
Methodology for the Construction of the Bicyclo[4.3.0]nonane Core

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Synthetic Strategies
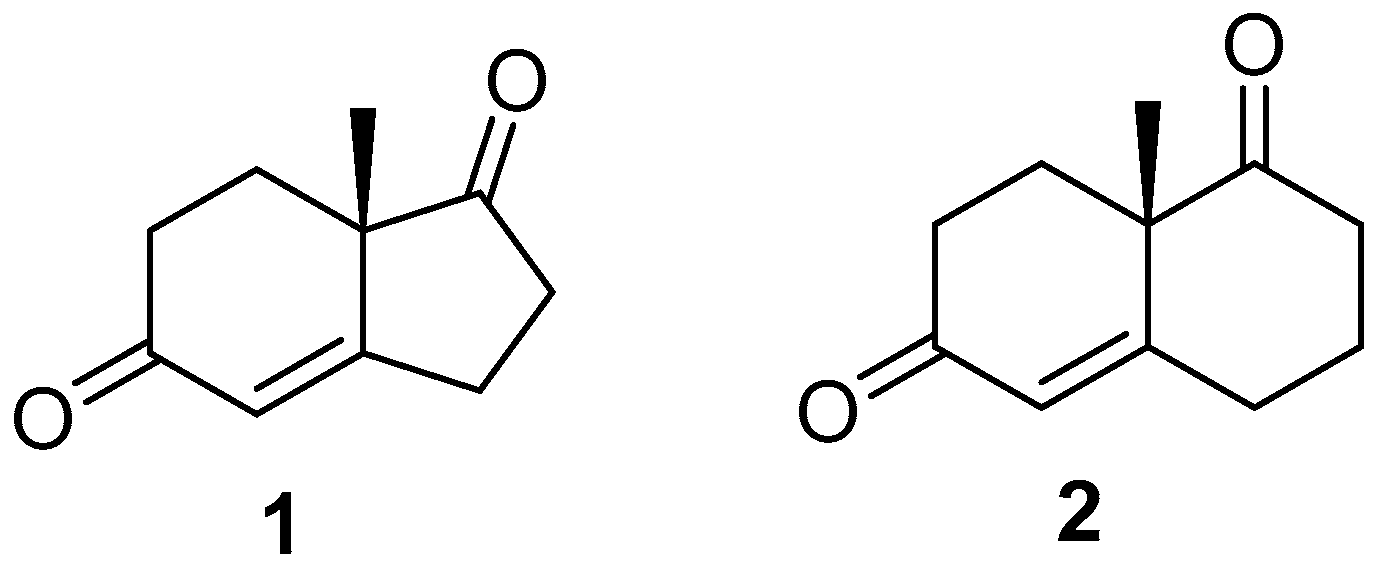
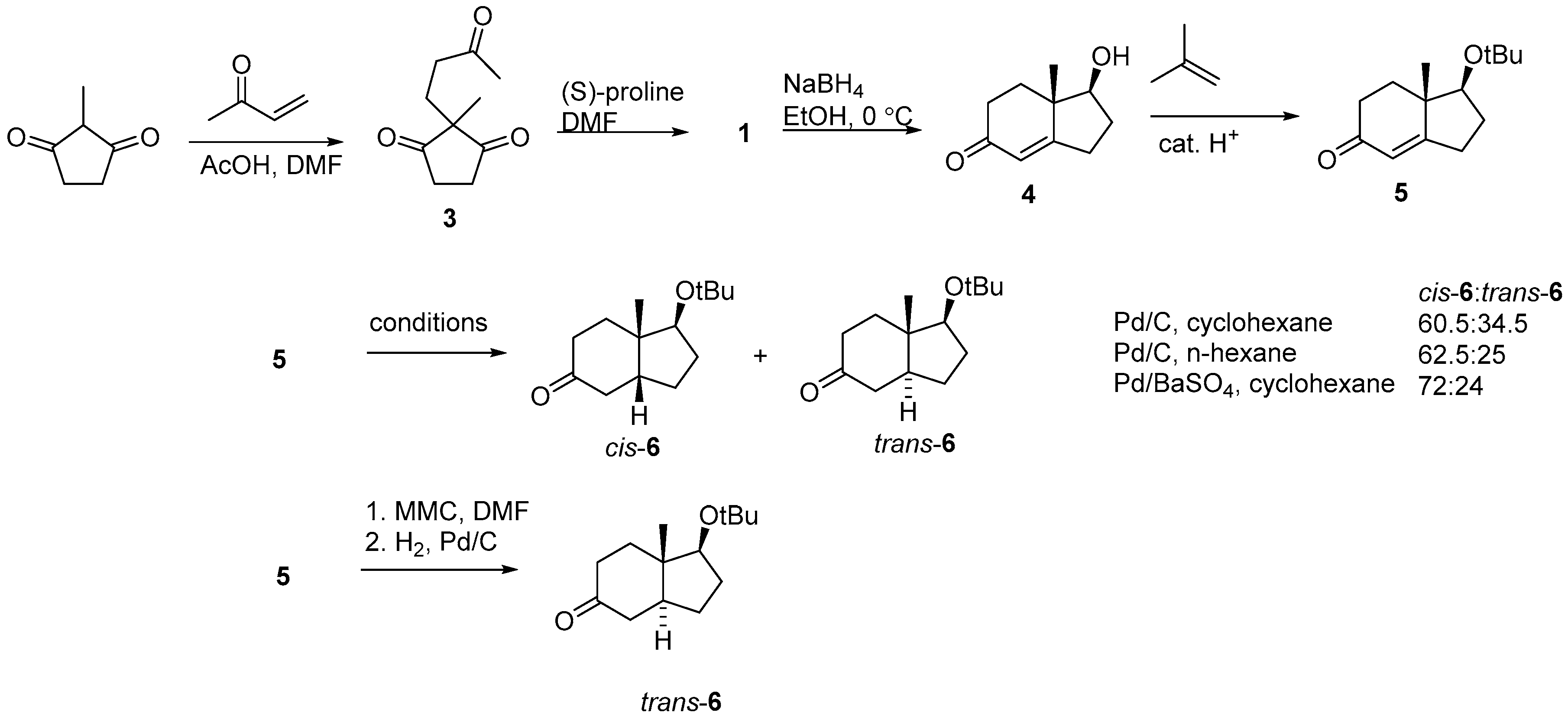
2.1. Hajos–Parrish Dione
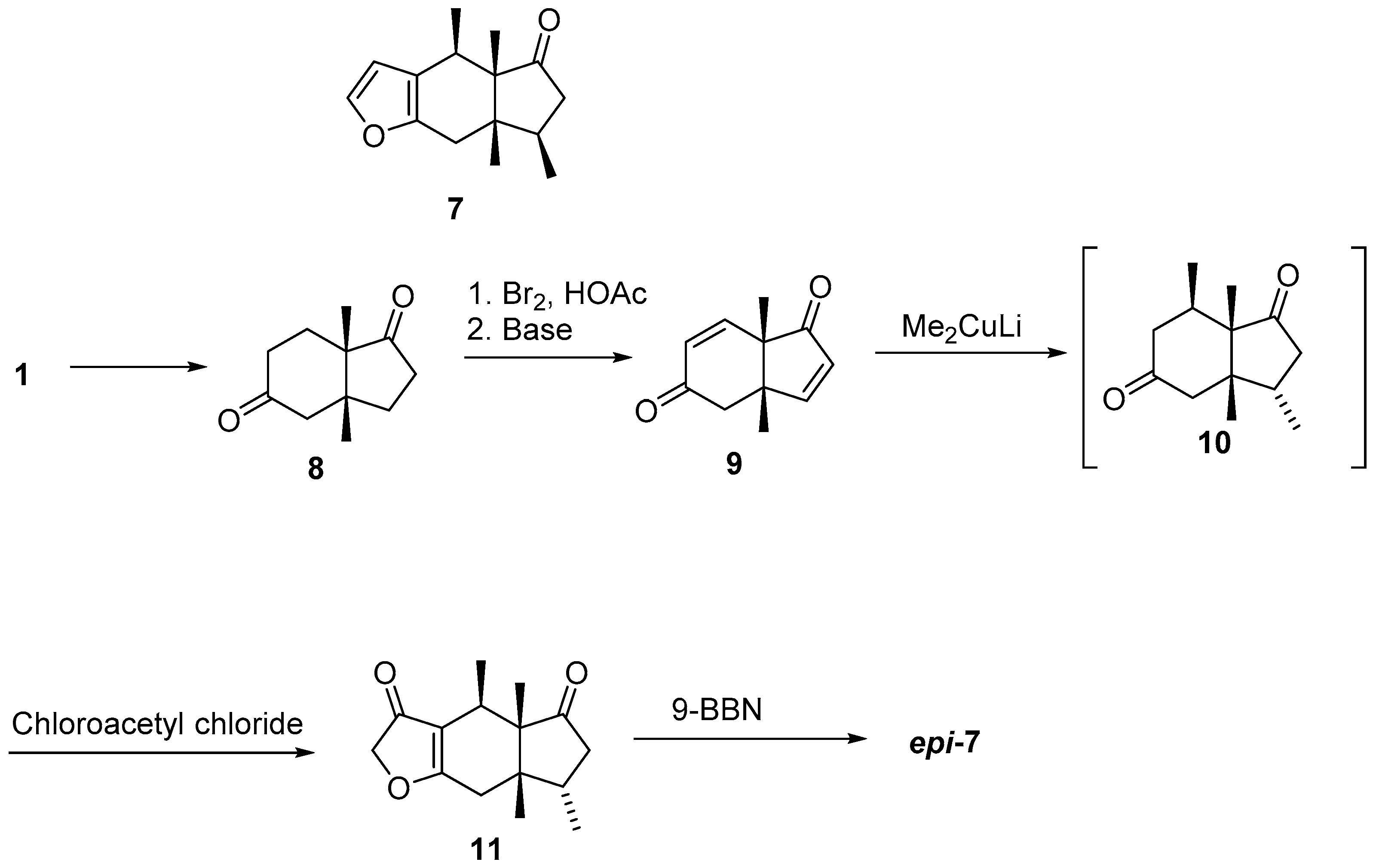
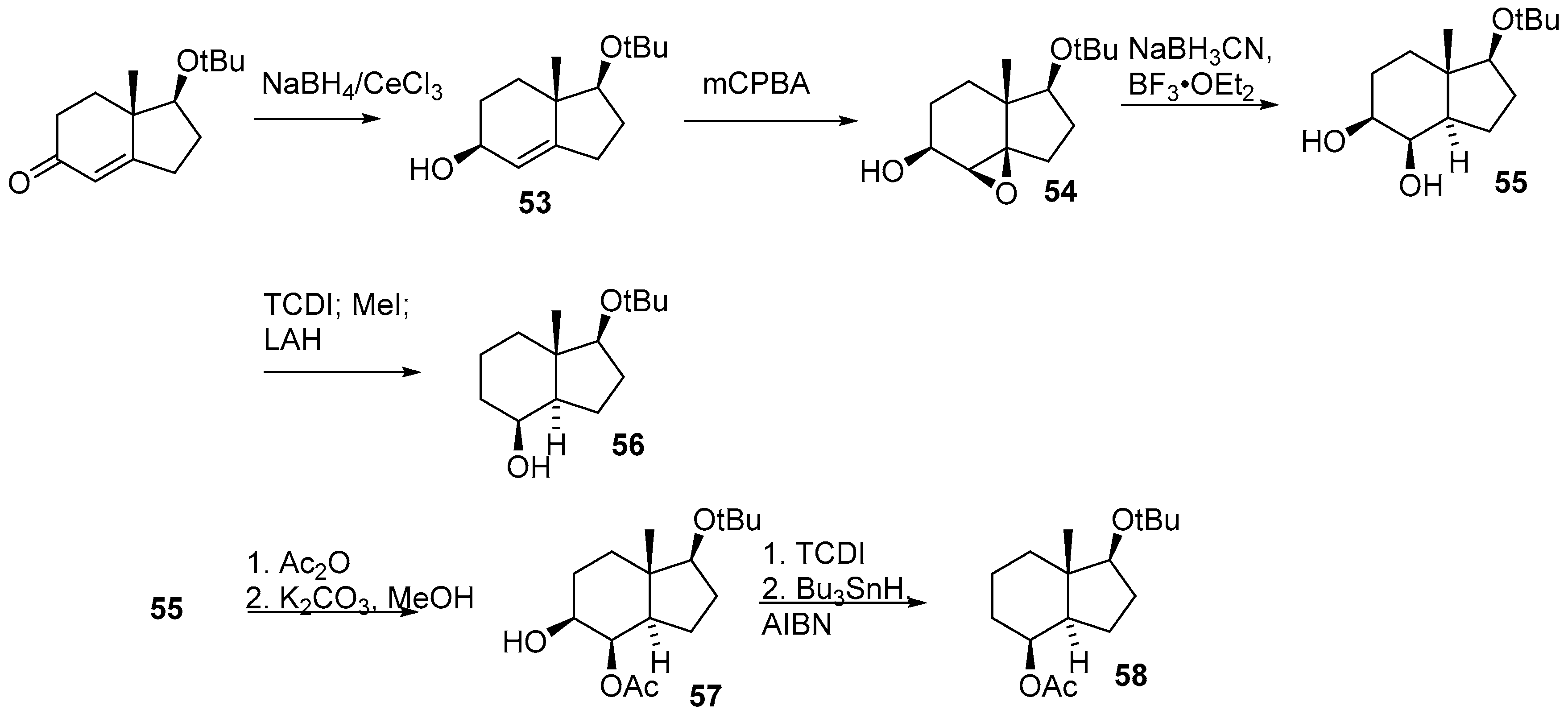
2.2. Cyclization Strategies to Hydrindane Cores
3. Summary and Outlook
Acknowledgments
Conflicts of Interest
References
- Jankowski, P.; Marczak, S.; Wicha, J. Methods for the construction of trans-hydrindane rings and their origins in steroid chemistry. Vitamin D total synthesis. Tetrahedron 1998, 54, 12071–12150. [Google Scholar]
- Hong, B.-C.; Sarshar, S. Recent advances in the synthesis of indan systems: A review. Org. Prep. Proced. Int. 1999, 31, 1–86. [Google Scholar] [CrossRef]
- Chapelon, A.-S.; Moraleda, D.; Rodriguez, R.; Ollivier, C.; Santelli, M. Enantioselective synthesis of steroids. Tetrahedron 2007, 2007, 11511–11616. [Google Scholar] [CrossRef]
- Xu, X.-M.; Guan, Y.-Z.; Shan, S.-M.; Luo, J.-G.; Kong, L.-Y. Withaphysalin-type withanolides from Physalis minima. Phytochem. Lett. 2016, 15, 1–6. [Google Scholar] [CrossRef]
- Xiang, L.; Wang, Y.; Yi, X.; Feng, J.; He, X. Furospirostanol and spirostanol saponins from the rhizome of Tupistra chinesis and their cytotoxic and anti-inflammatory activities. Tetrahedron 2016, 72, 134–141. [Google Scholar] [CrossRef]
- Srivastava, P.K.; Gupta, M.R.; Khare, N.K. Two novel steroidal derivatives from chloroform-soluble extract of hoya longifolia. Nat. Prod. Res. 2016, 30, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.-X.; Lin, J.-Q.; Wang, H.-W.; Chen, J.-X.; Chen, J.-C.; Chen, L.; Zhou, L.; Qiu, M.-H. Identification and antifeedant activities of limonoids from azadirachta indica. Chem. Biodivers. 2015, 12, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Fu, Z.; Zhang, Q.; Xu, L.; Liao, L. Identificatrion and structural elucidation of steroidal saponins from the root of Paris polyphylla by HPLC-ESI-QTOF-MS/MS. Nat. Prod. Res. 2015, 29, 1798–1803. [Google Scholar] [CrossRef] [PubMed]
- Li, K.-L.; Zhang, P.; Li, X.-N.; Guo, J.; Hu, H.-B.; Xiao, C.-F.; Xie, X.-Q.; Xu, Y.-K. Cyototoxic limonids from Trichilia americana leaves. Phytochemistry 2015, 118, 61–67. [Google Scholar]
- Sevillano, L.G.; Melero, C.P.; Boya, M.; Lopez, J.L.; Tome, F.; Caballero, E.; Carron, R.; Montero, M.J.; Medarde, M.; San Feliciano, A. Synthesis an inotropic activity of hydroindene derivatives. Bioorg. Med. Chem. 1999, 7, 2991–3001. [Google Scholar] [CrossRef]
- Schweiger, E.J.; Joullie, M.M.; Weisz, P.B. Synthesis of a C,D-ring analog of 17-a-hydroxyprogesterone. Tetrahedron Lett. 1997, 38, 6127–6130. [Google Scholar] [CrossRef]
- Shan, X.-Q.; Peng, S.-L.; Shi, H.-L.; Wang, X.-L.; Ding, L.-S.; Liao, X. Panthogenins A and B, two novel norergostanol steroids from Dioscorea panthaica. Chin. Chem. Lett. 2014, 25, 1256–1258. [Google Scholar] [CrossRef]
- Wang, Z.-B.; Zhai, Y.-D.; Ma, Z.-P.; Yang, C.-J.; Pan, R.; Yu, J.-L.; Wang, Q.-H.; Yang, B.-Y.; Kuang, H.-X. Triterpenoids and Flavonoids from the leaves of astragalus membranaceus and their inhibitory effects on nitric oxide production. Chem. Biodivers. 2015, 12, 1575–1574. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-X.; Shi, Y.-M.; Wang, W.-G.; Li, X.-N.; Du, X.; Liu, M.; Li, Y.; Xue, Y.-B.; Zhang, Y.-H.; Pu, J.-X.; et al. Kadcoccinones A–F, new biogenetically related lanostane-type triterpenoids with diverse skeletons from kadsura coccinea. Org. Lett. 2015, 17, 4616–4619. [Google Scholar] [CrossRef] [PubMed]
- Hajos, Z.G.; Parrish, D.R. Asymmetric synthesis of bicyclic intermediates of natural product chemistry. J. Org. Chem. 1974, 39, 1615–1621. [Google Scholar] [CrossRef]
- Micheli, R.A.; Hajos, Z.G.; Cohen, N.; Parrish, D.R.; Portland, L.A.; Sciamanna, W.; Scott, M.A.; Wehrli, P.A. Total synthesis of optically active 19-norsteroids. (+)-Estr-4-ene-3,17-dione and (+)-13b-ethylgon-4-ene-3,17-dione. J. Org. Chem. 1974, 40, 675–681. [Google Scholar] [CrossRef]
- Hajos, Z.G.; Parrish, D.R. The stereocontrolled synthesis of trans-hydrindan steroidal intermediates. J. Org. Chem. 1973, 38, 3239–3243. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, S.; Ferrari, M.; Gariboldi, P.; Jommi, G.; Sisti, M.; Destro, R. Synthetic study of pinguisane terpenoids. J. Chem. Soc. Perkin Trans. 1 1981. [Google Scholar] [CrossRef]
- Nassim, B.; Schlemper, E.O.; Crabbe, P. Total synthesis of dinordrin and analogues. J. Chem. Soc. Perkin Trans. 1 1983. [Google Scholar] [CrossRef]
- Paquette, L.A.; Sugimura, T. Enantiospecific total synthesis and absolute configurational assignment of (−)-punctatin A (antibiotic M95464). J. Am. Chem. Soc. 1986, 108, 3841–3842. [Google Scholar] [CrossRef]
- Daniewski, A.R.; Kiegiel, J. A simple way to (3aR,4S,7aS)-(Z)-ethylideneoctahydro-7a-methyl-1H-4-indenol, a synthon for total synthesis of vitamins D. J. Org. Chem. 1988, 53, 5534–5535. [Google Scholar] [CrossRef]
- Daniewski, A.R.; Liu, W. A novel silylcopper catalyst for the reduction bromination of Hajos dione. Improved preparation of a CD synthon for the synthesis of vitamin D. J. Org. Chem. 2001, 66, 626–628. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, B.; Perez, J.A.M.; Granja, J.R.; Castedo, L.; Mourino, A. Synthesis of hydrindan derivatives related to vitamin D. J. Org. Chem. 1992, 57, 3173–3178. [Google Scholar] [CrossRef]
- Paquette, L.A.; Wang, T.-Z.; Sivik, M.R. Enantioselective synthesis of natural (−)-austalide B, an unusual ortho ester metabolite produced by toxigenic cultures of aspergillus ustus. J. Am. Chem. Soc. 1994, 116, 2665–2666. [Google Scholar] [CrossRef]
- Paquette, L.A.; Wang, T.-Z.; Sivik, M.R. Total synthesis of (−)-austalide B. A generic solution to elaboration of the pyran/p-cresol/butenolide triad. J. Am. Chem. Soc. 1994, 116, 11323–11334. [Google Scholar] [CrossRef]
- Lugar, C.W.; Magee, D.; Adrian, M.D.; Shetler, P.; Bryant, H.U.; Dodge, J.A. B-ring unsaturated estrogens: Biological evaluation of 17a-dihydroequilein and novel b-nor-6-thiaequilenins as tissue selective estrogens. Bioorg. Med. Chem. Lett. 2003, 13, 4281–4284. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.S.; Sadnia, H.; Anderson, J.M.; Durst, T.; Asim, M.; El-Salfiti, M.; Choueiri, C.; Pratt, M.A.C.; Ruddy, S.C.; Lau, R.; et al. A-CD estrogens. I. Substitutent effects, hormone, potency, and receptor subtype selectivity in a new family of flexible estrogenic compounds. J. Med. Chem. 2011, 54, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Van Gool, M.; Zhao, X.; Sabbe, K.; Vandewalle, M. Synthesis of 14,20-bis-epi-1a,25-dihydroxy-19-norvitamin D3 and analogues. Eur. J. Org. Chem. 1999, 1999, 2241–2248. [Google Scholar] [CrossRef]
- Di Filippo, M.; Izzo, I.; Vece, A.; De Riccardis, F.; Sodano, G. Enantioselective synthesis of a trans-ethenyl-hydrindene, a useful steroid CD-ring diene precursor. Tetrahedron Lett. 2001, 42, 1155–1157. [Google Scholar] [CrossRef]
- Shi, H. Facile method for stereoselective synthesis of new chiral (1R, 4aR, 8aR)-1,3,4,4a,5,7,8,8a-octahydro-2-methylene-naphthalene-6-one-1-propanenitrile, an important precursor for solanapyrones. Synth. Commun. 2006, 36, 237–248. [Google Scholar] [CrossRef]
- Chochrek, P.; Kurek-Tyrlik, A.; Michalak, K.; Wicha, J. A new approach to a vitamin D ring C/D building block from the Hajos dione, involving epoxide opening at the more substituted carbon atom. Tetrahedron Lett. 2006, 47, 6017–6020. [Google Scholar] [CrossRef]
- Chochrek, P.; Wicha, J. An expedited approach to the vitamin D trans-hydrindane building block from the Hajos dione. Org. Lett. 2006, 8, 2551–2553. [Google Scholar] [CrossRef] [PubMed]
- Kotoku, N.; Tamada, N.; Hayashi, A.; Kobayashi, M. Synthesis of BC-ring model of globostellatic acid X methyl ester, an anti-angiogenic substance from marine sponge. Bioorg. Med. Chem. Lett. 2008, 18, 3532–3535. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Iso, K.; Hirama, M. A concise synthesis of the pentacyclic framework of cortistatins. Org. Lett. 2008, 10, 3414–3415. [Google Scholar] [CrossRef] [PubMed]
- Frie, J.L.; Jeffrey, C.S.; Sorensen, E.J. A hypervalent iodine-induced double annulation enables a concise synthesis of the pentacyclic core structure of the cortistatins. Org. Lett. 2009, 11, 5394–5397. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Peng, X.-S.; Sun, Y.-P.; Polet, D.; Zou, B.; Lim, C.S.; Chen, D.Y.-K. Total synthesis and biological evaluation of cortistatins A and J and analogues thereof. J. Am. Chem. Soc. 2009, 131, 10587–10597. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.C.; Ndungu, J.M.; Sarpong, R. Parallel kinetic resolution approach to the cyathane and cyanthiwigin diterpenes using a cyclopropanation/Cope rearrangement. Angew. Chem. Int. Ed. 2008, 48, 2398–2402. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.H.; Angeles, A.R.; Lee, J.H.; Danishefsky, S.J. Concise synthesis of the xenibellols core. Tetrahedron Lett. 2009, 50, 6440–6441. [Google Scholar] [CrossRef] [PubMed]
- Defaut, B.; Parsons, T.B.; Spencer, N.; Male, L.; Kariuki, B.M.; Grainger, R.S. Synthesis of the trans-hydrindane core of dictyoxetane. Org. Biomol. Chem. 2012, 10, 4926–4932. [Google Scholar] [CrossRef] [PubMed]
- Hog, D.T.; Mayer, P.; Trauner, D. A unified approach to trans-hydrindane sesterterpenoids. J. Org. Chem. 2012, 77, 5838–5843. [Google Scholar] [CrossRef] [PubMed]
- Hog, D.T.; Huber, F.M.E.; Jimenez-Oses, G.; Mayer, P.; Houk, K.N.; Trauner, D. Evolution of a unified strategy for complex sesterterpenoids: Progress toward astellatol and the total synthesis of (−)-nitidasin. Chem. Eur. J. 2015, 21, 13646–13665. [Google Scholar] [CrossRef] [PubMed]
- Minato, D.; Li, B.; Zhou, D.; Shigeta, Y.; Toyooka, N.; Sakurai, H.; Sugimoto, K.; Nemoto, H.; Matsuya, Y. Synthesis and antitumor activity of des-AB analogue of steroidal saponin OSW-1. Tetrahedron 2013, 69, 8019–8024. [Google Scholar] [CrossRef]
- Mehta, G.; Yaragorla, S. A concise, enantiospecific, Hajos–Parrish ketone based model approach toward the tetracyclic core of complex shiartane-type nortriterpenoid natural products. Tetrahedron Lett. 2013, 54, 549–552. [Google Scholar] [CrossRef]
- Fallis, A.G. The intramolecular Diels-Alder reaction: Recent advances and synthetic applications. Can. J. Chem. 1984, 62, 183–234. [Google Scholar] [CrossRef]
- Takao, K.; Munakata, R.; Tadano, K. Recent advances in natural product synthesis by using intramolecular Diels-Alder reactions. Chem. Rev. 2005, 105, 4779–4807. [Google Scholar] [CrossRef] [PubMed]
- Bear, B.R.; Sparks, S.M.; Shea, K.J. The type 2 intramolecular Diels-Alder reaction: Synthesis and chemistry of bridgehead alkenes. Angew. Chem. Int. Ed. 2001, 40, 820–849. [Google Scholar] [CrossRef]
- Juhl, M.; Tanner, D. Recent applications of intramolecular Diels-Alder reactions to natural products. Chem. Soc. Rev. 2009, 38, 2983–2992. [Google Scholar] [CrossRef] [PubMed]
- Reber, K.P.; Tilley, S.D.; Sorenson, E.J. Bond formations by intermolecular and intramolecular trappings of acetylketenes and their application to organic synthesis. Chem. Soc. Rev. 2009, 38, 3022–3034. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, R.A.; Okamura, W.H. A short enantioselective synthesis of (+)-sterpurene: Complete intramolecular transfer of central to axial to central chiral elements. J. Am. Chem. Soc. 1988, 110, 4062–4063. [Google Scholar] [CrossRef]
- Gibbs, R.A.; Bartles, K.; Lee, R.W.K.; Okamura, W.H. An enantioselective central-axial-central chiral element transfer process leading to a concise synthesis of (+)-sterpurene: Intramolecular Diels-Alder reactions of vinylallene sulfoxides. J. Am. Chem. Soc. 1989, 111, 3717–3725. [Google Scholar] [CrossRef]
- Urabe, H.; Kusaka, K.; Suzuki, D.; Sato, F. Chiral 1,3-butadiene-2-carboxylates for an efficient asymmetric Diels-Alder reaction. Tetrahedron Lett. 2002, 43, 285–289. [Google Scholar] [CrossRef]
- Chang, J.; Paquette, L.A. Studies aimed at the total synthesis of the antitumor antibiotic cochleamycin A. An enantioselective biosynthesis-based pathway to the AB cyclic core. Org. Lett. 2002, 4, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Ghorai, B.K.; Herndon, J.W.; Lam, Y.-F. One-step convergent synthesis of the steroid ring system via the coupling of g,d-unsaturated Fischer carbene complexes with o-ethynylbenzaldehyde. Org. Lett. 2001, 3, 3535–3538. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.F.; Yadav, R.N.; Mondal, S.; Jana, A.; Ghosh, S. Intramolecular Diels-Alder route to angularly oxygenated hydrindanes. Synthesis of the functionalized bicyclic skeleton present in galiellalactone. Tetrahedron 2013, 69, 7956–7963. [Google Scholar] [CrossRef]
- Constantino, M.G.; de Oliveira, K.T.; Polo, E.C.; da Silva, G.V.J.; Brocksom, T.J. Core structure of eremophilanes and bakkanes through niobium catalyzed Diels-Alder reaction: Synthesis of (+/−)-bakkenolide. A J. Org. Chem. 2006, 71, 9880–9883. [Google Scholar] [CrossRef] [PubMed]
- Back, T.G.; Nava-Salgado, V.O.; Payne, J.E. Synthesis of (+/−)-bakkenolide A and its C-7, C-10, and C-7,10 epimers by means of an intramolecular Diels-Alder reaction. J. Org. Chem. 2001, 66, 4361–4368. [Google Scholar] [CrossRef] [PubMed]
- Handore, K.L.; Seetharamsingh, B.; Reddy, D.S. Ready access to functionally embellished cis-hydrindanes and cis-decalins: Protecting group-free total syntheses of (+/−)-nootkatone and (+/−)-noreremophilane. J. Org. Chem. 2013, 78, 8149–8154. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Gray, D.L.F.; Tae, J. Total Synthesis of Hamigerans and analogues thereof. Photochemical generation and Diels-Alder trapping of hydroxy-o-quinodimethanes. J. Am. Chem. Soc. 2004, 126, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Zhu, L.; Gao, Y.; Lee, C.-S. A highly convergent cascade cyclization to cis-hydrindanes with all-carbon quaternary centers and its application in the synthesis of the aglycon of dendronobiloside. A Org. Lett. 2011, 13, 588–591. [Google Scholar] [CrossRef] [PubMed]
- Stork, G.; Nakamura, E. A simplified total synthesis of cytochalasins via an intramolecular Diels-Alder reaction. J. Am. Chem. Soc. 1983, 105, 5510–5512. [Google Scholar] [CrossRef]
- Hudlicky, T.; Fleming, A.; Radesca, L. [2+3] and [3+4] Annulation of enones. Enantiocontrolled total synthesis of (−)-retigeranic acid. J. Am. Chem. Soc. 1989, 111, 6691–6707. [Google Scholar] [CrossRef]
- Uenishi, J.; Kawahara, R.; Yonemitsu, O. Total synthesis of (−)-ircinianin and (+)-wistarin. J. Org. Chem. 1997, 62, 1691–1701. [Google Scholar] [CrossRef]
- Evans, D.A.; Johnson, J.S. Chiral C2 symmetric Cu(II) complexes as catalysts for enantioselective intramolecular Diels-Alder reactions. Asymmetric synthesis of (−)-isopulo'upone. J. Org. Chem. 1997, 62, 786–787. [Google Scholar] [CrossRef]
- Vosburg, D.A.; Vanderwal, C.D.; Sorenson, E.J. A synthesis of (+)-FR182877, featuring tandem transannular Diels-Alder reactions inspired by a postulated biogenesis. J. Am. Chem. Soc. 2002, 124, 4552–4553. [Google Scholar] [CrossRef] [PubMed]
- Heckrodt, T.J.; Mulzer, J. Total synthesis of elisabethinin A: Intramolecular Diels-Alder reaction under biomimetic conditions. J. Am. Chem. Soc. 2003, 125, 4680–4681. [Google Scholar] [CrossRef] [PubMed]
- Shiina, J.; Nishiyama, S. Intramolecular Diels-Alder reaction leading to tricyclic derivatives as intermediates of natural product synthesis. Tetrahedron 2003, 59, 6039–6044. [Google Scholar] [CrossRef]
- Shiina, J.; Nishiyama, S. A new approach to the bicyclo[4.3.0] ring system of natural products from the liverwort: Total synthesis of (+/−)-chiloscyphone and (+/−)-isochiloscyphone. Tetrahedron Lett. 2004, 45, 9033–9036. [Google Scholar] [CrossRef]
- Akai, S.; Tanimoto, K.; Kita, Y. Lipase-catalyzed domino kinetic resolution of racemic 3-vinylcyclohex-2-en-1-ols/intramolecular Diels-Alder reaction: One-pot synthesis of optically active polysubstituted decalins. Angew. Chem. 2004, 116, 1431–1434. [Google Scholar] [CrossRef]
- Zapf, C.W.; Harrison, B.A.; Drahl, C.; Sorenson, E.J. A Diels-Alder macrocyclizatioin enables an efficient asymmetric synthesis of the antibacterial natural product abyssomicin C. Angew. Chem. 2005, 117, 6691–6695. [Google Scholar] [CrossRef]
- Crimmins, M.T.; Brown, B.H. An intramolecular Diels-Alder approach to the eunicelins: Enantioselective total synthesis of ophirin B. J. Am. Chem. Soc. 2004, 126, 10264–10266. [Google Scholar] [CrossRef] [PubMed]
- Nelson, H.M.; Stoltz, B.M. Progress toward the synthesis of the basiliolides and transtaganolides: An intramolecular pyrone Diels-Alder entry into a novel class of natural products. Org. Lett. 2008, 10, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Ghosh, S. Olefin metathesis—Application in the synthesis of natural products and related organic compounds. Proc. Indian Nat. Sci. Acad. 2014, 80, 37–54. [Google Scholar] [CrossRef]
- Kurteva, V.B.; Afonso, C.A.M. Synthesis of cyclopentitols by ring-closing approaches. Chem. Rev. 2009, 109, 6809–6857. [Google Scholar] [CrossRef] [PubMed]
- Minger, T.L.; Phillips, A.J. Ring-opening-ring-closing metathesis of bicyclo[2.2.2]octenes: A novel synthesis of decalins and hydrindanes. Tetrahedron Lett. 2002, 43, 5357–5359. [Google Scholar] [CrossRef]
- Holtsclaw, J.; Koreeda, M. A ring-rearrangement metathesis approach toward the synthesis of cyclopenta- and cyclohexa[c]indene systems. Org. Lett. 2004, 6, 3719–3722. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, M.W.B.; Phillips, A.J. Total synthesis of (+)-cyanthiwigin U. J. Am. Chem. Soc. 2005, 127, 5334–5335. [Google Scholar] [CrossRef] [PubMed]
- Ramharter, J.; Mulzer, J. Total synthesis of valerenic acid, a potent GABA receptor modulator. Org. Lett. 2009, 11, 1151–1153. [Google Scholar] [CrossRef] [PubMed]
- Foucher, V.; Guizzardi, B.; Groen, M.B.; Light, M.; Linclau, B. A novel, versatile D → BCD steroid construction strategy, illustrated by the enantioselective total synthesis of estrone. Org. Lett. 2010, 12, 680–683. [Google Scholar] [CrossRef] [PubMed]
- Granger, K.; Snapper, M.L. Concise synthesis of norrisolide. Eur. J. Org. Chem. 2012. [Google Scholar] [CrossRef]
- Trost, B.M.; Krische, M.J. Transition metal catalyzed cycloisomerizations. Synlett 1998. [Google Scholar] [CrossRef]
- Semeril, D.; Bruneau, C.; Dixneuf, P.H. Imidazolium and imidazolinium salts as carbene precursors or solvent for ruthenium catalyzed diene and enyne metathesis. Adv. Synth. Catal. 2002, 344, 585–595. [Google Scholar] [CrossRef]
- Poulsen, C.S.; Madsen, R. Enyne metathesis catalyzed by ruthenium carbene complexes. Synthesis 2003. [Google Scholar] [CrossRef]
- Diver, S.T.; Giessert, A.J. Enyne metathesis (enyne bond reorganization). Chem. Rev. 2004, 104, 1317–1382. [Google Scholar] [CrossRef] [PubMed]
- Mori, M. Synthesis of natural products and related compounds using enyne metathesis. Adv. Synth. Catal. 2007, 349, 121–135. [Google Scholar] [CrossRef]
- Hato, Y.; Oonishi, Y.; Yamamoto, Y.; Nakajima, K.; Sato, Y. Stereselective construction of spiro-fused tricyclic frameworks by sequential reaction of enynes, imines, and diazoalkenes with Rh(I) and Rh(II) catalysts. J. Org. Chem. 2016, 81, 7847–7854. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Oberhuber, C.; Lopez, S.; Echavarren, A.M. Intramolecular [4+2] cycloadditions of 1,3-enynes of arylalkynes with alkenes with highly reactive cationic phosphine Au(I) complexes. J. Am. Chem. Soc. 2005, 127, 6178–6179. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Oberhuber, C.; Perez-Galan, P.; Herrero-Gomez, E.; Lauterbach, T.; Rodriguez, C.; Lopez, S.; Bour, C.; Rosellon, A.; Cardenas, D.J.; Echavarren, A.M. Gold(I)-catalyzed intramolecular [4+2] cycloadditions of arylalkynes or 1,3-enynes with alkenes: Scope and mechanism. J. Am. Chem. Soc. 2008, 130, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Trost, M.K. Intramolecular enyne metathesis reaction. Route to bridged bicycles with bridgehead olefins. J. Am. Chem. Soc. 1991, 113, 1850–1852. [Google Scholar] [CrossRef]
- Trost, B.M.; Yanai, M.; Hoogsteen, K. A Pd-catalyzed [2+2] cycloaddition. Mechanism of a Pd-catalyzed enyne metathesis. J. Am. Chem. Soc. 1993, 115, 5294–5295. [Google Scholar] [CrossRef]
- Monnier, F.; Bray, C.V.-L.; Castillo, D.; Aubert, V.; Derien, S.; Dixneuf, P.H.; Toupet, L.; Ienco, A.; Mealli, C. Selective ruthenium-catalyzed transformations of enynes with diazoalkanes into alkenylbicyclo[3.1.0]hexanes. J. Am. Chem. Soc. 2007, 129, 6037–6049. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Yu, S.; Gu, Z. Gold-catalyzed cyclization of enynes. Angew. Chem. Int. Ed. 2006, 45, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.; Munoz, M.P.; Nevado, C.; Cardenas, D.J.; Echavarren, A.M. Cyclizations of enynes catalyzed by PtCl2 or other transition metal chlorides: Divergent reaction pathways. J. Am. Chem. Soc. 2001, 123, 10511–10520. [Google Scholar] [CrossRef] [PubMed]
- Furstner, A.; Szillat, H.; Gabor, B.; Mynott, R. Platinum- and acid-catalyzed enyne metathesis reactions: Mechanistic studies and applications to the syntheses of streptorubin B and metacycloprodigiosin. J. Am. Chem. Soc. 1998, 120, 8305–8314. [Google Scholar] [CrossRef]
- Mori, M.; Sakakibara, N.; Kinoshita, A. Remarkable effect of ethylene gas in the intramolecular enyne metathesis of terminal alkynes. J. Org. Chem. 1998, 63, 6082–6083. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, A.; Mori, M. Total synthesis of (−)-stemoamide using ruthenium-catalyzed enyne metathesis reaction. J. Org. Chem. 1996, 61, 8356–8357. [Google Scholar] [CrossRef]
- Mendez, M.; Munoz, M.P.; Echavarren, A.M. Platinum-catalyzed alkoxy- and hydroxycyclization of enynes. J. Am. Chem. Soc. 2000, 122, 11549–11550. [Google Scholar] [CrossRef]
- Schramm, M.P.; Reddy, D.S.; Kozmin, S.A. Siloxyalkyne-alkene metathesis: Rapid access to highly functionalized enones. Angew. Chem. Int. Ed. 2001, 40, 4274–4277. [Google Scholar] [CrossRef]
- Mezailles, N.; Ricard, L.; Gagosz, F. Phosphine gold(I) bis-(trifluoromethanesulfonyl)imidate complexes as new highly efficient and air-stable catalysts for the cycloisomerization of enynes. Org. Lett. 2005, 7, 4133–4136. [Google Scholar] [CrossRef] [PubMed]
- Betkekar, V.V.; Panda, S.; Kaliappan, K.P. A tandem enyne/ring closing metathesis approach to 4-methylene-2-cyclohexenols: An efficient entry to otteliones and loloanolides. Org. Lett. 2012, 14, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Zuercher, W.J.; Scholl, M.; Grubbs, R.H. Ruthenium-catalyzed polycyclization reactions. J. Org. Chem. 1998, 63, 4291–4298. [Google Scholar] [CrossRef]
- Stork, G.; Shiner, C.S.; Winkler, J.D. Stereochemical control of the internal Michael reaction. A new construction of trans-hydrindane systems. J. Am. Chem. Soc. 1982, 104, 310–312. [Google Scholar] [CrossRef]
- Taber, D.F.; Malcolm, S.C. Synthesis of (−)-astrogorgiadiol. J. Org. Chem. 2001, 66, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Gorobets, E.; Stepanenko, V.; Wicha, J. Enantioselective total synthesis of a trans-hydrindane rings/side-chain building-block of vitamin D—Asymmetric induction an an acid-catalyzed conjugate-addition reaction. Eur. J. Org. Chem. 2004. [Google Scholar] [CrossRef]
- Danheiser, R.L.; Carini, D.J.; Basak, A. (Trimethylsilyl)cyclopentene annulation: A regiocontrolled approach to the synthesis of five-membered rings. J. Am. Chem. Soc. 1981, 103, 1604–1606. [Google Scholar] [CrossRef]
- Fleming, F.F.; Vu, V.A.; Shook, B.C.; Rahman, M.; Steward, O.W. Metalated nitriles: Chelation-controlled cyclizations to cis and trans-hydrindanes and decalins. J. Org. Chem. 2007, 72, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Fleming, F.F.; Zhang, Z.; Wang, Q.; Steward, O.W. Cyclic alkenenitriles: Synthesis, conjugate addition, and stereoselective annulation. J. Org. Chem. 2003, 68, 7646–7650. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.E.; Sun, J.; Fu, G.C. Stereoselective phosphine-catalyzed synthesis of highly functionalized diquinanes. Angew. Chem. Int. Ed. 2010, 49, 161–163. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jaunet, A.; Geoffroy, P.; Miesch, M. Phosphine-catalyzed reactions of activated olefins tethered to cycloalkanones. Substrate- and solvent-controlled synthesis of bicyclo[3.2.1]octanones, mixed acetals, and Morita-Baylis-Hillman products. Org. Lett. 2013, 15, 6198–6201. [Google Scholar] [CrossRef] [PubMed]
- Ressault, B.; Jaunet, A.; Geoffroy, P.; Goudedranche, S.; Miesch, M. Access to polyfunctionalized diquinanes, hyrindanes, and decalines via TiCl4 promoted Michael-Aldol and Baylis-Hillman reactions. Org. Lett. 2012, 14, 366–369. [Google Scholar] [CrossRef] [PubMed]




























© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eddy, N.A.; Ichalkaranje, P. Methodology for the Construction of the Bicyclo[4.3.0]nonane Core. Molecules 2016, 21, 1358. https://doi.org/10.3390/molecules21101358
Eddy NA, Ichalkaranje P. Methodology for the Construction of the Bicyclo[4.3.0]nonane Core. Molecules. 2016; 21(10):1358. https://doi.org/10.3390/molecules21101358
Chicago/Turabian StyleEddy, Nicholas A., and Pranjali Ichalkaranje. 2016. "Methodology for the Construction of the Bicyclo[4.3.0]nonane Core" Molecules 21, no. 10: 1358. https://doi.org/10.3390/molecules21101358
APA StyleEddy, N. A., & Ichalkaranje, P. (2016). Methodology for the Construction of the Bicyclo[4.3.0]nonane Core. Molecules, 21(10), 1358. https://doi.org/10.3390/molecules21101358