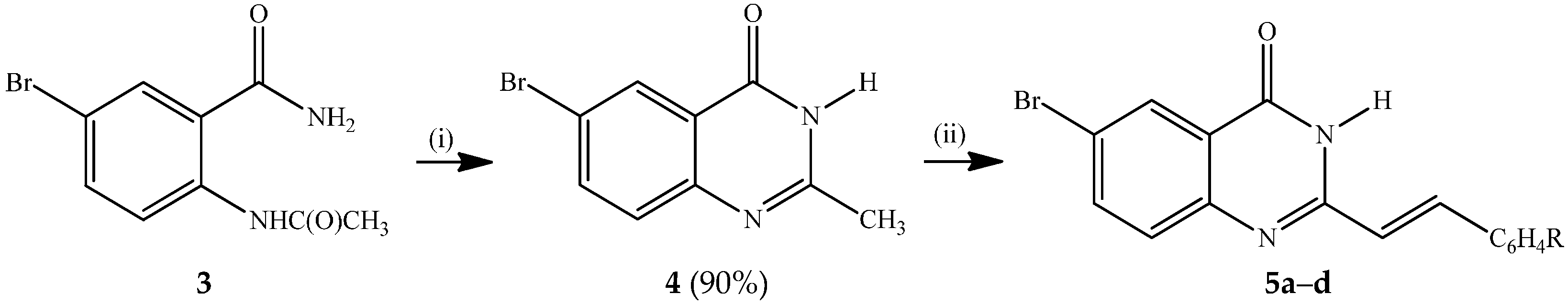
3.3. Typical Procedure for the Synthesis of the 6-Bromo-2-(styryl)Quinazolin-4(3H)-Ones 5a–d
A stirred mixture of 4 (1 equiv.) and benzaldehyde derivative (1.2 equiv.) in acetic acid (20 mL/mmol of 4) was refluxed for 6 h. The mixture was allowed to cool and then quenched with an ice-cold water. The resultant precipitate was filtered and washed with methanol to afford compound 5. The following compounds were prepared in this fashion:
(E)-6-Bromo-2-(phenylstyryl) quinazolin-4(3H)-one (
5a). Solid (0.30 g, 74%), mp. 328–330 °C (Lit. [
7] 334–338 °C); ν
max (ATR) 534, 827, 967, 1308, 1463, 1667, 3174 cm
−1; δ
H (300 MHz, DMSO-
d6) 6.98 (1H, d,
Jtrans = 16.0 Hz, H
a), 7.40–7.48 (3H, m, ArH), 7.61 (1H, d,
J = 8.7 Hz, 8-H), 7.64 (2H, dd,
J = 1.8 and 8.4 Hz, ArH), 7.92 (1H, dd,
J = 2.5 and 8.7 Hz, 7-H), 7.95 (1H, d,
Jtrans = 16.0 Hz, H
b), 8.16 (1H, d,
J = 2.5 Hz, 5-H), 12.49 (1H, s, NH); δ
C (75 MHz, DMSO-
d6) 119.0, 121.2, 123.2, 128.1, 128.4, 129.5, 129.9, 130.4, 135.3, 137.8, 139.3, 148.5, 152.4, 161.1;
m/
z 329 (100, MH
+); HRMS (ES): MH
+, found 329.0130. C
16H
12N
2O
79Br
+ requires 329.0133.
(E)-6-Bromo-2-(4-fluorostyryl)quinazolin-4(3H)-one (5b). Solid (0.34 g, 78.3%), mp. 332–334 °C; νmax (ATR) 506, 643, 817, 967, 1233, 1463, 1673, 3174 cm−1; δH (500 MHz, DMSO-d6) 6.91 (1H, d, Jtrans = 16.0 Hz, Ha), 7.28 (2H, t, J = 8.5 Hz, 3′,5′-H), 7.58 (1H, d, J = 8.0 Hz, 8-H), 7.71 (2H, t, J = 8.5 Hz, 2′,6′-H), 7.90 (1H, dd, J = 2.5 and 8.5 Hz, 7-H), 7.93 (1H, d, Jtrans = 16.0 Hz, Hb), 8.14 (1H, d, J = 2.5 Hz, 5-H), 12.48 (1H, s, NH); δC (125 MHz, DMSO-d6) 116.6 (d, 2JCF = 21.8 Hz), 119.0, 121.1, 123.1, 128.4, 129.9, 130.4 (d, 3JCF = 8.5 Hz), 131.9 (d, 4JCF = 3.1 Hz), 137.8, 138.1, 148.4, 152.4, 161.0, 163.4 (d, 1JCF = 246.5 Hz); m/z 345 (100, MH+); HRMS (ES): MH+, found 345.0036. C16H11N2O79BrF+ requires 345.0039.
(E)-6-Bromo-2-(4-chlorostyryl)quinazolin-4(3H)-one (
5c). Solid (0.34 g, 76%) mp. > 345 °C (Lit. [
31] > 280 °C); ν
max (ATR) 492, 624, 808, 831, 967, 1088, 1306, 1463, 1577, 1645, 1671, 3173 cm
−1; δ
H (500 MHz, DMSO-
d6) 6.98 (1H, d,
Jtrans = 16.0 Hz, H
a), 7.51 (2H, d,
J = 8.5 Hz, 3′,5′-H), 7.60 (1H, d,
J = 8.5 Hz, 8-H), 7.66, (2H, d,
J = 8.5 Hz, 2′,6′-H), 7.91 (1H, dd,
J = 2.5 and 8.5 Hz, 7-H), 7.92 (1H, d,
J = 16.0 Hz, H
b), 8.16 (1H, d,
J = 2.5 Hz, 5-H), 12.50 (1H, s, NH); δ
C (125 MHz, DMSO-
d6) 119, 122.1,123.1,128.4, 129.6, 129.8, 129.9, 134.3, 134.7, 137.9, 137.8, 148.3, 152.4, 161.1;
m/
z 361 (100, MH
+); HRMS (ES): MH
+, found 360.9746. C
16H
11N
2O
35Cl
79Br
+ requires 360.9743.
(E)-6-Bromo-2-(3-methoxystyryl)quinazolin-4(3H)-one (
5d). Solid (0.32 g, 71%), mp. 287–288 °C (Lit. [
31] 268–270 °C); ν
max (ATR) 535, 775, 890, 976, 1464, 1586, 1676, 3173 cm
−1; δ
H (500 MHz, DMSO-
d6) 3.80 (3H, s, -OCH
3), 6.98 (1H, dd,
J = 2.5 and 7.5 Hz, 4′-H), 6.99 (1H, d,
Jtrans = 16.5 Hz, H
a), 7.20 (1H, d,
J = 2.5 Hz, 2′-H), 7.22 (1H, d,
J = 7.5 Hz, 6′-H), 7.36 (1H, t,
J = 8.5 Hz, 5′-H), 7.59 (1H, d,
J = 8.5 Hz, 8-H), 7.91 (1H, d,
Jtrans = 16.0 Hz, H
b), 7.93 (1H, dd,
J = 2.5 and 8.5 Hz, 7-H), 8.16 (1H, d,
J = 2.5 Hz, 5-H), 12.47 (1H, s, NH); δ
C (125 MHz, DMSO-
d6) 55.6, 113.1, 116.2, 119.0, 120.5, 121.6, 128.4, 129.9, 130.6, 136.7, 137.8, 139.2, 148.4, 152.4, 160.1 (2xC), 161.0;
m/
z 357 (100, MH
+); HRMS (ES): MH
+, found 357.0235. C
17H
14N
2O
279Br
+ requires 357.0239.
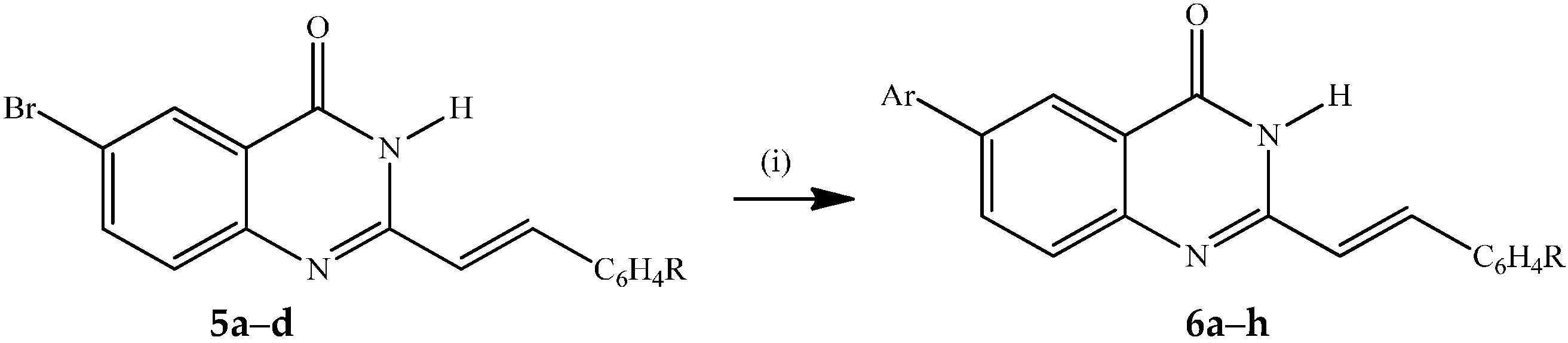
3.4. Typical Procedure for the Suzuki-Miyaura Cross-Coupling of 5a–d with Arylboronic Acids
(E)-6-Phenyl-2-styrylquinazolin-4(3H)-one (6a). A stirred mixture of 5a (0.20 g, 0.61 mmol), PdCl2(PPh3)2 (0.02 g, 0.03 mmol), PCy3 (0.017 g, 0.06 mmol) and K2CO3 (0.10 g, 0.73 mmol) in 3:1 dioxane–water (v/v; 10 mL) was purged with argon gas for 30 minutes. Phenylboronic acid (0.11 g, 0.91 mmol) was added to the mixture using a syringe. The reaction mixture was heated at 100 °C for 2 h and then quenched with an ice-cold water. The product was extracted into chloroform and the combined organic layers were washed with water, dried over Na2SO4 and filtered dry on a sintered funnel to afford 6a as a yellow solid (0.12 g, 61%), mp. 329 °C; νmax (ATR) 491, 695, 753, 794, 966, 1259, 1473, 1579, 1655 cm−1; δH (500 MHz, DMSO-d6) 7.01 (1H, d, Jtrans = 16.0 Hz, Ha), 7.40–7.52 (6H, m, ArH), 7.66 (2H, d, J = 7.5 Hz, ArH), 7.76 (3H, t, J = 7.5 Hz, ArH), 7.96 (1H, d, Jtrans = 16.0 Hz, Hb), 8.13 (1H, dd, J = 2.5 and 7.5 Hz, 7-H), 8.31 (1H, d, J = 2.5 Hz, 5-H), 12.41 (1H, s, NH); δC (125 MHz, DMSO-d6) 121.5, 121.9, 123.6, 127.2 (2xC), 128.1, 128.4, 128.6, 128.7, 130.3, 133.5, 135.4, 138.3, 138.9, 139.3, 148.8, 151.9, 162.3; m/z 325 (100, MH+); HRMS (E) (ES): MH+ found 325.1341. C22H17N2O+ requires 325.1339.
(E)-2-(4-Fluorostyryl)-6-phenylquinazolin-4(3H)-one (6b). Solid (0.13 g, 66%), mp. 345 °C; νmax (ATR) 499, 698, 761, 824, 968, 1157, 1552, 1660 cm−1; δH (300 MHz, CDCl3) 7.00 (1H, d, Jtrans = 16.0 Hz, Ha), 7.31 (2H, t, J = 9.0 Hz, 2′,6′-H), 7.43 (1H, d, J = 7.5 Hz, 8-H), 7.52 (2H, t, J = 9.0 Hz, 3′,5′-H), 7.71–7.80 (5H, m, Ph), 7.96 (1H, d, Jtrans = 16.0 Hz, Hb), 8.13 (1H, dd, J = 2.0 and 9.0 Hz, 7-H), 8.33 (1H, d, J = 2.0 Hz, 5-H), 12.39 (1H, s, NH); δC (75 MHz, DMSO-d6) 116.6 (d, 2JCF = 21.9 Hz), 118,6, 119.0, 121.5, 123.1, 128.2, 128.4, 129.5, 130.4 (d, 3JCF = 8.5 Hz), 132.0 (d, 4JCF = 3.2 Hz), 137.6, 137.8, 138.1, 148.4, 152.4, 161.1, 163.4 (d, 1JCF = 246.6 Hz); m/z 343 (100, MH+); HRMS (ES): MH+ found 343.1239. C22H16N2OF+ requires 343.1247.
(E)-2-(4-Chlorostyryl)-6-phenylquinazolin-4(3H)-one (6c). Solid (0.22 g, 71%), mp. 345 °C; νmax (ATR) 490, 697, 764, 793, 969, 1652 cm−1; δH (500 MHz, DMSO-d6) 7.01 (1H, d, Jtrans = 16.0 Hz, Ha), 7.40 (1H, d, J = 7.5 Hz, 8-H), 7.48–7.520 (4H, m, ArH), 7.67 (2H, d, J = 8.0 Hz, 3′,5′-H), 7.74 (1H, d, J 7.5 Hz, 8-H), 7.75 (2H, d, J = 8.5 Hz, 2′,6′-H), 7.93 (1H, d, Jtrans = 16.5 Hz, Hb), 8.12 (1H, dd, J = 2.0 and 7.5 Hz, 7-H), 8.32 (1H, d, J = 2.0 Hz, 5-H), 12.41 (1H, s, NH); m/z 359 (100, MH+); HRMS (E)(ES): MH+ found 359.094. C22H16N2O35Cl+ requires 359.095.
(E)-2-(3-Methoxystyryl)-6-phenylquinazolin-4(3H)-one (6d). Solid (0.23 g, 75%), mp. 268 °C; νmax (ATR) 529, 827, 871, 1156, 1226, 1310, 1474, 1582, 1661, 3176 cm−1; δH (500 MHz, DMSO-d6) 3.80 (3H, s, -OCH3), 6.98 (1H, dd, J = 1.5 and 7.5 Hz, 6'-H), 7.02 (1H, d, Jtrans = 16.0 Hz, Ha), 7.22 (1H, d, J = 1.5 Hz, 2'-H), 7.23 (1H, d, J = 7.5 Hz, 4′-H), 7.36 (1H, t, J = 9.0 Hz, 5′-H), 7.41 (1H, d, J = 7.5 Hz, 8-H), 7.50 (2H, t, J = 9.0 Hz, ArH), 7.73–7.77 (3H, m, ArH), 7.93 (1H, d, Jtrans = 16.0 Hz, Hb), 8.11 (1H, dd, J = 2.0 and 7.5 Hz, 7-H), 8.31 (1H, d, J = 2.0 Hz, 5-H), 12.38 (1H, s, NH); δC (125 MHz, DMSO-d6) 55.6, 113.1, 116.1, 120.5, 121.8, 121.9, 123.6, 127.2 (2xC), 128.3, 129.6, 130.5, 133.4, 136.9, 138.3, 138.7, 139.3, 148.7, 151.8, 160.2, 162.1; m/z 355 (100, MH+); HRMS (ES): MH+ found 355.1435. C23H19N2O2+ requires 355.1447.
(E)-6-(4-Fluorophenyl)-2-styrylquinazolin-4(3H)-one (6e). Solid (0.20 g, 66%), mp. 343 °C; νmax (ATR) 490, 528, 696, 755, 968, 1156, 1226, 1478, 1661 cm−1; δH (500 MHz, DMSO-d6) 7.01 (1H, d, Jtrans = 16.0 Hz, Ha), 7.33 (2H, t, J 7.5 Hz, 3′,5′-H), 7.40–7.447 (3H, m, ArH), 7.65 (2H, d, J = 7.8 Hz, ArH), 7.73 (1H, d, J = 7.8 Hz, 8-H), 7.82 (2H, t, J = 8.5 Hz, 2′,6′-H), 7.95 (1H, d, Jtrans = 16.0 Hz, Hb), 8.10 (1H, dd, J = 2.5 and 7.8 Hz, 7-H), 8.28 (1H, d, J = 2.5 Hz, 5-H), 12.40 (1H, s, NH); δC (125 MHz, DMSO-d6) 116.6 (d, 2JCF = 21.75 Hz), 121.1, 121.4, 128.6, 128.8 (d, 3JCF = 8.0 Hz), 129.0, 129.7, 131.3, 131.4, 131.9 (d, 4JCF = 2.8 Hz), 132.8, 134.9, 135.3, 136.7, 138.3, 151.5, 161.7, 162.0 (d, 1JCF = 243.6 Hz); m/z 343 (100, MH+); HRMS (ES): MH+. found 343.1247. C22H16N2OF+ requires 343.1238.
(E)-6-(4-Fluorophenyl)-2-(4-fluorostyryl)quinazolin-4(3H)-one (6f). Solid (0.20 g, 63%), mp. 345 °C; νmax (ATR) 508, 551, 823, 968, 1157, 1237, 1476, 1659, 3174 cm−1; δH (500 MHz, DMSO-d6) 6.96 (1H, d, Jtrans = 16.0 Hz, Ha), 7.29 (2H, t, J = 9.0 Hz, 3′,5′-H), 7.32 (2H, t, J = 9.0 Hz, 3″,5″-H), 7.72 (2H, t, J = 9.0 Hz, 2″,6″-H), 7.73 (1H, d, J 8.5 Hz, 8-H), 7.81 (2H, t, J = 9.0 Hz, 2',6'-H), 7.94 (1H, d, Jtrans = 16.0 Hz, Hb), 8.10 (1H, dd, J = 2.0 and 8.5 Hz, 7-H), 8.29 (1H, d, J = 2.0 Hz, 5-H), 12.40 (1H, br s, NH); δC (125 MHz, DMSO-d6) 116.4 (d, 2JCF = 21.7 Hz), 121.9, 122.3, 123.6, 128.3, 128.7, 129.3 (d, 3JCF = 8.5 Hz),129.6, 129.7, 133.5, 134.4, 134.6, 135.7 (d, 4JCF = 2.8 Hz), 137.3, 137.4, 152.6, 161.6, 163.1 (d, 1JCF = 243.0 Hz); m/z 361 (100, MH+); HRMS (ES): MH+ found 361.1143. C22H15N2OF2+ requires 361.1152.
(E)-2-(4-Chlorophenyl)-6-(4-fluorostyryl)quinazolin-4(3H)-one (6g). Solid (0.20 g, 65%), mp. 345 °C; νmax (ATR) 494, 523, 811, 967. 1092, 1241, 1474, 1490, 1518, 1665, 3173 cm−1; δH (500 MHz, DMSO-d6) 7.01 (1H, d, Jtrans = 16.0 Hz, Ha), 7.32 (2H, d, J = 9.0 Hz, 3″,5″-H-), 7.51 (2H, t, J 8.5 Hz, 3,5-H), 7.68 (2H, d, J = 8.5 Hz, 2″,6″-H), 7.73 (1H, d, J = 8.5 Hz, 8-H), 7.81 (2H, d, J = 9.0 Hz, 2,6-H), 7.93 (1H, d, Jtrans = 16.0 Hz, Hb), 8.10 (1H, dd, J = 2.0 and 8.5 Hz, 7-H), 8.29 (1H, d, J = 2.0 Hz, 5-H), 12.4 (1H, s, NH); m/z 377 (100, MH+); HRMS (ES): MH+ found 377.0852. C22H15N2O35ClF requires 377.0857.
(E)-6-(4-Fluorophenyl)-2-(3-methoxystyryl)quinazolin-4(3H)-one (6h). Solid (0.22 g, 72%), mp. 315 °C; νmax (ATR) 493, 672, 762, 841, 1249, 1433, 1669 cm−1; δH (500 MHz, DMSO-d6) 3.80 (3H, s, -OCH3), 6.98 (1H, dd, J 2.0 and 9.0 Hz, 6′-H), 7.02 (1H, d, Jtrans = 16.0 Hz, Ha), 7.21 (1H, d, J = 2.0 Hz, 2′-H), 7.22 (1H, d, J = 7.5 Hz, 4′-H), 7.32 (2H, t, J = 7.8 Hz, 3,5-H), 7.36 (1H, t, J = 7.5 Hz, 5′-H), 7.73 (1H, d, J = 8.5 Hz, 8-H), 7.81 (2H, t, J = 7.5 Hz, 2,6-H), 7.92 (1H, d, Jtrans = 16.0 Hz, Hb), 8.10 (1H, dd, J = 2.0 and 8.5 Hz, 7-H), 8.29 (1H, d, J = 2.0 Hz, 5-H), 12.37 (1H, s, NH); δC (125 MHz, DMSO-d6) 55.6, 113.1, 116.1, 116.4 (d, 2JCF = 20.9 Hz), 120.5, 121.8, 121.9, 123.6, 128.3, 129.3 (d, 3JCF = 7.5 Hz), 130.6, 133.4, 135.3 (d, 4JCF = 2.9 Hz), 136.7, 137.3, 137.8, 148.7, 151.9, 160.1, 162.1, 162.6 (d, 1JCF = 243.7 Hz); m/z 373 (100, MH+); HRMS (ES): MH+ found 373.1346. C23H18N2O2F+ requires 373.1352.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


