
Chemoenzymatic Synthesis of trans-β-Aryl-δ-hydroxy-γ-lactones and Enzymatic Kinetic Resolution of Their Racemic Mixtures


Abstract
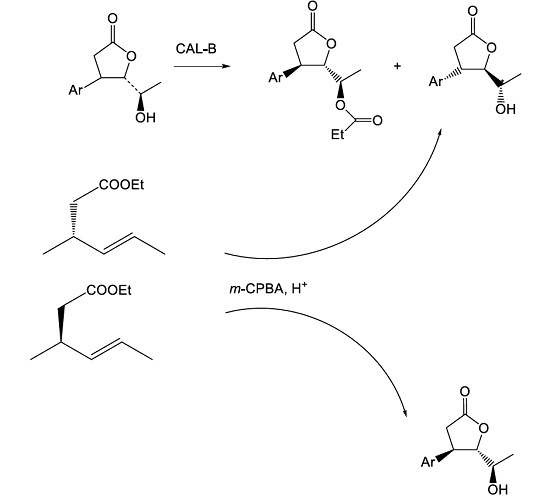
:
1. Introduction
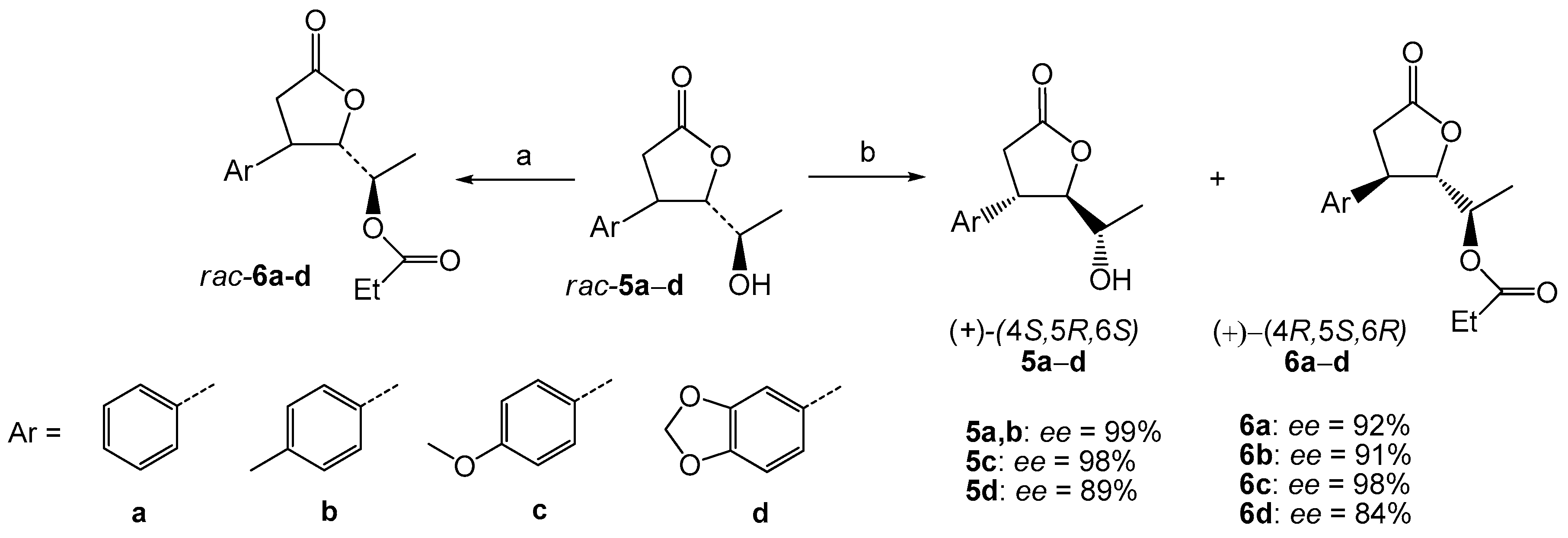
2. Results and Discussion
3. Experimental Section
3.1. General
3.2. Chemicals and Enzymes
3.3. General Procedure for the Synthesis of Racemic Esters 2, 3 and 6a–d
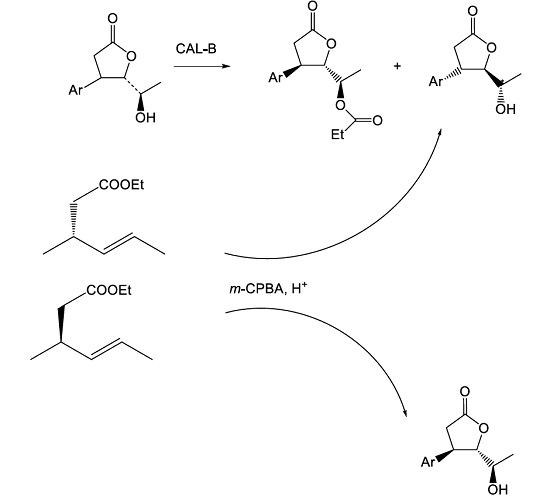
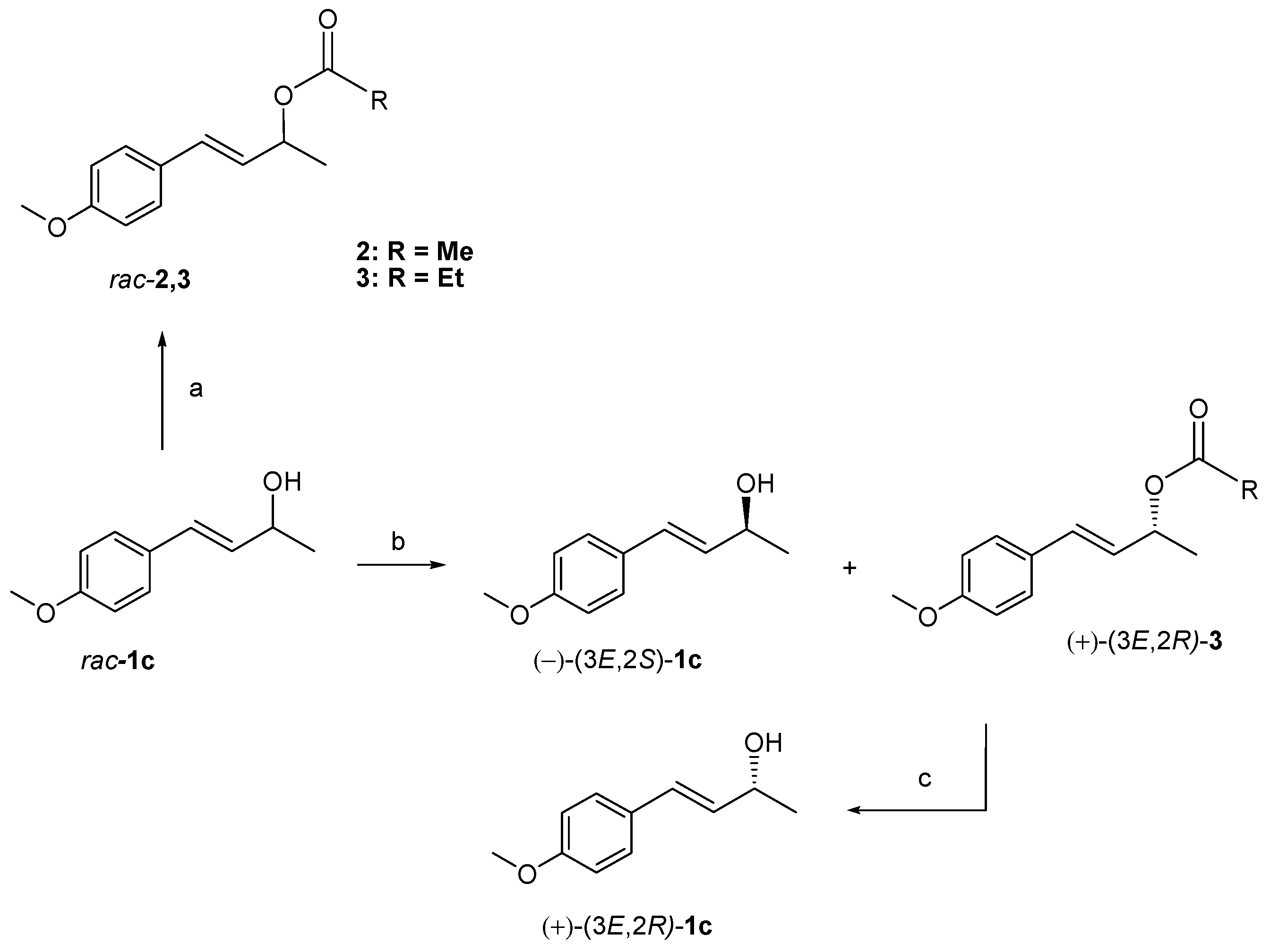
3.4. Enzymatic Resolution of Racemic Allyl Alcohol 1c
3.5. Johnson–Claisen Rearrangement of Enantiomerically Enriched Allyl Alcohols (−)-1c and (+)-1c
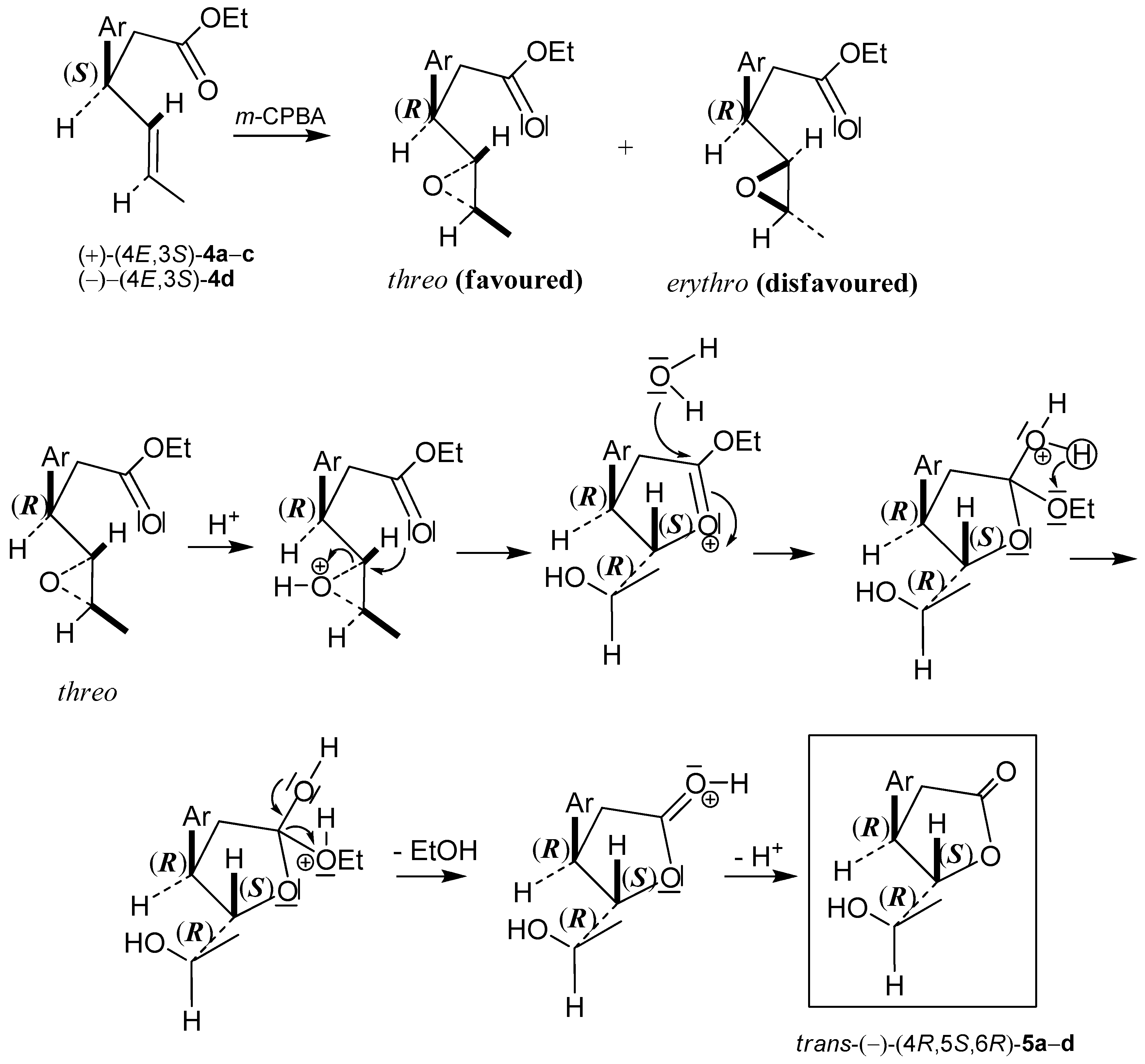
3.6. Lactonization of Enantiomerically Enriched γ,δ-Unsaturated Esters (S)-4a–d and (R)-4a–d
3.7. Enzymatic Resolution of Hydroxylactones 5a–d
3.7.1. Screening Procedure
3.7.2. Preparative Transesterification
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Willför, S.M.; Ahotupa, M.O.; Hemming, J.E.; Reunanen, M.H.T.; Eklund, P.C.; Sjöholm, R.E.; Eckerman, C.S.E.; Pohjamo, S.P.; Holmbom, B.H. Antioxidant activity of knotwood extractives and phenolic compounds of selected tree species. J. Agric. Food Chem. 2003, 51, 7600–7606. [Google Scholar] [CrossRef] [PubMed]
- Cornwell, T.; Cohick, W.; Raskin, I. Dietary phytoestrogens and health. Phytochemistry 2004, 65, 995–1016. [Google Scholar] [CrossRef] [PubMed]
- Raffaelli, B.; Hoikkala, A.; Leppälä, E.; Wähälä, K. Enterolignans. J. Chromatogr. B 2002, 777, 29–43. [Google Scholar] [CrossRef]
- Habrant, D.; Poigny, S.; Ségur-Derai, M.; Brunel, Y.; Heurtaux, B.; Le Gall, T.; Strehle, A.; Saladin, R.; Meunier, S.; Mioskowski, C.; et al. Evaluation of antioxidant properties of monoaromatic derivatives of pulvinic acids. J. Med. Chem. 2009, 52, 2454–2464. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Wong, H-F. Total synthesis of (±)-maculalactone A, maculalactone B and maculalactone C and the determination of the absolute configuration of natural (+)-maculalactone A by asymmetric synthesis. Tetrahedron 2004, 60, 5439–5451. [Google Scholar] [CrossRef]
- Pawar, V.U.; Ghosh, S.; Chopade, B.A.; Shinde, V.S. Design and synthesis of harzialactone analogues: Promising anticancer agents. Bioorg. Med. Chem. Lett. 2010, 20, 7243–7245. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, B.H.; Foroumadi, A.; Emami, S.; Khoobi, M.; Panah, F.; Ardestani, S.K.; Shafiee, A. Isochaihulactone analogues: Synthesis and anti-proliferative activity of novel dibenzylbutyrolactones. Eur. J. Med. Chem. 2010, 45, 5979–5984. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, S.; Hayashi, Y.; Nakashima, Y.; Kirikihira, T.; Yamada, K.; Masuda, T. Effect of benzylic oxygen on the antioxidant activity of phenolic lignans. J. Nat. Prod. 2005, 68, 1459–1470. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.K.; Srinivas, P.V.; Kumar, P.; Rao, J.M. Free radical scavenging active components from cedrus deodara. J. Agric. Food Chem. 2001, 49, 4642–4645. [Google Scholar] [CrossRef] [PubMed]
- Zapf, S.; Anke, T.; Sterner, O. Incrustoporin, a new antibiotic from Incrustoporia carneola (Bres.) Ryv. (Basidiomycetes). Acta Chem. Scand. 1995, 49, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Pour, M.; Ŝpulák, M.; Balŝánek, V.; Kuneŝ, J.; Kubanová, P.; Buchta, V. Synthesis and structure-antifungal activity relationships of 3-aryl-5-alkyl-2,5-dihydrofuran-2-ones and their carbanalogues: Further refinement of tentative pharmacophore group. Bioorg. Med. Chem. 2003, 11, 2843–2866. [Google Scholar] [CrossRef]
- Singh, S.B.; Jayasuriya, H.; Dewey, R.; Polishook, J.D.; Dombrowski, A.W.; Zink, D.L.; Guan, Z.; Collado, J.; Platas, G.; Pelaez, F.; et al. Isolation, structure, and HIV-1-integrase inhibitory activity of structurally diverse fungal metabolites. J. Ind. Microbiol. Biotechnol. 2003, 301, 721–731. [Google Scholar]
- Bourdreux, Y.; Bodio, E.; Willis, C.; Billaud, C.; le Gall, T.; Mioskowski, C. Synthesis of vulpinic and pulvinic acids from tetronic acid. Tetrahedron 2008, 64, 8930–8937. [Google Scholar] [CrossRef]
- Da Silva, R.; de Souza, G.H.B.; da Silva, A.A.; de Souza, V.A.; Pereira, A.C.; de A. Royo, V.; de Silva, M.L.A.; Donate, P.M.; de M. Araújo, A.L.S.; Carvalho, J.C.T.; et al. Synthesis and biological activity evaluation of lignin lactones derived from (–)-cubebin. Bioorg. Med. Chem. Lett. 2005, 15, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Harmatha, J.; Nawrot, J. Insect feeding deterrent activity of lignans and related phenylpropanoids with a methylenedioxyphenyl (piperonyl) structure moiety. Entomol. Exp. Appl. 2002, 104, 51–60. [Google Scholar] [CrossRef]
- Gonzales, E.B.; Bell-Horner, C.L.; de la Cruz, M.A.M.; Ferrendelli, J.A.; Covey, D.F.; Dillon, G.H. Enantioselectivity of α-benzyl-α-methyl-γ-butyrolactone-mediated modulation of anticonvulsant activity and GABAA receptor function. J. Pharmacol. Exp. Ther. 2010, 309, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Landete, J.M. Plant and mammalian lignans: A review of source, intake, metabolism, intestinal bacteria and health. Food Res. Int. 2012, 46, 410–424. [Google Scholar] [CrossRef]
- Cosentino, M.; Marino, F.; Ferrari, M.; Rasini, E.; Bombelli, R.; Luini, A.; Legnaro, M.; Delle Canne, M.D.; Luzzani, M.; Crema, F.; et al. Estrogenic activity of 7-hydroxymatairesinol potassium acetate (HMR/lignan™) from Norway spruce (Picea abies) knots and of its active metabolite enterolactone in MCF-7 cells. Pharm. Res. 2007, 56, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.S.C.; Chamberlin, A.R. Enantioselective preparation of β-alkyl-γ-butyrolactones from functionalized ketene dithioacetals. J. Org. Chem. 1993, 58, 2725–2737. [Google Scholar] [CrossRef]
- Kondaveti, L.; Al-Azemi, T.F.; Bisht, K.S. Lipase-catalyzed solvent-free kinetic resolution of substituted racemic ε-caprolactones. Tetrahedron-Asymmetry 2002, 13, 129–135. [Google Scholar] [CrossRef]
- Brenna, E.; Fuganti, C.; Serra, S. Enantioselective perception of chiral odorants. Tetrahedron Asymmetry 2003, 14, 1–42. [Google Scholar] [CrossRef]
- Nawrot, J.; Dams, I.; Wawrzeńczyk, C. Feeding deterrent activity of terpenoid lactones with a p-menthane system against stored-product pests. J. Stored Prod. Res. 2009, 45, 221–225. [Google Scholar] [CrossRef]
- Sefkow, M.; Kelling, A.; Schilde, U. Enantioselective synthesis of α-hydroxylated enterolactone and analogs. Tetrahedron Lett. 2001, 42, 5101–5104. [Google Scholar] [CrossRef]
- Pohmakotr, M.; Soorukram, D.; Tuchinda, P.; Prabpai, S.; Kongsaeree, P.; Reutrakul, V. Highly diastereoselective alkylation of vicinal dianions of chiralsuccinic acid derivatives: a new general strategy to (R)-β-arylmethyl-γ-butyrolactones. Tetrahedron Lett. 2004, 45, 4315–4318. [Google Scholar] [CrossRef]
- Donate, P.M.; Frederico, D.; da Silva, R.; Constantino, M.G.; del Ponte, G.; Bonatto, P.S. Asymmetric synthesis of γ-butyrolactones by enantioselective hydrogenation of butenolides. Tetrahedron Asymmetry 2003, 14, 3253–3256. [Google Scholar] [CrossRef]
- Caro, Y.; Masaguer, C.F.; Raviña, E. Synthesis of optically active β-benzyl-γ-butyrolactone through lipase-catalyzed kinetic resolution. Tetrahedron Asymmetry 2001, 12, 1723–1726. [Google Scholar] [CrossRef]
- Berti, F.; Forzato, C.; Furlan, G.; Nitti, P.; Pitacco, G.; Valentin, E.; Zangrando, E. Synthesis of optically active α-benzyl paraconic acids and their esters and assignment of their absolute configuration. Tetrahedron Asymmetry 2009, 20, 313–321. [Google Scholar] [CrossRef]
- Koul, S.; Singh, B.; Taneja, C.; Qazi, G.N. New chemo and chemo-enzymatic synthesis of β-benzyl-γ-butyrolactones. Tetrahedron 2003, 59, 3487–3491. [Google Scholar] [CrossRef]
- Ribeiro, J.B.; Sousa, L.M.A.; Fraga, C.A.M.; Leita, S.G.F.; Ramos, M.C.K.V.; de Aquino Neto, F.R.; Aguiar, L.C.S.; de Souza, R.O.M.A.; Antunes, O.A.C. Microbial reduction of alpha-substituted-alpha-acetyl-gamma-butyrolactones. Catal. Commun. 2008, 9, 1782–1786. [Google Scholar] [CrossRef]
- Wang, M.-X.; Zhao, S.-M. Synthesis of enantiomerically enriched (S)-(+)-2-aryl-4-pentenoic acids and (R)-(–)-2-aryl-4-pentenamides via microbial hydrolysis of nitriles, a chemoenzymatic approach to stereoisomers of α,γ-disubstituted γ-butyrolactones. Tetrahedron Asymmetry 2002, 13, 1695–1702. [Google Scholar] [CrossRef]
- Mazur, M.; Gładkowski, W.; Wawrzeńczyk, C. Synteza chlorowcolaktonów z podstawnikiem metoksyfenylowym. Przem. Chem. 2011, 90/5, 286–294. [Google Scholar]
- Gładkowski, W.; Skrobiszewski, A.; Mazur, M.; Siepka, M.; Pawlak, A.; Obmińska-Mrukowicz, B.; Białońska, A.; Poradowski, D.; Drynda, A.; Urbaniak, M. Synthesis and anticancer activity of novel halolactones with β-aryl substituents from simple aromatic aldehydes. Tetrahedron 2013, 69, 10414–10423. [Google Scholar] [CrossRef]
- Skrobiszewski, A.; Gładkowski, W.; Lis, M.; Gliszczyńska, A.; Maciejewska, G.; Klejdysz, T.; Obmińska-Mrukowicz, B.; Nawrot, J.; Wawrzeńczyk, C. Laktony. Cz. XLVa). Synteza hydroksylaktonów z pierścieniem aromatycznym oraz ocena ich aktywności antyfidantnej i antyproliferacyjnej. Przem. Chem. 2014, 93, 1637–1643. [Google Scholar]
- Mazur, M.; Skrobiszewski, A.; Gładkowski, W.; Podkowik, M.; Bania, J.; Nawrot, J.; Klejdysz, T.; Wawrzeńczyk, C. Lactones 47. Synthesis, antifeedant and antibacterial activity of γ-lactones with p-methoxyphenyl substituent. Pest. Manag. Sci. 2016, 72, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Skrobiszewski, A.; Gładkowski, W.; Walczak, P.; Gliszczyńska, A.; Maciejewska, G.; Klejdysz, T.; Nawrot, J.; Wawrzeńczyk, C. Synthesis of β-aryl-γ-lactones and relationship: Structure-antifeedant and antifungal activity. J. Chem. Sci. 2015, 127, 687–699. [Google Scholar] [CrossRef]
- Gładkowski, W.; Skrobiszewski, A.; Mazur, M.; Siepka, M.; Białońska, A. Convenient chemoenzymatic route to optically active β-aryl-δ-iodo-γ-lactones and β-aryl-γ-iodo-δ-lactones with the defined configurations of stereogenic centers. Eur. J. Org. Chem. 2015, 2015, 605–615. [Google Scholar] [CrossRef]
- Gładkowski, W.; Gliszczyńska, A.; Siepka, M.; Czarnecka, M.; Maciejewska, G. Kinetic resolution of (E)-4-(2ʹ,5ʹ-dimethylphenyl)-but-3-en-2-ol and (E)-4-(benzo[d][1ʹ,3ʹ]dioxol-5ʹ-yl)-but-3-en-2-ol through lipase-catalyzed transesterification. Tetrahedron-Asymmetry 2015, 26, 702–709. [Google Scholar] [CrossRef]
- Gładkowski, W.; Skrobiszewski, A.; Mazur, M.; Gliszczyńska, A.; Czarnecka, M.; Pawlak, A.; Obmińska-Mrukowicz, B.; Maciejewska, G.; Białońska, A. Chiral δ-iodo-γ-lactones derived from cuminaldehyde, 2,5-dimethylbenzaldehyde and piperonal: Chemoenzymatic synthesis and antiproliferative activity. Tetrahedron Asymmetry 2016, 27, 227–237. [Google Scholar] [CrossRef]
- GowriSankar, S.; Gon Lee, C.; Nyoung Kim, J. Facile synthesis of lactones and dihydronaphthalenes from methyl 2-isobutenyl (or 2-isopentenyl) cinnamates as the common intermediates. Tetrahedron Lett. 2004, 45, 6949–6953. [Google Scholar] [CrossRef]
- Akai, S.; Hanada, R.; Fujiwara, N.; Kita, Y.; Egi, M. One-pot synthesis of optically active allyl esters via lipase-vanadium combo catalysis. Org. Lett. 2010, 12, 4900–4903. [Google Scholar] [CrossRef] [PubMed]
- Olejniczak, T.; Nawrot, J.; Ciunik, Z.; Wawrzeńczyk, C. Synthesis of some terpenoid lactones from γ,δ-epoxy esters. Pol. J. Chem. 2000, 74, 673–680. [Google Scholar]
- Olejniczak, T.; Gawroński, J.; Wawrzeńczyk, C. Microbial lactonization of γ,δ-epoxy esters. Chirality 2001, 13, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskas, R.J.; Weissfloch, A.N.E. A structure-based rationalization of the enantiopreference of subtilisin toward secondary alcohols and isosteric primary amines. J. Mol. Catal. B Enzym. 1997, 3, 65–72. [Google Scholar] [CrossRef]
- Chojnacka, A.; Obara, R.; Wawrzeńczyk, C. Kinetic resolution of racemic secondary aliphatic allylic alcohols in lipase-catalyzed transesterification. Tetrahedron Asymmetry 2007, 18, 101–107. [Google Scholar] [CrossRef]
- He, P.; Liu, X.; Zheng, Z.; Li, W.; Lin, L.; Feng, X. Asymmetric 1,2-Reduction of Enones with Potassium Borohydride Catalyzed by Chiral N,Nʹ-Dioxide Scandium(III) Complexes. Org. Lett. 2012, 14, 5134–5137. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 5a−d are available from the authors.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Time (h) | Conversion a (mol %) | ee of (+)-Hydroxylactone 5a (%) | ee of (+)-Propionyloxylactone 6a (%) | E b |
|---|---|---|---|---|---|
| CAL-B | 1 | 49 | 96 | 99 | >200 |
| 2 | 51 | 99 | 96 | >200 | |
| 4 | 51 | 99 | 96 | >200 | |
| 6 | 51 | 99 | 96 | >200 | |
| CCL | 2 | 17 | 20 | 97 | 80 |
| 4 | 34 | 42 | 80 | 13 | |
| 6 | 44 | 55 | 71 | 10 | |
| Lipozyme TL IM | 2 | 11 | 12 | 99 | >200 |
| 4 | 43 | 68 | 90 | 38 | |
| 6 | 53 | 91 | 80 | 28 | |
| Amano Lipase PS | 2 | 11 | 12 | 99 | >200 |
| 4 | 25 | 34 | 99 | >200 | |
| 6 | 40 | 65 | 99 | >200 |
| Substrate | Conversion a (mol %) | ee of (+)-Hydroxylactone (%) | ee of (+)-Propionyloxylactone (%) | E b |
|---|---|---|---|---|
| 5b | 52 | 99 (5b) | 91 (6b) | 111 |
| 5c | 50 | 98 (5c) | 98 (6c) | >200 |
| 5d | 51 | 89 (5d) | 84 (6d) | 34 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skrobiszewski, A.; Gładkowski, W.; Maciejewska, G.; Wawrzeńczyk, C. Chemoenzymatic Synthesis of trans-β-Aryl-δ-hydroxy-γ-lactones and Enzymatic Kinetic Resolution of Their Racemic Mixtures. Molecules 2016, 21, 1552. https://doi.org/10.3390/molecules21111552
Skrobiszewski A, Gładkowski W, Maciejewska G, Wawrzeńczyk C. Chemoenzymatic Synthesis of trans-β-Aryl-δ-hydroxy-γ-lactones and Enzymatic Kinetic Resolution of Their Racemic Mixtures. Molecules. 2016; 21(11):1552. https://doi.org/10.3390/molecules21111552
Chicago/Turabian StyleSkrobiszewski, Andrzej, Witold Gładkowski, Gabriela Maciejewska, and Czesław Wawrzeńczyk. 2016. "Chemoenzymatic Synthesis of trans-β-Aryl-δ-hydroxy-γ-lactones and Enzymatic Kinetic Resolution of Their Racemic Mixtures" Molecules 21, no. 11: 1552. https://doi.org/10.3390/molecules21111552
APA StyleSkrobiszewski, A., Gładkowski, W., Maciejewska, G., & Wawrzeńczyk, C. (2016). Chemoenzymatic Synthesis of trans-β-Aryl-δ-hydroxy-γ-lactones and Enzymatic Kinetic Resolution of Their Racemic Mixtures. Molecules, 21(11), 1552. https://doi.org/10.3390/molecules21111552