
Synthesis and Cytotoxic Effect of Some Novel 1,2-Dihydropyridin-3-carbonitrile and Nicotinonitrile Derivatives

Abstract
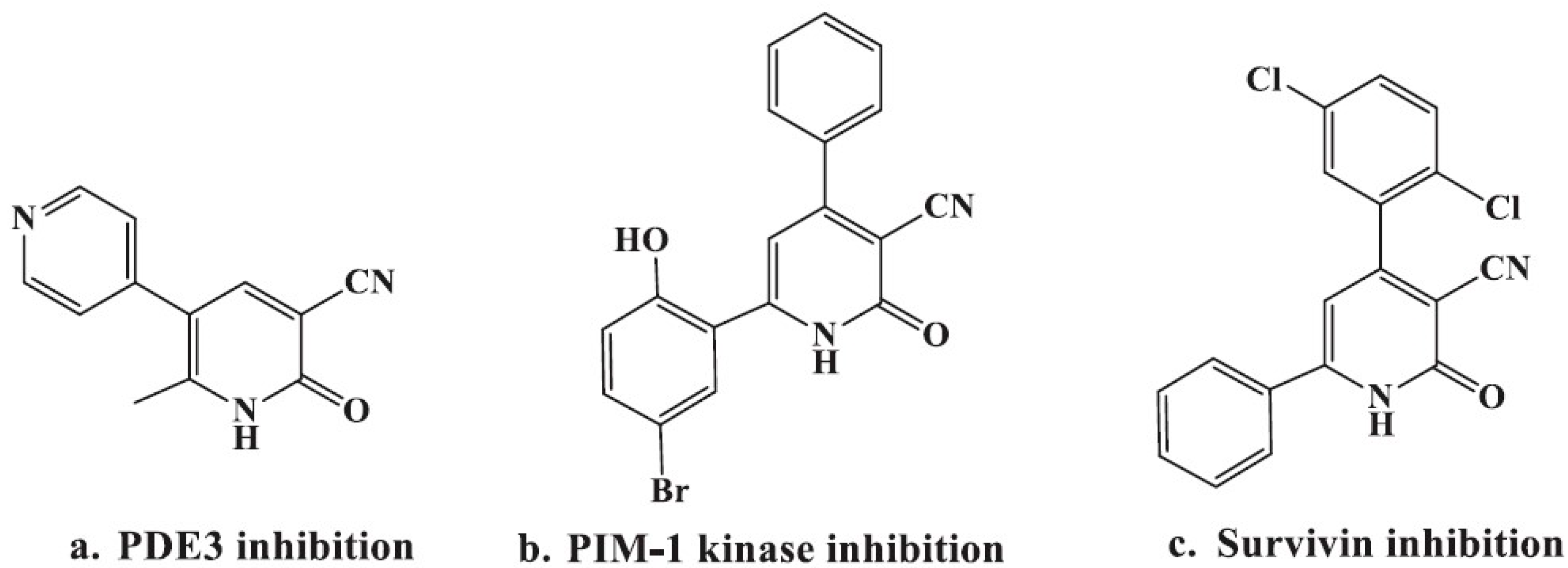
:1. Introduction

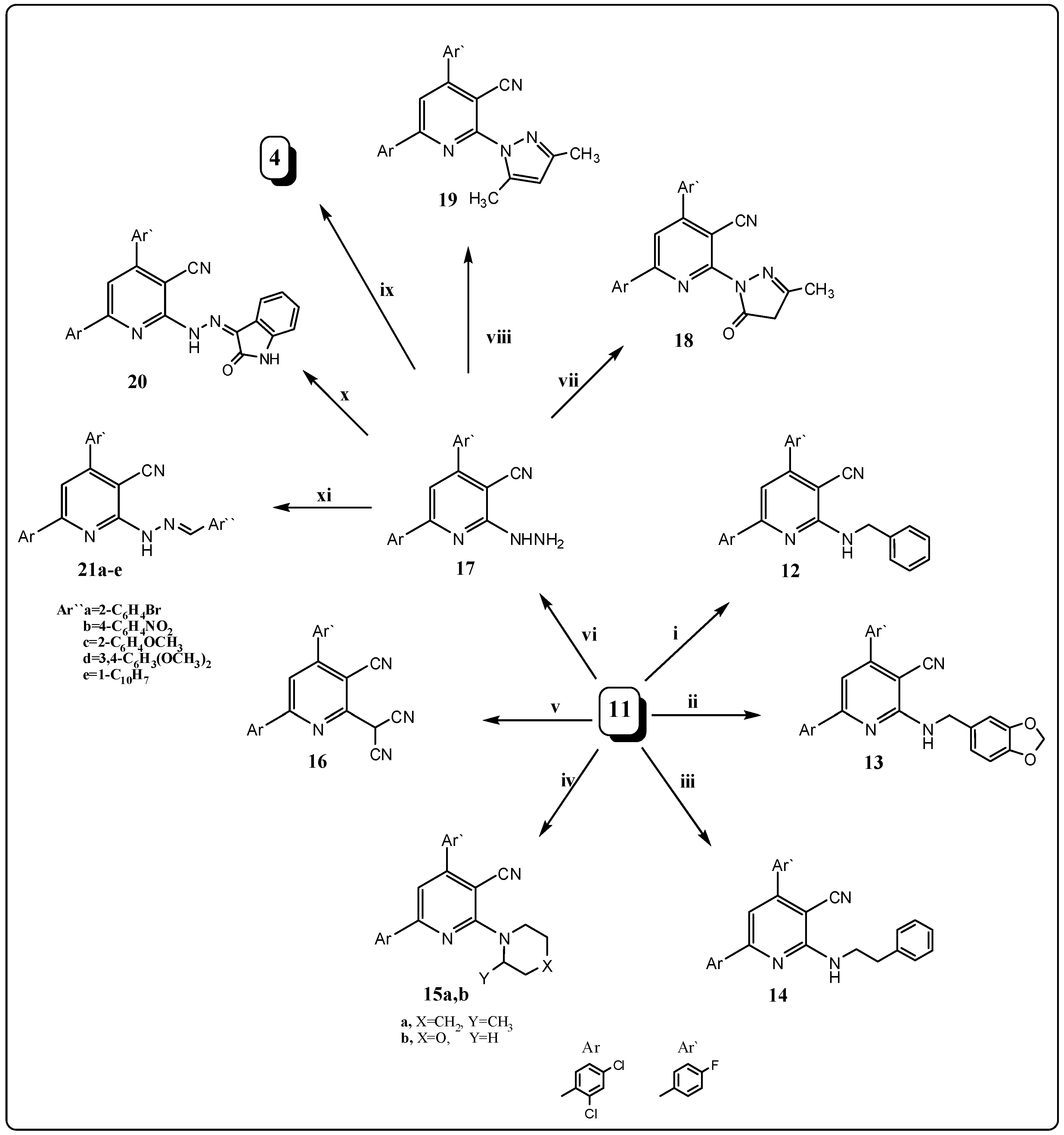
2. Results and Discussion
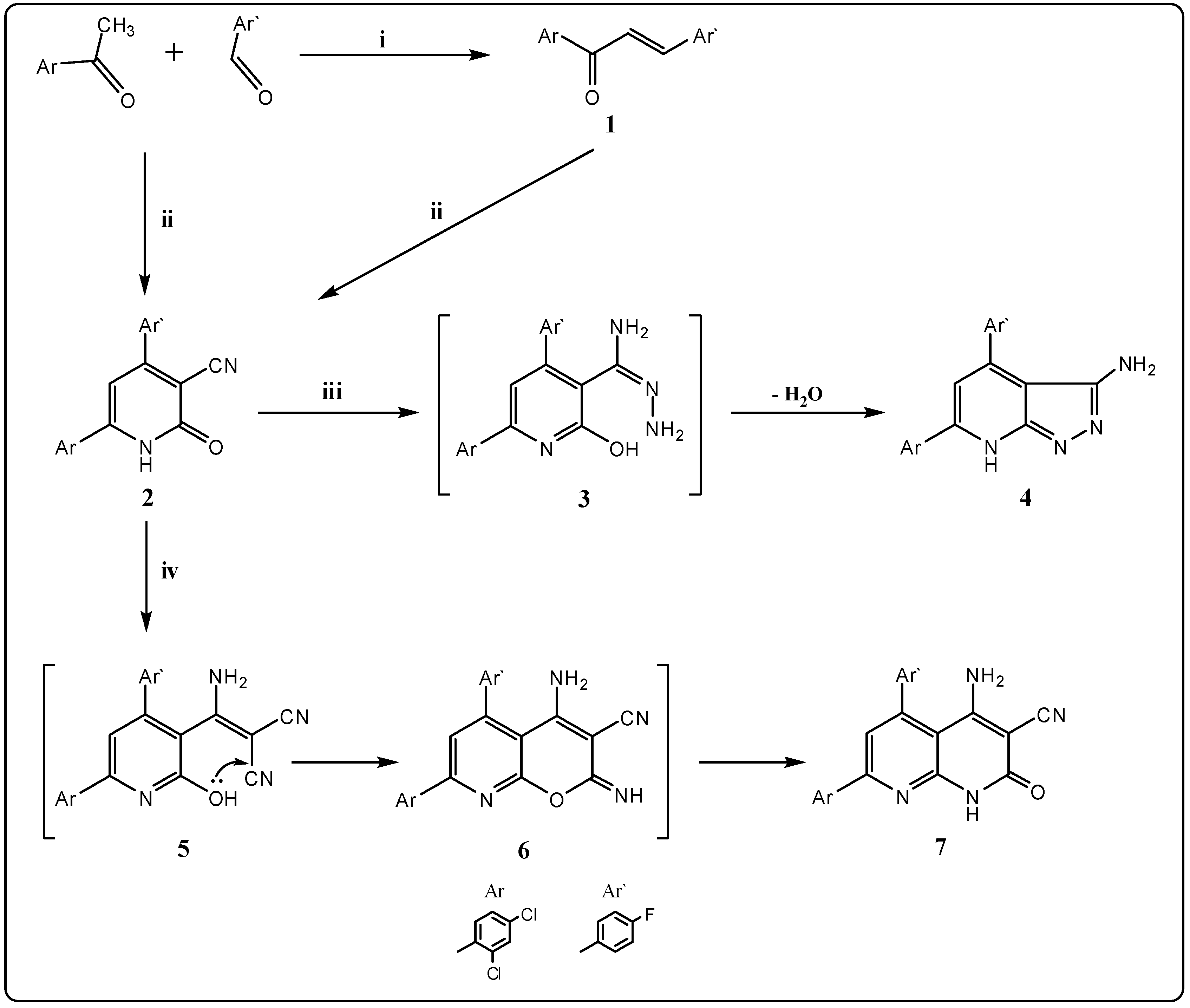
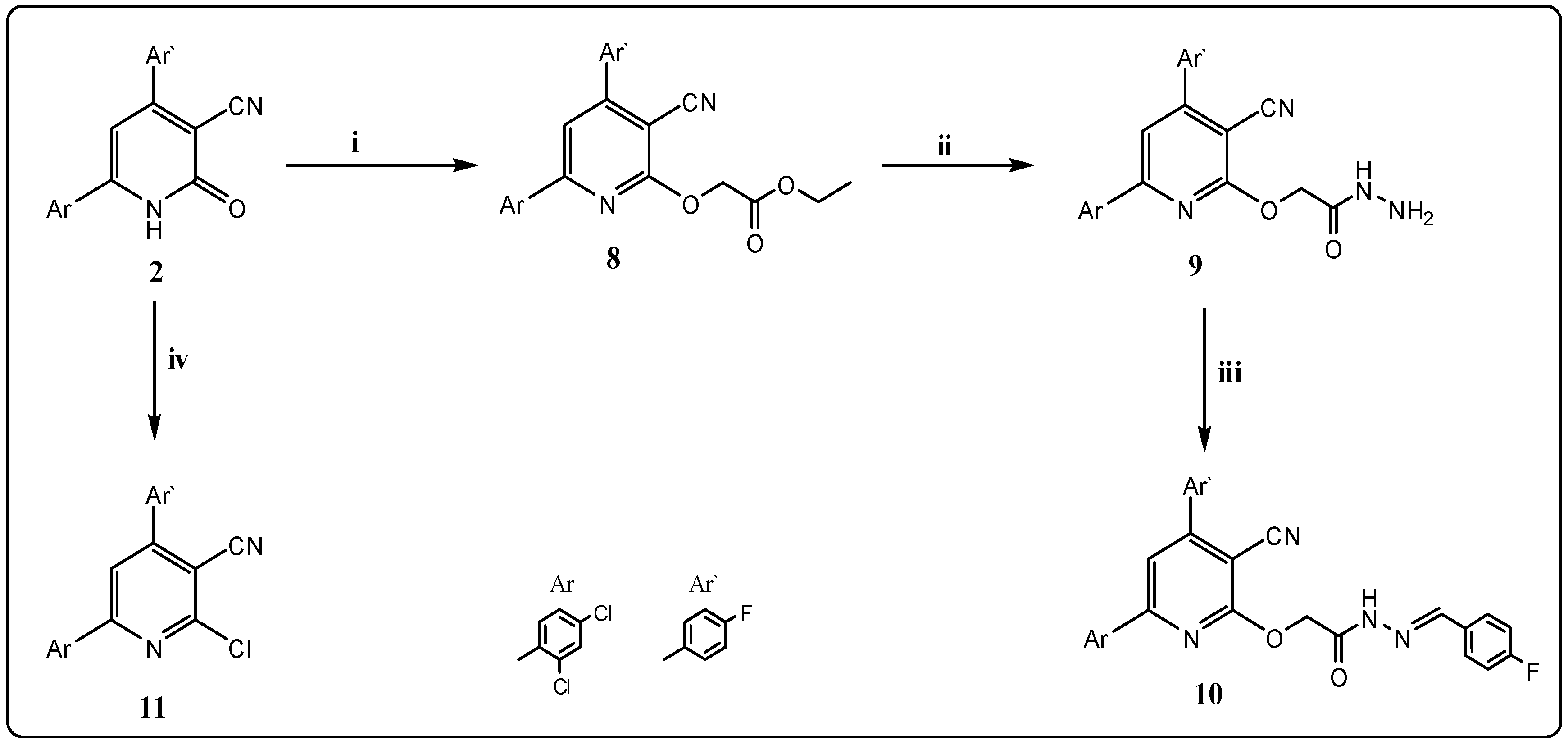
2.1. Chemistry



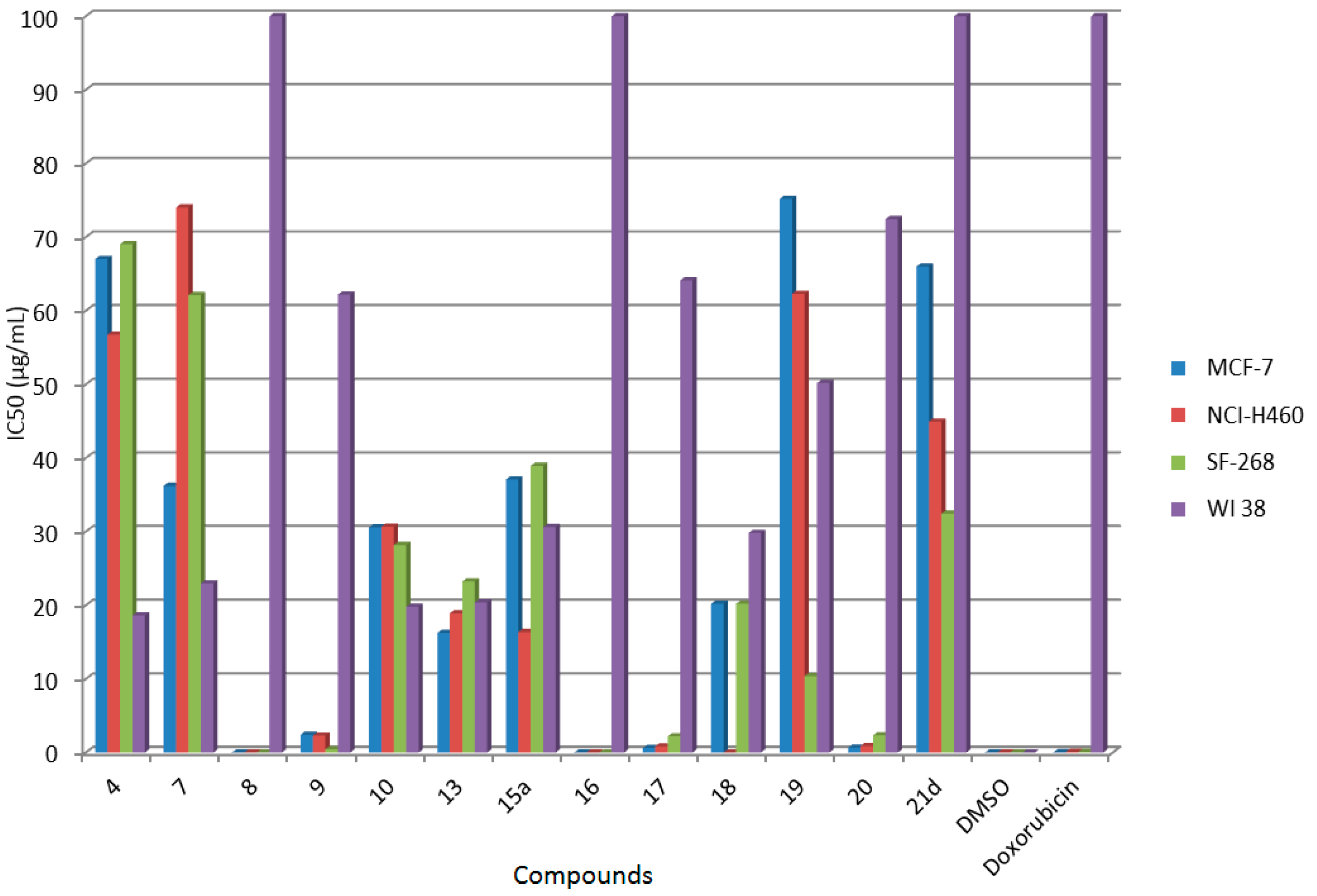
2.2. In Vitro Anticancer Screening

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. No. | IC50 (μg/mL) | |||
|---|---|---|---|---|
| MCF-7 | NCI-H460 | SF-268 | WI 38 | |
| 4 | 67.04 ± 6.23 c | 56.75 ± 8.20 c | 69.05 ± 9.15 c | 18.62 ± 1.21 |
| 7 | 36.22 ± 2.14 c | 74.03 ± 3.65 c | 62.13 ± 3.61 c | 22.97 ± 8.2 |
| 8 | 0.02 ± 0.002 a | 0.01 ± 0.002 a | 0.02 ± 0.045 a | non-cytotoxic |
| 9 | 2.41 ± 1.24 a | 2.30 ± 2.86 a | 0.46 ± 0.06 a | 62.19 ± 2.02 |
| 10 | 30.58 ± 1.10 b | 30.67 ± 1.64 b | 28.18 ± 8.83 b | 19.80 ± 2.68 |
| 13 | 16.26 ± 1.87 b | 18.92 ± 1.03 b | 23.24 ± 4.12 b | 20.38 ± 4.99 |
| 15a | 37.07 ± 7.34 c | 16.37 ± 2.32 b | 38.94 ± 2.63 c | 30.62 ± 6.21 |
| 16 | 0.01 ± 0.002 a | 0.02 ± 0.001 a | 0.01 ± 0.003 a | non-cytotoxic |
| 17 | 0.61 ± 0.082 a | 0.86 ± 0.02 a | 2.19 ± 0.83 a | 64.11 ± 1.22 |
| 18 | 20.22 ± 2.26 b | 0.01 ± 0.003 a | 20.20 ± 3.26 b | 29.82 ± 4.88 |
| 19 | 75.20 ± 13.86 c | 62.30 ± 10.35 c | 10.39 ± 4.19 a | 50.20 ± 10.22 |
| 20 | 0.66 ± 0.21 a | 0.90 ± 0.12 a | 2.34 ± 0.51 a | 72.45 ± 2.40 |
| 21d | 66.02 ± 8.25 c | 44.95 ± 10.46 c | 32.45 ± 6.04 b | non-cytotoxic |
| DMSO | 0 | 0 | 0 | 0 |
| Doxorubicin | 0.04 ± 0.008 | 0.09 ± 0.008 | 0.09 ± 0.007 | non-cytotoxic |

3. Experimental Section
3.1. General Information
3.2. Synthetic Procedures
3.2.1. 6-(2,4-Dichlorophenyl)-4-(4-fluorophenyl)-2-oxo-1,2-dihydropyridin-3-carbonitrile (2)
3.2.2. 6-(2,4-Dichlorophenyl)-4-(4-fluorophenyl)-7H-pyrazolo[3,4-b]pyridin-3-amine (4)
3.2.3. 4-Amino-7-(2,4-dichlorophenyl)-5-(4-fluorophenyl)-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carbonitrile (7)
3.2.4. Ethyl 2-[3-cyano-6-(2,4-dichlorophenyl)-4-(4-fluorophenyl)pyridin-2-yloxy]acetate (8)
3.2.5. 2-[3-Cyano-6-(2,4-dichlorophenyl)-4-(4-fluorophenyl)pyridin-2-yloxy]acetohydrazide (9)
3.2.6. 2-[3-Cyano-6-(2,4-dichlorophenyl)-4-(4-fluorophenyl)pyridin-2-yloxy]-N′-(4-fluorobenzylidene)-acetohydrazide (10)
3.2.7. 2-Chloro-6-(2,4-dichlorophenyl)-4-(4-fluorophenyl)nicotinonitrile (11)
3.2.8. General procedure for the synthesis of 2-(benzylamino)-6-(2,4-dichlorophenyl)-4-(4-fluoro-phenyl)-nicotinonitrile (12), 2-(benzo[d][1,3]dioxol-5-ylmethylamino)-6-(2,4-dichlorophenyl)-4-(4-fluorophenyl)nicotinonitrile (13), 6-(2,4-dichlorophenyl)-4-(4-fluorophenyl)-2-(1-phenylethyl-amino)nicotinonitrile (14), and 6-(2,4-dichlorophenyl)-4-(4-fluorophenyl)-2-(2-substituted-1-yl)nicotinonitriles 15a,b
3.2.9. 2-[3-Cyano-6-(2,4-dichlorophenyl)-4-(4-fluorophenyl)pyridin-2-yl]malononitrile (16)
3.2.10. 6-(2,4-Dichlorophenyl)-4-(4-fluorophenyl)-2-hydrazinylnicotinonitrile (17)
3.2.11. General procedure for the synthesis of 6-(2,4-dichlorophenyl)-4-(4-fluorophenyl)-2-(3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)nicotinonitrile (18) and 6-(2,4-dichlorophenyl)-2-(3,5-dimethyl-1H-pyrazol-1-yl)-4-(4-fluorophenyl)nicotinonitrile (19)
3.2.12. 6-(2,4-Dichlorophenyl)-4-(4-fluorophenyl)-2-[2-(2-oxoindolin-3-ylidene)hydrazinyl]nicotinonitrile (20)
3.2.13. General procedure for the synthesis of 6-(2,4-dichlorophenyl)-4-(4-fluorophenyl)-2-[2-(2-substiutedbenzylidene)hydrazinyl]nicotinonitriles (1a–e)
3.3. Anticancer Activity
3.3.1. Cell Cultures
3.3.2. Cancer Cell Growth Assay
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Zhang, J.Y. Apoptosis-based anticancer drugs. Nat. Rev. Drug Disccov. 2002, 1, 101–102. [Google Scholar] [CrossRef]
- Ali, A.; Fergus, K.; Wright, F.C.; Pritchard, K.I.; Kiss, A.; Warner, E. The impact of a breast cancer diagnosis in young women on their relationship with their mothers. Breast 2014, 23, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.W.; Jimenez, C.R.; Boven, E. Breast cancer classification by proteomic technologies: Current state of knowledge. Cancer 2014, 40, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Hassan, G.S.; Kadry, H.H.; Abou-Seri, S.M.; Ali, M.M.; Mahmoud, A.E.E. Synthesis and in vitro cytotoxic activity of novel pyrazolo[3,4-d]pyrimidines and related pyrazole hydrazones toward breast adenocarcinoma MCF-7 cell line. Bioorg. Med. Chem. 2011, 19, 6808–6817. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Georgey, H.H.; El-Subbagh, H.I. Novel 1,3,4-heterodiazole analogues: Synthesis and in vitro antitumor activity. Eur. J. Med. Chem. 2012, 47, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Carmen, A.J.; Carlos, M. Medicinal Chemistry of Anticancer Drugs, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 1–8. [Google Scholar]
- Borowski, E.; Bontemps-Gracz, M.M.; Piwkowska, A. Strategies for overcoming ABC-transporters-mediated multidrug resistance (MDR) of tumor cells. Acta Biochim. Pol. 2005, 52, 609–627. [Google Scholar] [PubMed]
- Avila, H.P.; Smania, E.F.; Monache, F.D.; Smania, A. Structure-activity relationship of antibacterial chalcones. Bioorg. Med. Chem. 2008, 16, 9790–9794. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, X.; Yin, D.; Yuan, F. Syntheses and biological activity of chalcones-imidazole derivatives. Res. Chem. Intermed. 2013, 39, 1037–1048. [Google Scholar] [CrossRef]
- Sortino, M.; Delgado, P.; Juarez, S.; Quiroga, J.; Abonia, R.; Insuasty, B.; Nogueras, M.; Rodero, L.; Garibotto, F.M.; Enriz, R.D.; et al. Synthesis and antifungal activity of (Z)-5-arylidenerhodanines. Bioorg. Med. Chem. 2007, 15, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Lopez, S.N.; Castelli, M.V.; Zacchino, S.A.; Dominguez, J.N.; Lobo, G.; Charris-Charris, J.; Cortes, J.C.; Ribas, J.C.; Devia, C.; Rodriguez, A.M.; et al. In vitro antifungal evaluation and structure-activity relationships of a new series of chalcone derivatives and synthetic analogues with inhibitory properties against polymers of the fungal cell wall. Bioorg. Med. Chem. 2001, 8, 1999–2013. [Google Scholar] [CrossRef]
- Cheng, J.H.; Hung, C.F.; Yang, S.C.; Wang, J.P.; Won, S.J.; Lin, C.N. Synthesis and cytotoxic, anti-inflammatory, and anti-oxidant activities of 2′,5′-dialkoxylchalcones as cancer chemopreventive agents. Bioorg. Med. Chem. 2008, 16, 7270–7276. [Google Scholar] [CrossRef] [PubMed]
- Katsori, A.M.; Hadjipavlou-Litina, D. Chalcones in cancer: Understanding their role in terms of QSAR. Curr. Med. Chem. 2009, 16, 1062–1081. [Google Scholar] [CrossRef] [PubMed]
- Modzelewska, A.; Pettit, C.; Achanta, G.; Davidson, N.E.; Huang, P.; Khan, S.R. Anticancer activities of novel chalcone and bis-chalcone derivatives. Bioorg. Med. Chem. 2006, 14, 3491–3495. [Google Scholar] [CrossRef] [PubMed]
- Abdelhafez, O.M.; Abdel-Latif, N.A.; Badria, F.A. DNA, Antiviral activities and cytotoxicity of new furochromone and benzofuran derivatives. Arch. Pharm. Res. 2011, 34, 1623–1632. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Latif, N.A. Synthesis and antidepressant activity of some new coumarin derivatives. Sci. Pharm. 2005, 74, 173–216. [Google Scholar]
- Son, J.K.; Zhao, L.X.; Basnet, A.; Thapa, P.; Karki, R.; Na, Y.; Jahng, Y.; Jeong, T.C.; Jeong, B.S.; Lee, C.S.; et al. Synthesis of 2,6-diaryl-substituted pyridines and their antitumor activities. Eur. J. Med. Chem. 2008, 43, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Amr, A.G.; Abdulla, M.M. Anti-inflammatory profile of some synthesized heterocyclic pyridone and pyridine derivatives fused with steroidal structure. Bioorg. Med. Chem. 2006, 14, 4341–4352. [Google Scholar] [CrossRef] [PubMed]
- Hammam, A.G.; Abdel Hafez, N.A.; Midura, W.H.; Mikolajczyk, M.Z. Chemistry of seven-membered heterocycles, VI. Synthesis of novel bicyclic heterocyclic compounds as potential anticancer and anti-HIV agents. Z. Naturforsch. 2000, 55, 417–424. [Google Scholar] [CrossRef]
- Kotb, E.R.; Anwar, M.M.; Abbas, H.A.S.; Abd El-Moez, S.I. A concise synthesis and antimicrobial activity of a novel series of naphthylpyridine-3-carbonitrile compounds. Acta Pol. Pharm. Drug Res. 2013, 70, 667–679. [Google Scholar]
- Sayed, H.H.; Morsy, E.M.; Flefel, E.M. Synthesis and reactions of some novel nicotinonitrile, thiazolotriazole, and imidazolotriazole derivatives for antioxidant evaluation. Synth. Commun. 2010, 40, 1360–1370. [Google Scholar] [CrossRef]
- Akira, M.; Aya, N.; Shigeki, I.; Motoki, T.; Kazuo, S. JBIR-54, a new 4-pyridinone derivative isolated from Penicillium daleae Zaleski fE50. J. Antibiot. 2009, 62, 705–706. [Google Scholar]
- Al-Omar, M.A.; Amr, A.E.; A.l-Salahi, R.A. Anti-inflamatory, analgesic, anticonvulsant and antiparkinsonian activities of some pyridine derivatives using 2,6-disubstituted isonicotinic acid hydrazides. Archiv. Phaem. 2010, 343, 648–656. [Google Scholar]
- Martin, C.; Göggel, R.; dal Piaz, V.; Vergelli, C.; Giovannoni, P.; Ernst, M.; Uhlig, S. Airway relaxant and anti-inflammatory properties of a PDE4 inhibitor with low affinity for the high-affinity rolipram binding site. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2002, 365, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Amr, A.E.; Sayed, H.H.; Abdulla, M.A. Synthesis and reactions of some new substituted pyridine and pyrimidine derivatives as analgesic, anticonvulsant and antiparkinsonian agents. Arch. Pharm. Chem. Life Sci. 2005, 338, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Al-Abdullah, E.S. Synthesis and anticancer activity of some novel tetralin-6-yl-pyrazoline, 2-thioxopyrimidine, 2-oxopyridine, 2-thioxo-pyridine and 2-iminopyridine derivatives. Molecules 2011, 16, 3410–3419. [Google Scholar] [CrossRef] [PubMed]
- Abo-Ghalia, M.; Abdulla, M.M.Z.; Amr, A.E. Synthesis of some new (Nα-dipicolinoyl)-bis-l-leucyl-dl-norvalyl linear tetra and cyclic octa bridged peptides as new antiinflammatory agents. Z. Naturforsch. 2003, 58b, 903–910. [Google Scholar]
- Kotb, E.R.; El-Hashash, M.A.; Salama, M.A.; Kalf, H.S.; Abdel Wahed, N.A.M. Synthesis and reactions of some novel nicotinonitrile derivatives for anticancer and antimicrobial evaluation. Acta Chim. Slov. 2009, 56, 908–919. [Google Scholar]
- Kumar, S.; Das, S.; Dey, S.; Maity, P.; Guha, M.; Choubey, V.; Panda, G.; Bandyopadhyay, V. Antiplasmodial activity of [(aryl)arylsulfanylmethyl]pyridine. Antimicrob. Agents Chemother. 2008, 52, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.S.; Manna, K.; Banik, U.; Das, M.; Sarkar, P. Synthetic strategies and pharmacology of 2-oxo-3-cyanopyridine derivatives: A review. Int. J Pharm. Pharm. Sci. 2014, 6, 39–42. [Google Scholar]
- Abbas, H.-A.S.; El Sayed, W.A.; Fathy, N.M. Synthesis and antitumor activity of new dihydropyridine thioglycosides and their corresponding dehydrogenated forms. Eur. J. Med. Chem. 2010, 45, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Al-Mutairi, M.S.; Al-Abdullah, E.S.; Haiba, M.E.; Khedr, M.A.; Zaghary, W.A. Synthesis, molecular docking and preliminary in vitro cytotoxic evaluation of some substituted tetrahydronaphthalene (2′,3′,4′,6′-Tetra-O-Acetyl-β-d-Gluco-/Galactopyranosyl) derivatives. Molecules 2012, 17, 4717–4732. [Google Scholar] [CrossRef] [PubMed]
- Kotb, E.R.; Abbas, H.-A.S.; Flefel, E.M.; Sayed, H.H.; Abdel Wahed, N.A.M. Utility of hantzsch ester in synthesis of some 3,5-bisdihydropyridine derivatives and studying their biological evaluation. J. Heterocycl. Chem. 2015, 52, 1531–1539. [Google Scholar] [CrossRef]
- Sayed, H.H.; Flefel, E.M.; Abd El-Fatah, A.M.; El-Sofany, W.I. Focus on the synthesis and reactions of some new pyridine carbonitrile derivatives as antimicrobial and antioxidant agents. Egypt J. Chem. 2010, 53, 17–35. [Google Scholar]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenne, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Zhang, R.; Li, V.; Liu, X.; Sun, R.W.Y.; Che, C.M.; Wong, K.K.Y. Enhancement of anticancer efficacy using modified lipophilic nanoparticle drug encapsulation. Int. J. Nanomed. 2012, 7, 731–737. [Google Scholar] [Green Version]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paul, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
M. Flefel, E.; S. Abbas, H.-A.; E. Abdel Mageid, R.; A. Zaghary, W. Synthesis and Cytotoxic Effect of Some Novel 1,2-Dihydropyridin-3-carbonitrile and Nicotinonitrile Derivatives. Molecules 2016, 21, 30. https://doi.org/10.3390/molecules21010030
M. Flefel E, S. Abbas H-A, E. Abdel Mageid R, A. Zaghary W. Synthesis and Cytotoxic Effect of Some Novel 1,2-Dihydropyridin-3-carbonitrile and Nicotinonitrile Derivatives. Molecules. 2016; 21(1):30. https://doi.org/10.3390/molecules21010030
Chicago/Turabian StyleM. Flefel, Eman, Hebat-Allah S. Abbas, Randa E. Abdel Mageid, and Wafaa A. Zaghary. 2016. "Synthesis and Cytotoxic Effect of Some Novel 1,2-Dihydropyridin-3-carbonitrile and Nicotinonitrile Derivatives" Molecules 21, no. 1: 30. https://doi.org/10.3390/molecules21010030
APA StyleM. Flefel, E., S. Abbas, H. -A., E. Abdel Mageid, R., & A. Zaghary, W. (2016). Synthesis and Cytotoxic Effect of Some Novel 1,2-Dihydropyridin-3-carbonitrile and Nicotinonitrile Derivatives. Molecules, 21(1), 30. https://doi.org/10.3390/molecules21010030