
Development and Applications of the Copper-Catalyzed Azide-Alkyne Cycloaddition (CuAAC) as a Bioorthogonal Reaction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
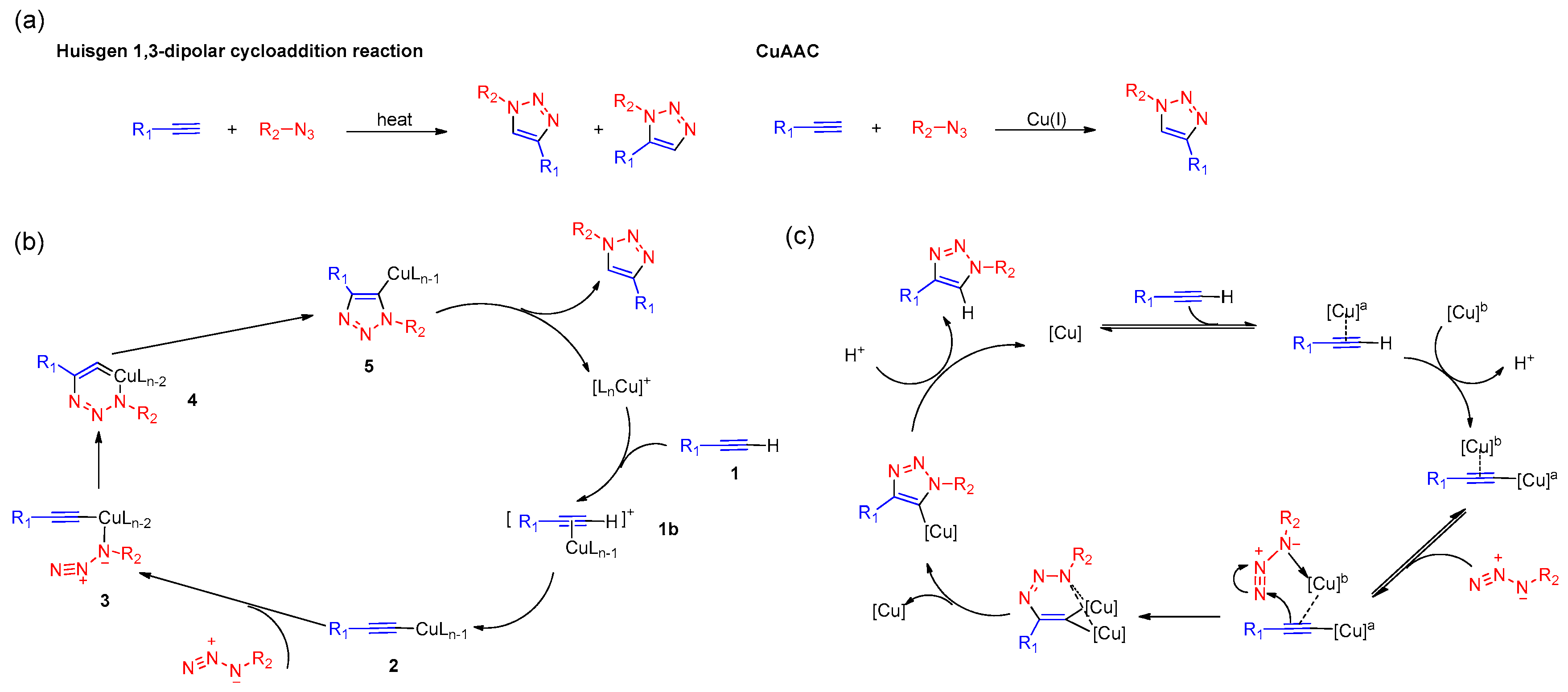
2. History of CuAAC
3. Mechanistic Studies of CuAAC
4. Early Bioorthogonal Applications of CuAAC and Its Limitations
5. Ligand-Assisted CuAAC and Its Applications as a Bioorthogonal Reaction
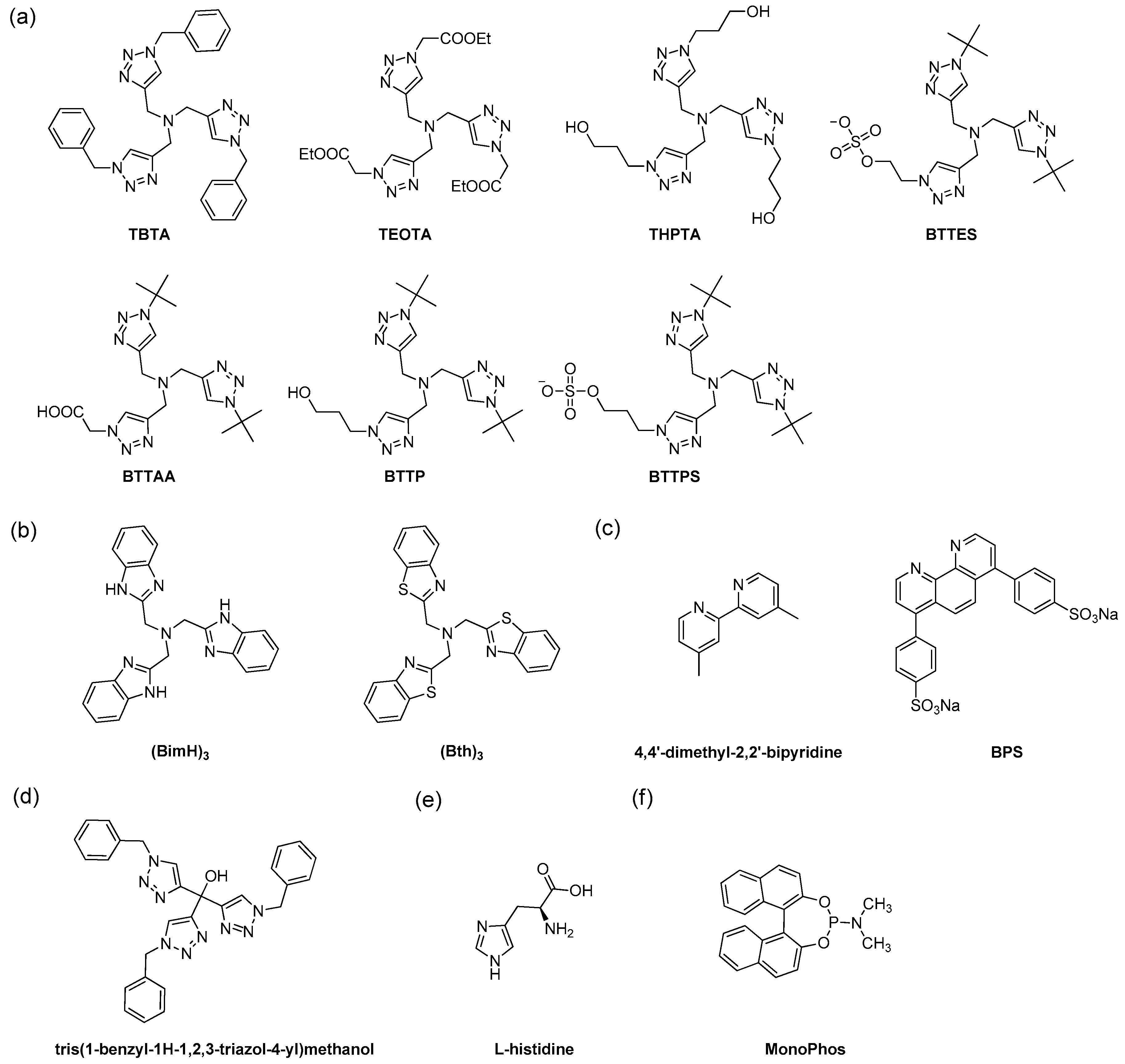
5.1. Development of Different Classes of Ligands
5.2. Kinetic and Mechanism Studies of Ligand Assisted CuAAC
5.3. Biocompatible Ligands
5.4. Bioorthogonal Applications of Ligand Assisted CuAAC
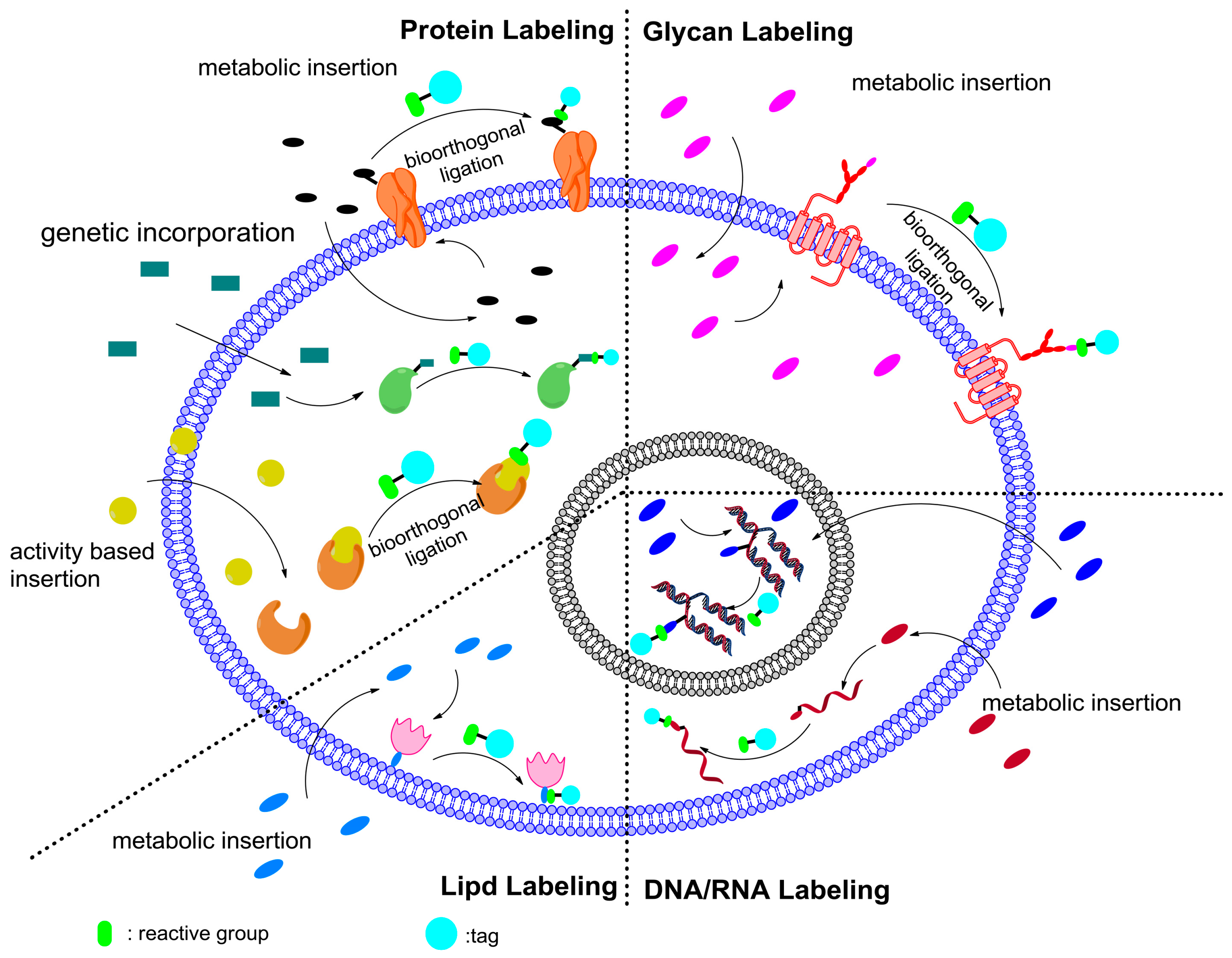
5.4.1. Protein Labeling
5.4.2. Glycan Labeling
5.4.3. Lipid Labeling
5.4.4. Nucleic Acid Labeling
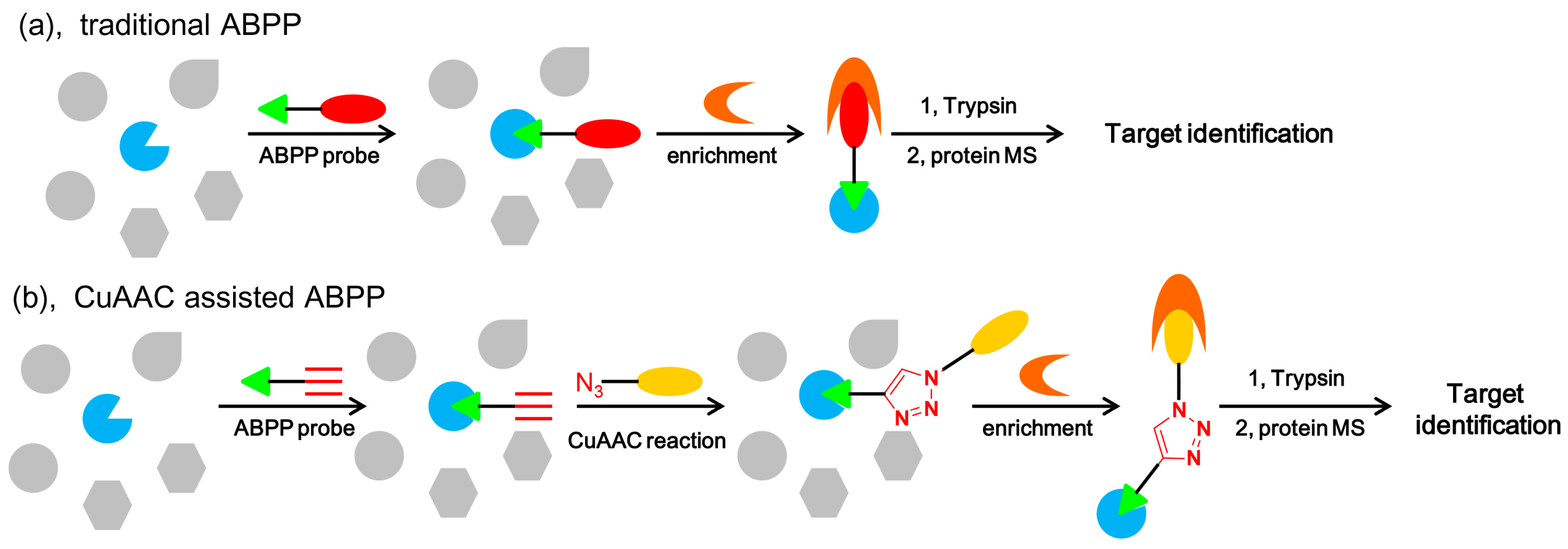
5.4.5. Activity-Based Protein Profiling
5.4.6. Protein-Protein Coupling
6. Chelation-Assisted CuAAC and Its Applications as a Bioorthogonal Reaction
6.1. Development of Chelation-Assisted CuAAC
6.2. Bioorthogonal Applications of Chelation Assisted CuAAC
7. Strain-Promoted Azide-Alkyne Cycloaddition (SPAAC)
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tsien, R.Y. The green fluorescent protein. Ann. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef] [PubMed]
- Lippincott-Schwartz, J.; Patterson, G.H. Development and use of fluorescent protein markers in living cells. Science 2003, 300, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Giepmans, B.N.; Adams, S.R.; Ellisman, M.H.; Tsien, R.Y. The fluorescent toolbox for assessing protein location and function. Science 2006, 312, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Prescher, J.A.; Bertozzi, C.R. Chemistry in living systems. Nat. Chem. Biol. 2005, 1, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D.M.; Nazarova, L.A.; Prescher, J.A. Finding the right (bioorthogonal) chemistry. ACS Chem. Biol. 2014, 9, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, J.; Chen, P.R. Transition metal-mediated bioorthogonal protein chemistry in living cells. Chem. Soc. Rev. 2014, 43, 6511–6526. [Google Scholar] [CrossRef] [PubMed]
- Ramil, C.P.; Lin, Q. Bioorthogonal chemistry: Strategies and recent developments. Chem. Commun. 2013, 49, 11007–11022. [Google Scholar] [CrossRef] [PubMed]
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. 2009, 48, 6974–6998. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.; Chin, J.W. Bioorthogonal reactions for labeling proteins. ACS Chem. Biol. 2014, 9, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Hoyle, C.E.; Bowman, C.N. Thiol-ene click chemistry. Angew. Chem. Int. Ed. 2010, 49, 1540–1573. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-X.; Triola, G.; Waldmann, H. Bioorthogonal chemistry for site-specific labeling and surface immobilization of proteins. Acc. Chem. Res. 2011, 44, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Mahal, L.K.; Yarema, K.J.; Bertozzi, C.R. Engineering chemical reactivity on cell surfaces through oligosaccharide biosynthesis. Science 1997, 276, 1125–1128. [Google Scholar] [CrossRef] [PubMed]
- Jencks, W.P. Studies on the mechanism of oxime and semicarbazone formation1. J. Am. Chem. Soc. 1959, 81, 475–481. [Google Scholar] [CrossRef]
- Yarema, K.J.; Mahal, L.K.; Bruehl, R.E.; Rodriguez, E.C.; Bertozzi, C.R. Metabolic delivery of ketone groups to sialic acid residues application to cell surface glycoform engineering. J. Biol. Chem. 1998, 273, 31168–31179. [Google Scholar] [CrossRef] [PubMed]
- Hudak, J.E.; Yu, H.H.; Bertozzi, C.R. Protein glycoengineering enabled by the versatile synthesis of aminooxy glycans and the genetically encoded aldehyde tag. J. Am. Chem. Soc. 2011, 133, 16127–16135. [Google Scholar] [CrossRef] [PubMed]
- Nauman, D.A.; Bertozzi, C.R. Kinetic parameters for small-molecule drug delivery by covalent cell surface targeting. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2001, 1568, 147–154. [Google Scholar] [CrossRef]
- Saxon, E.; Bertozzi, C.R. Cell surface engineering by a modified staudinger reaction. Science 2000, 287, 2007–2010. [Google Scholar] [CrossRef] [PubMed]
- Vocadlo, D.J.; Hang, H.C.; Kim, E.-J.; Hanover, J.A.; Bertozzi, C.R. A chemical approach for identifying O-GlcNac-modified proteins in cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9116–9121. [Google Scholar] [CrossRef] [PubMed]
- Prescher, J.A.; Dube, D.H.; Bertozzi, C.R. Chemical remodelling of cell surfaces in living animals. Nature 2004, 430, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, T.; Ishii, R.; Fukunaga, R.; Kobayashi, T.; Sakamoto, K.; Yokoyama, S. Multistep engineering of pyrrolysyl-tRNA synthetase to genetically encode N ɛ-(O-azidobenzyloxycarbonyl) lysine for site-specific protein modification. Chem. Biol. 2008, 15, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Tsao, M.L.; Tian, F.; Schultz, P.G. Selective staudinger modification of proteins containing p-azidophenylalanine. ChemBioChem 2005, 6, 2147–2149. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.L.; Hoyt, H.M.; Van Halbeek, H.; Bergman, R.G.; Bertozzi, C.R. Mechanistic investigation of the staudinger ligation. J. Am. Chem. Soc. 2005, 127, 2686–2695. [Google Scholar] [CrossRef] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper (i)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. 2002, 114, 2708–2711. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase:[1,2,3]-triazoles by regiospecific copper (i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Appukkuttan, P.; Dehaen, W.; Fokin, V.V.; van der Eycken, E. A microwave-assisted click chemistry synthesis of 1,4-disubstituted 1,2,3-triazoles via a copper (i)-catalyzed three-component reaction. Org. Lett. 2004, 6, 4223–4225. [Google Scholar] [CrossRef] [PubMed]
- Blackman, M.L.; Royzen, M.; Fox, J.M. Tetrazine ligation: Fast bioconjugation based on inverse-electron-demand diels-alder reactivity. J. Am. Chem. Soc. 2008, 130, 13518–13519. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.C.; McKay, C.S.; Legault, M.C.; Danielson, D.C.; Blake, J.A.; Pegoraro, A.F.; Stolow, A.; Mester, Z.; Pezacki, J.P. Cellular consequences of copper complexes used to catalyze bioorthogonal click reactions. J. Am. Chem. Soc. 2011, 133, 17993–18001. [Google Scholar] [CrossRef] [PubMed]
- Huisgen, R. 1,3-dipolar cycloadditions. Past and future. Angew. Chem. Int. Edit. 1963, 2, 565–598. [Google Scholar] [CrossRef]
- Repasky, M.P.; Jorgensen, W.L. Ab initio and monte carlo study of solvent eÜects on a 1,3–dipolar cycloaddition. Faraday Discuss. 1998, 110, 379–389. [Google Scholar] [CrossRef]
- Booth, C.A.; Philp, D. Efficient recognition-induced acceleration of a [3 + 2] dipolar cycloaddition reaction. Tetrahedron Lett. 1998, 39, 6987–6990. [Google Scholar] [CrossRef]
- Howell, S.J.; Spencer, N.; Philp, D. Recognition-mediated regiocontrol of a dipolar cycloaddition reaction. Tetrahedron 2001, 57, 4945–4954. [Google Scholar] [CrossRef]
- Chen, J.; Rebek, J. Selectivity in an encapsulated cycloaddition reaction. Org. Lett. 2002, 4, 327–329. [Google Scholar] [CrossRef] [PubMed]
- Bock, V.D.; Hiemstra, H.; Van Maarseveen, J.H. Cui-catalyzed alkyne-azide “click” cycloadditions from a mechanistic and synthetic perspective. Eur. J. Org. Chem. 2006, 2006, 51–68. [Google Scholar] [CrossRef]
- Meldal, M.; Tornøe, C.W. Cu-catalyzed azide-alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef] [PubMed]
- Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V.V.; Noodleman, L.; Sharpless, K.B.; Fokin, V.V. Copper(i)-catalyzed synthesis of azoles. Dft study predicts unprecedented reactivity and intermediates. J. Am. Chem. Soc. 2005, 127, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, V.O.; Fokin, V.V.; Finn, M. Mechanism of the ligand-free cui-catalyzed azide-alkyne cycloaddition reaction. Angew. Chem. 2005, 117, 2250–2255. [Google Scholar] [CrossRef]
- Worrell, B.; Malik, J.; Fokin, V. Direct evidence of a dinuclear copper intermediate in Cu (i)-catalyzed azide-alkyne cycloadditions. Science 2013, 340, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Özkılıç, Y.; Tüzün, N.S. A dft study on the binuclear CUAAC reaction: Mechanism in light of new experiments. Organometallics 2016, 35, 2589–2599. [Google Scholar] [CrossRef]
- Haldon, E.; Nicasio, M.C.; Pérez, P.J. Copper-catalysed azide-alkyne cycloadditions (CUAAC): An update. Org. Biomol. Chem. 2015, 13, 9528–9550. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.V.; Mitchell, M.L.; Huang, S.-J.; Fokin, V.V.; Sharpless, K.B.; Wong, C.-H. A potent and highly selective inhibitor of human α-1,3-fucosyltransferase via click chemistry. J. Am. Chem. Soc. 2003, 125, 9588–9589. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Lutz, J.F. 1,3-dipolar cycloadditions of azides and alkynes: A universal ligation tool in polymer and materials science. Angew. Chem. Int. Ed. 2007, 46, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Breinbauer, R.; Köhn, M. Azide-alkyne coupling: A powerful reaction for Bioconjugate Chemistry. ChemBioChem 2003, 4, 1147–1149. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chan, T.R.; Hilgraf, R.; Fokin, V.V.; Sharpless, K.B.; Finn, M. Bioconjugation by copper (i)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003, 125, 3192–3193. [Google Scholar] [CrossRef] [PubMed]
- Link, A.J.; Tirrell, D.A. Cell surface labeling of escherichia c oli via copper (i)-catalyzed [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003, 125, 11164–11165. [Google Scholar] [CrossRef] [PubMed]
- Link, A.J.; Vink, M.K.; Tirrell, D.A. Presentation and detection of azide functionality in bacterial cell surface proteins. J. Am. Chem. Soc. 2004, 126, 10598–10602. [Google Scholar] [CrossRef] [PubMed]
- Deiters, A.; Cropp, T.A.; Mukherji, M.; Chin, J.W.; Anderson, J.C.; Schultz, P.G. Adding amino acids with novel reactivity to the genetic code of saccharomyces cerevisiae. J. Am. Chem. Soc. 2003, 125, 11782–11783. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Cravatt, B.F. Mechanism-based profiling of enzyme families. Chem. Rev. 2006, 106, 3279–3301. [Google Scholar] [CrossRef] [PubMed]
- Speers, A.E.; Adam, G.C.; Cravatt, B.F. Activity-based protein profiling in vivo using a copper (i)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003, 125, 4686–4687. [Google Scholar] [CrossRef] [PubMed]
- Speers, A.E.; Cravatt, B.F. Profiling enzyme activities in vivo using click chemistry methods. Chem. Biol. 2004, 11, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Soares, E.V.; Hebbelinck, K.; Soares, H.M. Toxic effects caused by heavy metals in the yeast saccharomyces cerevisiae: A comparative study. Can. J. Microbiol. 2003, 49, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J. Risks of copper and iron toxicity during aging in humans. Chem. Res. Toxicol. 2009, 23, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.R.; Hilgraf, R.; Sharpless, K.B.; Fokin, V.V. Polytriazoles as copper (i)-stabilizing ligands in catalysis. Org. Lett. 2004, 6, 2853–2855. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, P.S.; Zanatta, S.D.; Zammit, S.C.; White, J.M.; Williams, S.J. “Click” cycloaddition catalysts: Copper (i) and copper (ii) tris (triazolylmethyl) amine complexes. Chem. Commun. 2008, 2459–2461. [Google Scholar] [CrossRef] [PubMed]
- Hong, V.; Presolski, S.I.; Ma, C.; Finn, M. Analysis and optimization of copper-catalyzed azide-alkyne cycloaddition for bioconjugation. Angew. Chem. Int. Ed. 2009, 48, 9879–9883. [Google Scholar] [CrossRef] [PubMed]
- Besanceney-Webler, C.; Jiang, H.; Zheng, T.; Feng, L.; Soriano del Amo, D.; Wang, W.; Klivansky, L.M.; Marlow, F.L.; Liu, Y.; Wu, P. Increasing the efficacy of bioorthogonal click reactions for bioconjugation: A comparative study. Angew. Chem. Int. Ed. 2011, 50, 8051–8056. [Google Scholar] [CrossRef] [PubMed]
- Soriano del Amo, D.; Wang, W.; Jiang, H.; Besanceney, C.; Yan, A.C.; Levy, M.; Liu, Y.; Marlow, F.L.; Wu, P. Biocompatible copper (i) catalysts for in vivo imaging of glycans. J. Am. Chem. Soc. 2010, 132, 16893–16899. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Fahrni, C.J. A fluorogenic probe for the copper (i)-catalyzed azide-alkyne ligation reaction: Modulation of the fluorescence emission via 3 (n, π*)-1 (π, π*) inversion. J. Am. Chem. Soc. 2004, 126, 8862–8863. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Hong, S.; Tran, A.; Jiang, H.; Triano, R.; Liu, Y.; Chen, X.; Wu, P. Sulfated ligands for the copper (i)-catalyzed azide-alkyne cycloaddition. Chem. Asian J. 2011, 6, 2796–2802. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, V.O.; Presolski, S.I.; Gardinier, S.; Lim, Y.-H.; Finn, M. Benzimidazole and related ligands for Cu-catalyzed azide-alkyne cycloaddition. J. Am. Chem. Soc. 2007, 129, 12696–12704. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, V.O.; Presolski, S.I.; Díaz Díaz, D.; Fokin, V.V.; Finn, M. Ligand-accelerated Cu-catalyzed azide-alkyne cycloaddition: A mechanistic report. J. Am. Chem. Soc. 2007, 129, 12705–12712. [Google Scholar] [CrossRef] [PubMed]
- Presolski, S.I.; Hong, V.; Cho, S.-H.; Finn, M. Tailored ligand acceleration of the cu-catalyzed azide-alkyne cycloaddition reaction: Practical and mechanistic implications. J. Am. Chem. Soc. 2010, 132, 14570–14576. [Google Scholar] [CrossRef] [PubMed]
- Lewis, W.G.; Magallon, F.G.; Fokin, V.V.; Finn, M. Discovery and characterization of catalysts for azide-alkyne cycloaddition by fluorescence quenching. J. Am. Chem. Soc. 2004, 126, 9152–9153. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.S.; Kuzelka, J.; Singh, P.; Lewis, W.G.; Manchester, M.; Finn, M. Accelerated bioorthogonal conjugation: A practical method for the ligation of diverse functional molecules to a polyvalent virus scaffold. Bioconj. Chem. 2005, 16, 1572–1579. [Google Scholar] [CrossRef] [PubMed]
- Özçubukçu, S.; Ozkal, E.; Jimeno, C.; Pericas, M.A. A highly active catalyst for huisgen 1,3-dipolar cycloadditions based on the tris (triazolyl) methanol-Cu(i) structure. Org. Lett. 2009, 11, 4680–4683. [Google Scholar] [CrossRef] [PubMed]
- Ozkal, E.; Özçubukçu, S.; Jimeno, C.; Pericas, M.A. Covalently immobilized tris-(triazolyl) methanol-Cu(i) complexes: Highly active and recyclable catalysts for cuaac reactions. Catal. Sci. Technol. 2012, 2, 195–200. [Google Scholar] [CrossRef]
- Campbell-Verduyn, L.S.; Mirfeizi, L.; Dierckx, R.A.; Elsinga, P.H.; Feringa, B.L. Phosphoramidite accelerated copper (i)-catalyzed [3 + 2] cycloadditions of azides and alkynes. Chem. Commun. 2009, 2139–2141. [Google Scholar] [CrossRef] [PubMed]
- Gonda, Z.; Novák, Z. Highly active copper-catalysts for azide-alkyne cycloaddition. Dalton Trans. 2010, 39, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Beatty, K.E.; Liu, J.C.; Xie, F.; Dieterich, D.C.; Schuman, E.M.; Wang, Q.; Tirrell, D.A. Fluorescence visualization of newly synthesized proteins in mammalian cells. Angew. Chem. 2006, 118, 7524–7527. [Google Scholar] [CrossRef]
- Beatty, K.E.; Xie, F.; Wang, Q.; Tirrell, D.A. Selective dye-labeling of newly synthesized proteins in bacterial cells. J. Am. Chem. Soc. 2005, 127, 14150–14151. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Zhang, Z.; Xu, H.; Li, L.; Chen, S.; Li, J.; Hao, Z.; Chen, P.R. Site-specific incorporation of photo-cross-linker and bioorthogonal amino acids into enteric bacterial pathogens. J. Am. Chem. Soc. 2011, 133, 20581–20587. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Song, Y.; Lin, S.; Yang, M.; Liang, Y.; Wang, J.; Chen, P.R. A readily synthesized cyclic pyrrolysine analogue for site-specific protein “click” labeling. Chem. Commun. 2011, 47, 4502–4504. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Fan, X.; Chen, P.R. A genetically encoded multifunctional unnatural amino acid for versatile protein manipulations in living cells. Chem. Sci. 2016. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, S.; Fast, W. A click chemistry mediated in vivo activity probe for dimethylarginine dimethylaminohydrolase. J. Am. Chem. Soc. 2009, 131, 15096–15097. [Google Scholar] [CrossRef] [PubMed]
- Garcia, F.J.; Carroll, K.S. Redox-based probes as tools to monitor oxidized protein tyrosine phosphatases in living cells. Eur. J. Med. Chem. 2014, 88, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.V.; Chen, X.; Smyrniotis, C.; Xenakis, A.; Hu, T.; Bertozzi, C.R.; Wu, P. Metabolic labeling of sialic acids in living animals with alkynyl sugars. Angew. Chem. Int. Ed. 2009, 48, 4030–4033. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Jiang, H.; Gros, M.; Soriano del Amo, D.; Sundaram, S.; Lauvau, G.; Marlow, F.; Liu, Y.; Stanley, P.; Wu, P. Tracking N-acetyllactosamine on cell-surface glycans in vivo. Angew. Chem. Int. Ed. 2011, 50, 4113–4118. [Google Scholar] [CrossRef] [PubMed]
- Hong, V.; Steinmetz, N.F.; Manchester, M.; Finn, M. Labeling live cells by copper-catalyzed alkyne-azide click chemistry. Bioconj. Chem. 2010, 21, 1912–1916. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Hong, S.; Rong, J.; You, Q.; Dai, P.; Huang, R.; Tan, Y.; Hong, W.; Xie, C.; Zhao, J. Bifunctional unnatural sialic acids for dual metabolic labeling of cell-surface sialylated glycans. J. Am. Chem. Soc. 2013, 135, 9244–9247. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Du, Y.; Zhu, Y.; Chen, X. A cis-membrane fret-based method for protein-specific imaging of cell-surface glycans. J. Am. Chem. Soc. 2014, 136, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Hanson, S.R.; Hsu, T.-L.; Weerapana, E.; Kishikawa, K.; Simon, G.M.; Cravatt, B.F.; Wong, C.-H. Tailored glycoproteomics and glycan site mapping using saccharide-selective bioorthogonal probes. J. Am. Chem. Soc. 2007, 129, 7266–7267. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.; Han, J.; Dong, L.; Tan, Y.; Yang, H.; Feng, L.; Wang, Q.-W.; Meng, R.; Zhao, J.; Wang, S.-Q. Glycan imaging in intact rat hearts and glycoproteomic analysis reveal the upregulation of sialylation during cardiac hypertrophy. J. Am. Chem. Soc. 2014, 136, 17468–17476. [Google Scholar] [CrossRef] [PubMed]
- Jao, C.Y.; Roth, M.; Welti, R.; Salic, A. Metabolic labeling and direct imaging of choline phospholipids in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 15332–15337. [Google Scholar] [CrossRef] [PubMed]
- Charron, G.; Zhang, M.M.; Yount, J.S.; Wilson, J.; Raghavan, A.S.; Shamir, E.; Hang, H.C. Robust fluorescent detection of protein fatty-acylation with chemical reporters. J. Am. Chem. Soc. 2009, 131, 4967–4975. [Google Scholar] [CrossRef] [PubMed]
- Rangan, K.J.; Yang, Y.-Y.; Charron, G.; Hang, H.C. Rapid visualization and large-scale profiling of bacterial lipoproteins with chemical reporters. J. Am. Chem. Soc. 2010, 132, 10628–10629. [Google Scholar] [CrossRef] [PubMed]
- Salic, A.; Mitchison, T.J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 2415–2420. [Google Scholar] [CrossRef] [PubMed]
- Neef, A.B.; Luedtke, N.W. Dynamic metabolic labeling of DNA in vivo with arabinosyl nucleosides. Proc. Natl. Acad. Sci. USA 2011, 108, 20404–20409. [Google Scholar] [CrossRef] [PubMed]
- Rao, H.; Sawant, A.A.; Tanpure, A.A.; Srivatsan, S.G. Posttranscriptional chemical functionalization of azide-modified oligoribonucleotides by bioorthogonal click and staudinger reactions. Chem. Commun. 2012, 48, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Rudd, P.M.; Elliott, T.; Cresswell, P.; Wilson, I.A.; Dwek, R.A. Glycosylation and the immune system. Science 2001, 291, 2370–2376. [Google Scholar] [CrossRef] [PubMed]
- Gouyer, V.; Leteurtre, E.; Zanetta, J.; Lesuffleur, T.; Delannoy, P.; Huet, G. Inhibition of the glycosylation and alteration in the intracellular trafficking of mucins and other glycoproteins by galnacalpha-O-bn in mucosal cell lines: An effect mediated through the intracellular synthesis of complex galnacalpha-O-bn oligosaccharides. Front. Biosci. 2001, 6, 1235–1244. [Google Scholar] [CrossRef]
- Wells, L.; Vosseller, K.; Hart, G.W. Glycosylation of nucleocytoplasmic proteins: Signal transduction and O-glcnac. Science 2001, 291, 2376–2378. [Google Scholar] [CrossRef] [PubMed]
- Slawson, C.; Hart, G.W. Dynamic interplay between O-glcnac and O-phosphate: The sweet side of protein regulation. Curr. Opin. Struct. Biol. 2003, 13, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Orntoft, T.F.; Vestergaard, E.M. Clinical aspects of altered glycosylation of glycoproteins in cancer. Electrophoresis 1999, 20, 362–371. [Google Scholar] [CrossRef]
- Lowe, J.B. Glycan-dependent leukocyte adhesion and recruitment in inflammation. Curr. Opin. Cell Biol. 2003, 15, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Sell, S. Cancer-associated carbohydrates identified by monoclonal antibodies. Hum. Pathol. 1990, 21, 1003–1019. [Google Scholar] [CrossRef]
- Jørgensen, T.; Berner, A.; Kaalhus, O.; Tveter, K.J.; Danielsen, H.E.; Bryne, M. Up-regulation of the oligosaccharide sialyl lewisx: A new prognostic parameter in metastatic prostate cancer. Cancer Res. 1995, 55, 1817–1819. [Google Scholar] [PubMed]
- Maier, O.; Oberle, V.; Hoekstra, D. Fluorescent lipid probes: Some properties and applications (a review). Chem. Phys. Lipids 2002, 116, 3–18. [Google Scholar] [CrossRef]
- Cravatt, B.F.; Wright, A.T.; Kozarich, J.W. Activity-based protein profiling: From enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem. 2008, 77, 383–414. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.; Sieber, S. Chemical proteomics approaches for identifying the cellular targets of natural products. Nat. Prod. Rep. 2016, 33, 681–708. [Google Scholar] [CrossRef] [PubMed]
- Staub, I.; Sieber, S.A. Β-lactams as selective chemical probes for the in vivo labeling of bacterial enzymes involved in cell wall biosynthesis, antibiotic resistance, and virulence. J. Am. Chem. Soc. 2008, 130, 13400–13409. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T.; Schmuck, K.; Tietze, L.F.; Sieber, S.A. Duocarmycin analogues target aldehyde dehydrogenase 1 in lung cancer cells. Angew. Chem. 2012, 124, 2928–2931. [Google Scholar] [CrossRef]
- Wang, G.; Han, T.; Nijhawan, D.; Theodoropoulos, P.; Naidoo, J.; Yadavalli, S.; Mirzaei, H.; Pieper, A.A.; Ready, J.M.; McKnight, S.L. P7c3 neuroprotective chemicals function by activating the rate-limiting enzyme in nad salvage. Cell 2014, 158, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zhang, C.-J.; Chen, G.Y.; Yao, S.Q. Cell-based proteome profiling of potential dasatinib targets by use of affinity-based probes. J. Am. Chem. Soc. 2012, 134, 3001–3014. [Google Scholar] [CrossRef] [PubMed]
- Schenone, M.; Dančík, V.; Wagner, B.K.; Clemons, P.A. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 2013, 9, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.I.; Kwon, I. Bioconjugation of therapeutic proteins and enzymes using the expanded set of genetically encoded amino acids. Crit. Rev. Biotechnol. 2015, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, X. Click to join peptides/proteins together. Chem. Asian J. 2011, 6, 2606–2616. [Google Scholar] [CrossRef]
- Xiao, J.; Tolbert, T.J. Synthesis of n-terminally linked protein dimers and trimers by a combined native chemical ligation-CUAAC click chemistry strategy. Org. Lett. 2009, 11, 4144–4147. [Google Scholar] [CrossRef] [PubMed]
- Foot, J.S.; Lui, F.E.; Kluger, R. Hemoglobin bis-tetramers via cooperative azide-alkyne coupling. Chem. Commun. 2009, 7315–7317. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Dubinsky-Davidchik, I.S.; Yang, Y.; Kluger, R. Subunit-directed click coupling via doubly cross-linked hemoglobin efficiently produces readily purified functional bis-tetrameric oxygen carriers. Org. Biomol. Chem. 2015, 13, 11118–11128. [Google Scholar] [CrossRef] [PubMed]
- Bundy, B.C.; Swartz, J.R. Site-specific incorporation of p-propargyloxyphenylalanine in a cell-free environment for direct protein-protein click conjugation. Bioconj. Chem. 2010, 21, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Brotherton, W.S.; Michaels, H.A.; Simmons, J.T.; Clark, R.J.; Dalal, N.S.; Zhu, L. Apparent copper (ii)-accelerated azide-alkyne cycloaddition. Org. Lett. 2009, 11, 4954–4957. [Google Scholar] [CrossRef] [PubMed]
- Kuang, G.-C.; Michaels, H.A.; Simmons, J.T.; Clark, R.J.; Zhu, L. Chelation-assisted, copper (ii)-acetate-accelerated azide-alkyne cycloaddition. J. Org. Chem. 2010, 75, 6540–6548. [Google Scholar] [CrossRef] [PubMed]
- Kuang, G.-C.; Guha, P.M.; Brotherton, W.S.; Simmons, J.T.; Stankee, L.A.; Nguyen, B.T.; Clark, R.J.; Zhu, L. Experimental investigation on the mechanism of chelation-assisted, copper (ii) acetate-accelerated azide-alkyne cycloaddition. J. Am. Chem. Soc. 2011, 133, 13984–14001. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, V.; King, M.; Chaumontet, M.; Nothisen, M.; Gabillet, S.; Buisson, D.; Puente, C.; Wagner, A.; Taran, F. Copper-chelating azides for efficient click conjugation reactions in complex media. Angew. Chem. Int. Ed. 2014, 53, 5872–5876. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Li, L.; Wang, H.; Wang, X.; Zhang, Z. All-in-one azides: Empowered click reaction for in vivo labeling and imaging of biomolecules. Chem. Commun. 2016, 52, 2185–2188. [Google Scholar] [CrossRef] [PubMed]
- Uttamapinant, C.; Tangpeerachaikul, A.; Grecian, S.; Clarke, S.; Singh, U.; Slade, P.; Gee, K.R.; Ting, A.Y. Fast, cell-compatible click chemistry with copper-chelating azides for biomolecular labeling. Angew. Chem. Int. Ed. 2012, 51, 5852–5856. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zheng, T.; Lopez-Aguilar, A.; Feng, L.; Kopp, F.; Marlow, F.L.; Wu, P. Monitoring dynamic glycosylation in vivo using supersensitive click chemistry. Bioconj. Chem. 2014, 25, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Agard, N.J.; Prescher, J.A.; Bertozzi, C.R. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 2004, 126, 15046–15047. [Google Scholar] [CrossRef] [PubMed]
- Agard, N.J.; Baskin, J.M.; Prescher, J.A.; Lo, A.; Bertozzi, C.R. A comparative study of bioorthogonal reactions with azides. ACS Chem. Biol. 2006, 1, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Codelli, J.A.; Baskin, J.M.; Agard, N.J.; Bertozzi, C.R. Second-generation difluorinated cyclooctynes for copper-free click chemistry. J. Am. Chem. Soc. 2008, 130, 11486–11493. [Google Scholar] [CrossRef] [PubMed]
- Sletten, E.M.; Bertozzi, C.R. A hydrophilic azacyclooctyne for Cu-free click chemistry. Org. Lett. 2008, 10, 3097–3099. [Google Scholar] [CrossRef] [PubMed]
- Varga, B.R.; Kállay, M.; Hegyi, K.; Béni, S.; Kele, P. A non-fluorinated monobenzocyclooctyne for rapid copper-free click reactions. Chem. Eur. J. 2012, 18, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.; Guo, J.; Wolfert, M.A.; Boons, G.J. Visualizing metabolically labeled glycoconjugates of living cells by copper-free and fast huisgen cycloadditions. Angew. Chem. Int. Ed. 2008, 47, 2253–2255. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, A.; Poloukhtine, A.; Wolfert, M.A.; Popik, V.V. Surface functionalization using catalyst-free azide-alkyne cycloaddition. Bioconj. Chem. 2010, 21, 2076–2085. [Google Scholar] [CrossRef] [PubMed]
- Debets, M.F.; van Berkel, S.S.; Schoffelen, S.; Rutjes, F.P.; van Hest, J.C.; van Delft, F.L. Aza-dibenzocyclooctynes for fast and efficient enzyme pegylation via copper-free [3 + 2] cycloaddition. Chem. Commun. 2010, 46, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Jewett, J.C.; Sletten, E.M.; Bertozzi, C.R. Rapid cu-free click chemistry with readily synthesized biarylazacyclooctynones. J. Am. Chem. Soc. 2010, 132, 3688–3690. [Google Scholar] [CrossRef] [PubMed]
- Dommerholt, J.; Schmidt, S.; Temming, R.; Hendriks, L.J.; Rutjes, F.P.; van Hest, J.; Lefeber, D.J.; Friedl, P.; van Delft, F.L. Readily accessible bicyclononynes for bioorthogonal labeling and three-dimensional imaging of living cells. Angew. Chem. Int. Ed. 2010, 49, 9422–9425. [Google Scholar] [CrossRef] [PubMed]
- Tummatorn, J.; Batsomboon, P.; Clark, R.J.; Alabugin, I.V.; Dudley, G.B. Strain-promoted azide-alkyne cycloadditions of benzocyclononynes. J. Org. Chem. 2012, 77, 2093–2097. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, G.; Sletten, E.M.; Nakamura, H.; Palaniappan, K.K.; Bertozzi, C.R. Thiacycloalkynes for copper-free click chemistry. Angew. Chem. 2012, 124, 2493–2497. [Google Scholar] [CrossRef]
- King, M.; Wagner, A. Developments in the field of bioorthogonal bond forming reactions past and present trends. Bioconj. Chem. 2014, 25, 825–839. [Google Scholar] [CrossRef] [PubMed]
- Debets, M.F.; van Hest, J.C.; Rutjes, F.P. Bioorthogonal labelling of biomolecules: New functional handles and ligation methods. Org. Biomol. Chem. 2013, 11, 6439–6455. [Google Scholar] [CrossRef] [PubMed] [Green Version]









© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Zhang, Z. Development and Applications of the Copper-Catalyzed Azide-Alkyne Cycloaddition (CuAAC) as a Bioorthogonal Reaction. Molecules 2016, 21, 1393. https://doi.org/10.3390/molecules21101393
Li L, Zhang Z. Development and Applications of the Copper-Catalyzed Azide-Alkyne Cycloaddition (CuAAC) as a Bioorthogonal Reaction. Molecules. 2016; 21(10):1393. https://doi.org/10.3390/molecules21101393
Chicago/Turabian StyleLi, Li, and Zhiyuan Zhang. 2016. "Development and Applications of the Copper-Catalyzed Azide-Alkyne Cycloaddition (CuAAC) as a Bioorthogonal Reaction" Molecules 21, no. 10: 1393. https://doi.org/10.3390/molecules21101393
APA StyleLi, L., & Zhang, Z. (2016). Development and Applications of the Copper-Catalyzed Azide-Alkyne Cycloaddition (CuAAC) as a Bioorthogonal Reaction. Molecules, 21(10), 1393. https://doi.org/10.3390/molecules21101393