
Screening for Triterpenoid Saponins in Plants Using Hyphenated Analytical Platforms

 ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
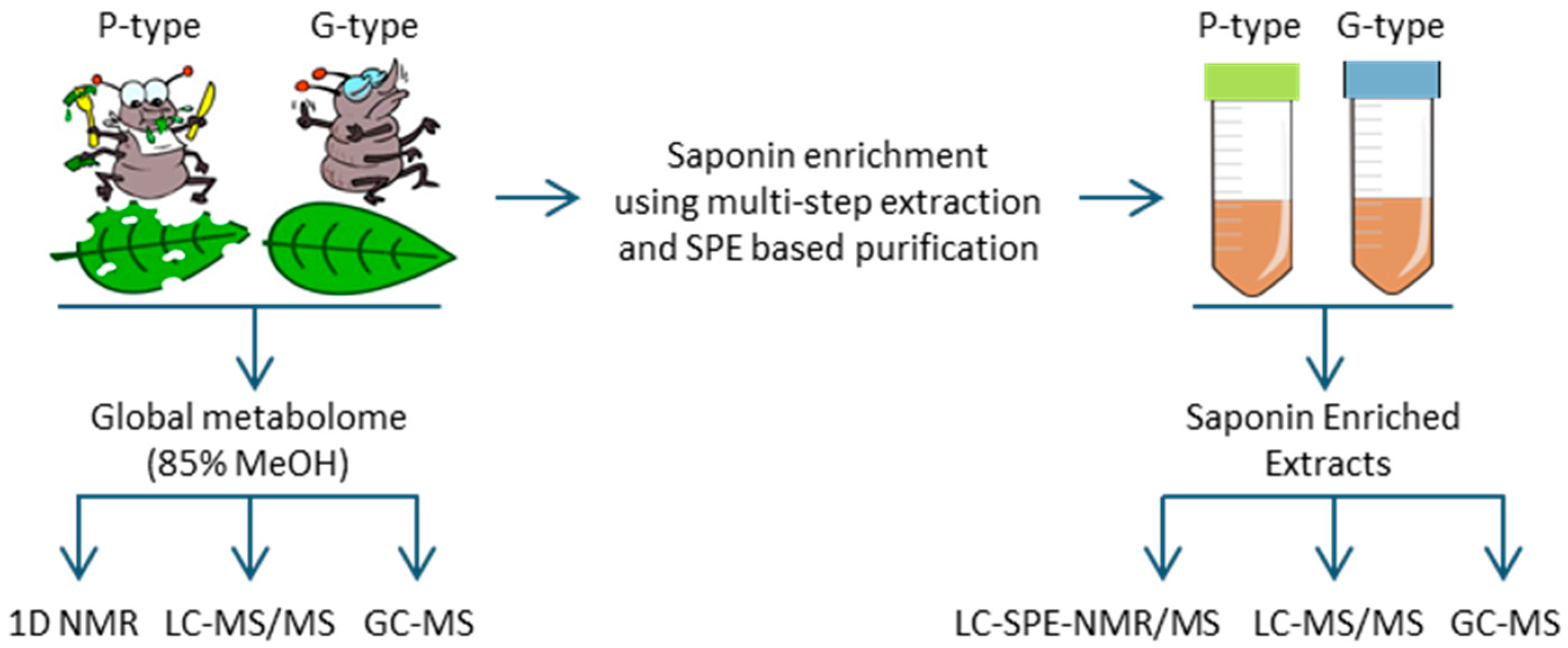
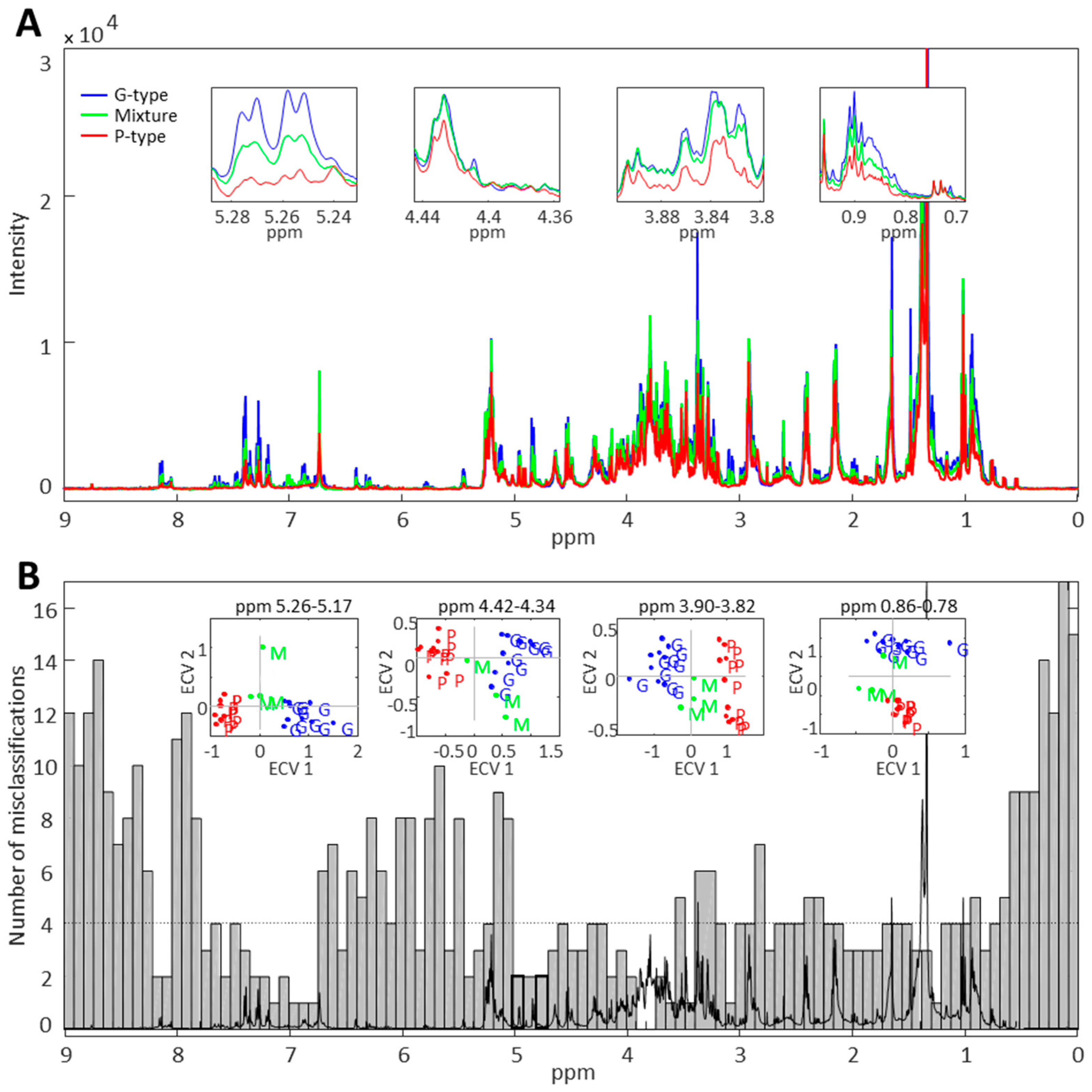
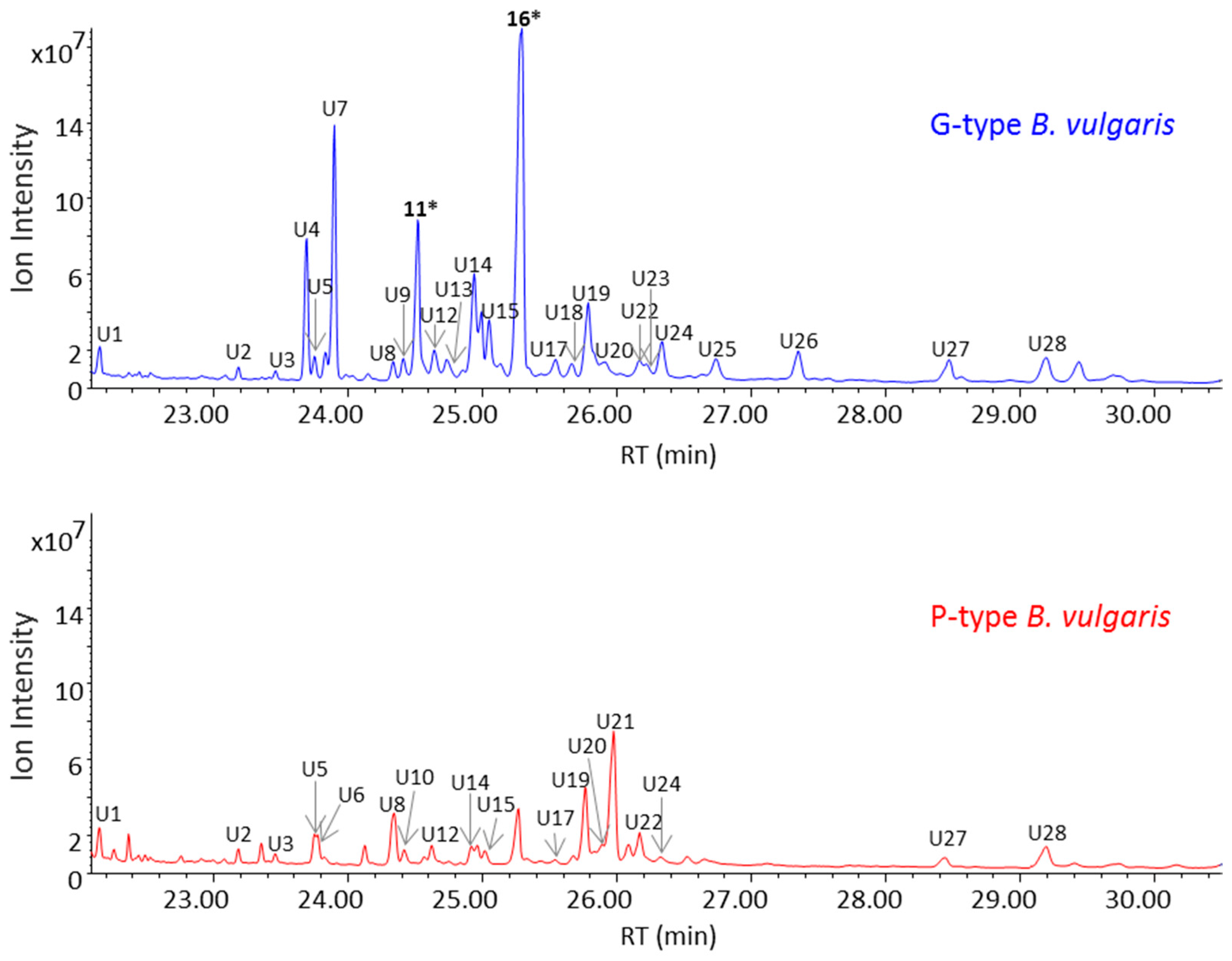
2.1. Untargeted Metabolomics for Analysis of Triterpenoid Saponins in B. vulgaris
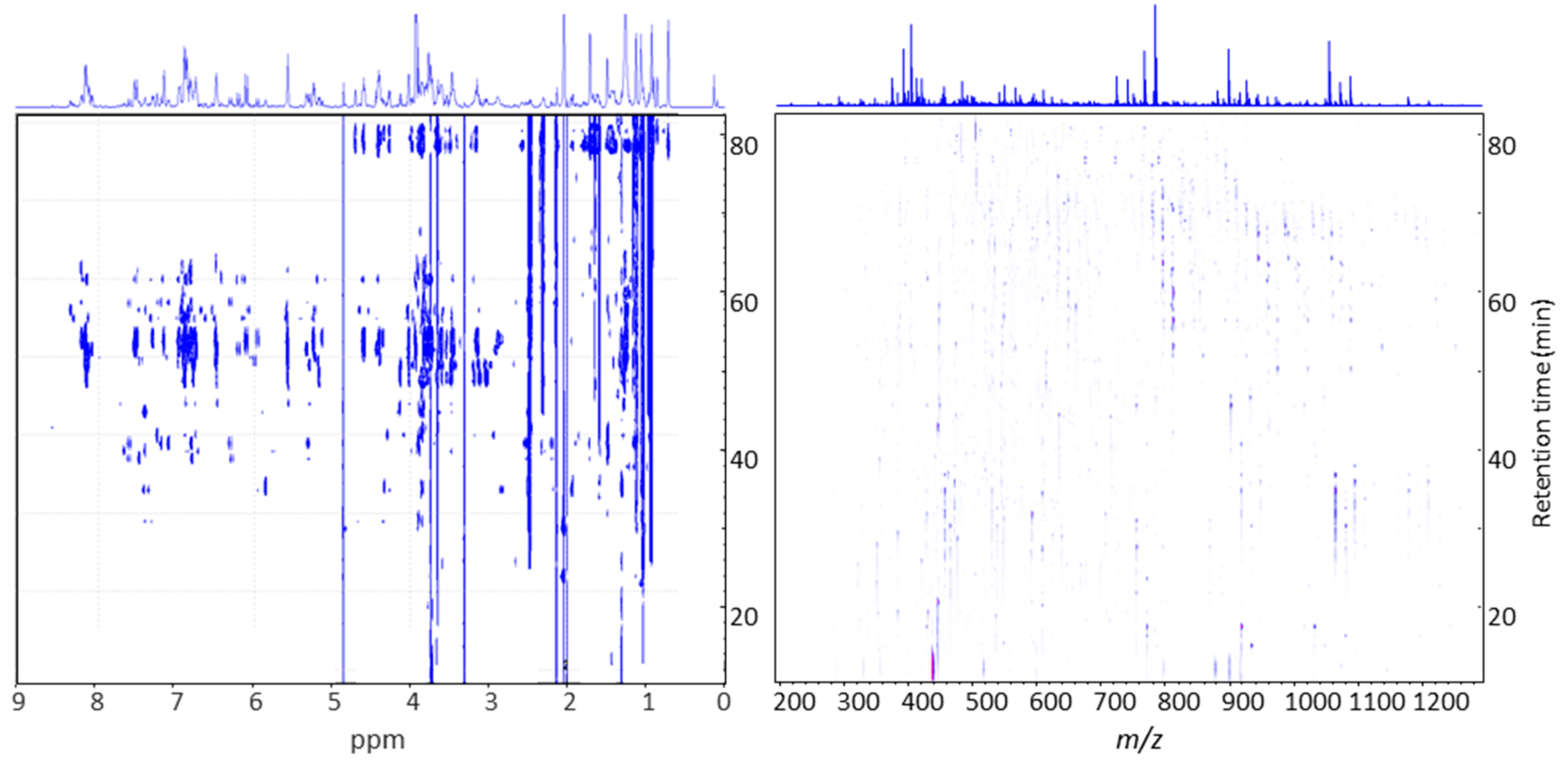
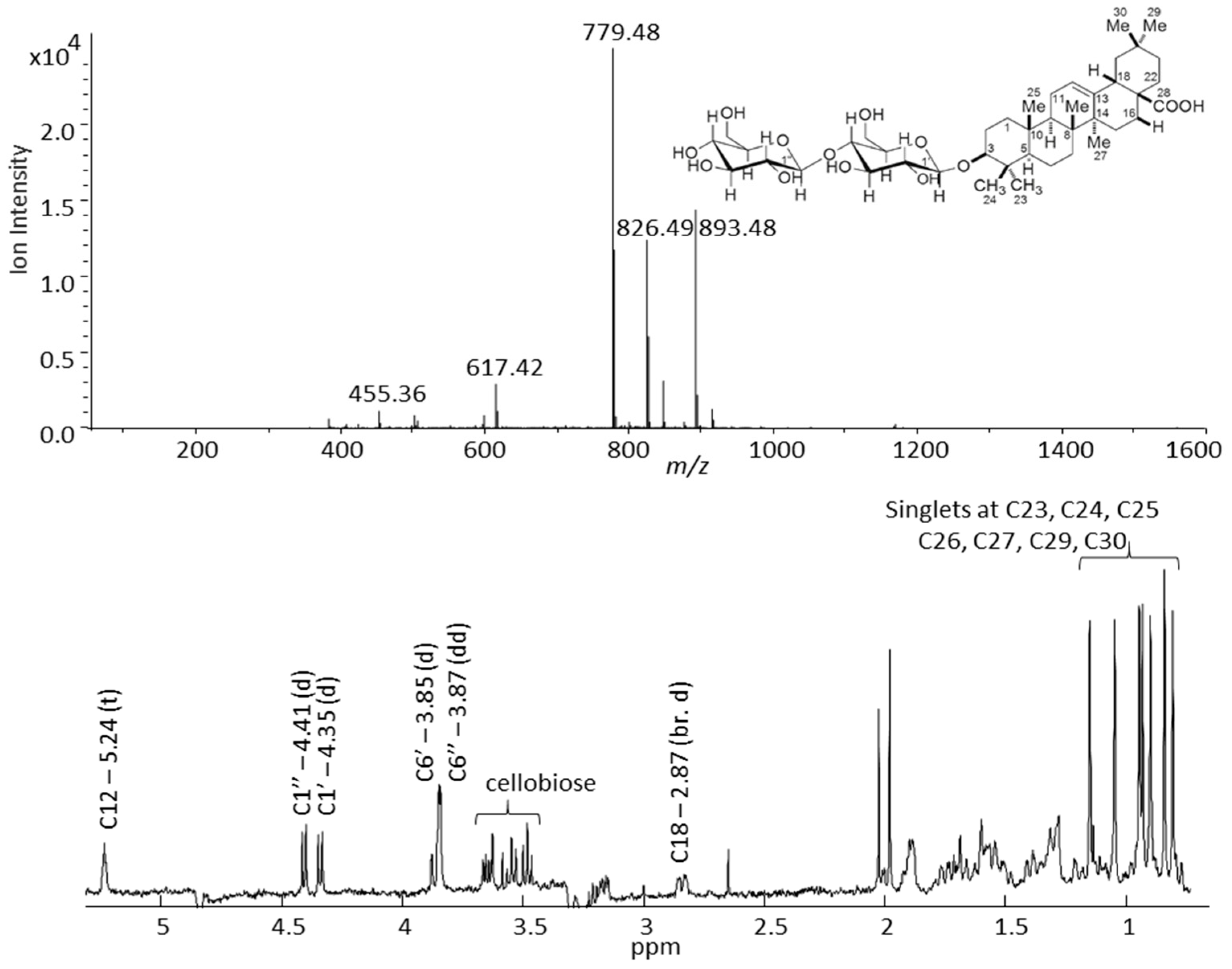
2.2. Time Slice LC-SPE-NMR/MS Experiment on Saponin Enriched Extracts from G- and P-Type B. vulgaris
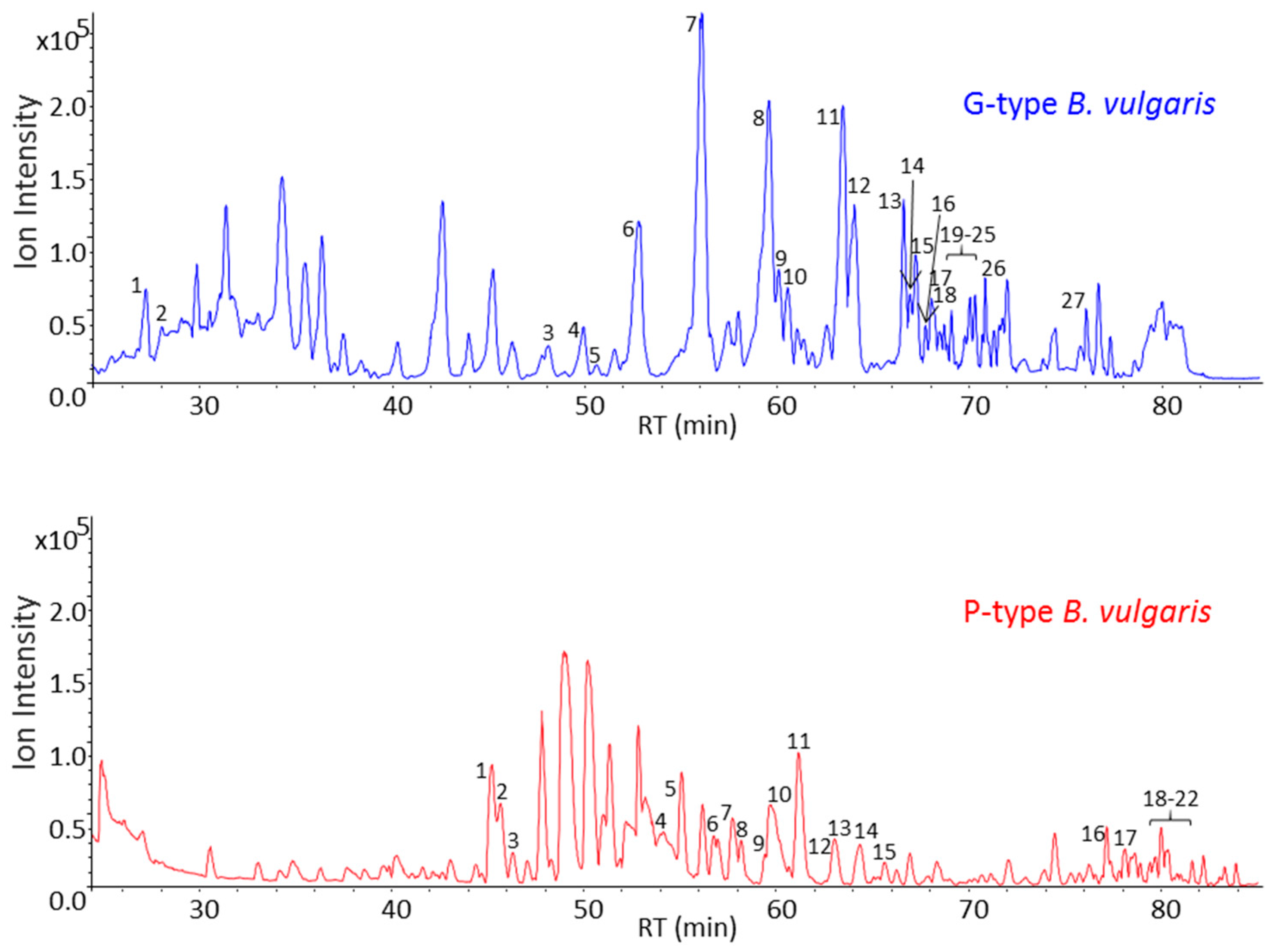
2.3. GC-MS Analysis of Saponin Enriched Extracts of G- and P-Types B. vulgaris
3. Experimental Section
3.1. Plant Materials and Metabolite Extraction
3.2. GC-MS Analysis
3.3. NMR Analysis
3.4. LC-SPE-NMR/MS Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Augustin, J.M.; Kuzina, V.; Andersen, S.B.; Bak, S. Molecular activities, biosynthesis and evolution of triterpenoid saponins. Phytochemistry 2011, 72, 435–457. [Google Scholar] [CrossRef] [PubMed]
- Kuzina, V.; Ekstrom, C.T.; Andersen, S.B.; Nielsen, J.K.; Olsen, C.E.; Bak, S. Identification of Defense Compounds in Barbarea vulgaris against the Herbivore Phyllotreta nemorum by an Ecometabolomic Approach. Plant Physiol. 2009, 151, 1977–1990. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, T.; Nagao, T.; Nakayama, M.; Serizawa, H.; Koshioka, M.; Okabe, H.; Kawai, A. Identification of a triterpenoid saponin from a crucifer, Barbarea vulgaris, as a feeding deterrent to the diamondback moth, Plutella xylostella. J. Chem. Ecol. 2002, 28, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Agerbirk, N.; Olsen, C.E.; Bibby, B.M.; Frandsen, H.O.; Brown, L.D.; Nielsen, J.K.; Renwick, J.A.A. A saponin correlated with variable resistance of Barbarea vulgaris to the diamondback moth Plutella xylostella. J. Chem. Ecol. 2003, 29, 1417–1433. [Google Scholar] [CrossRef] [PubMed]
- Di Fabio, G.; Romanucci, V.; de Marco, A.; Zarrelli, A. Triterpenoids from Gymnema sylvestre and Their Pharmacological Activities. Molecules 2014, 19, 10956–10981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asl, M.N.; Hosseinzadeh, H. Review of pharmacological effects of Glycyrrhiza sp. and its bioactive compounds. Phytother. Res. 2008, 22, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Liu, J. Pharmacology of oleanolic acid and ursolic acid. J. Ethnopharmacol. 1995, 49, 57–68. [Google Scholar] [CrossRef]
- Shah, M.R.; Ishtiaq; Hizbullah, S.M.; Habtemariam, S.; Zarrelli, A.; Muhammad, A.; Collina, S.; Khan, I. Protein tyrosine phosphatase 1B inhibitors isolated from Artemisia roxburghiana. J. Enzym. Inhib. Med. Chem. 2016, 31, 563–567. [Google Scholar]
- Augustin, J.M.; Drok, S.; Shinoda, T.; Sanmiya, K.; Nielsen, J.K.; Khakimov, B.; Olsen, C.E.; Hansen, E.H.; Kuzina, V.; Ekstrom, C.T.; et al. UDP-Glycosyltransferases from the UGT73C Subfamily in Barbarea vulgaris Catalyze Sapogenin 3-O-Glucosylation in Saponin-Mediated Insect Resistance. Plant Physiol. 2012, 160, 1881–1895. [Google Scholar] [CrossRef] [PubMed]
- Khakimov, B.; Kuzina, V.; Erthmann, P.O.; Fukushima, E.O.; Augustin, J.M.; Olsen, C.E.; Scholtalbers, J.; Volpin, H.; Andersen, S.B.; Hauser, T.P.; et al. Identification and genome organization of saponin pathway genes from a wild crucifer, and their use for transient production of saponins in Nicotiana benthamiana. Plant J. 2015, 84, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Geisler, K.; Hughes, R.K.; Sainsbury, F.; Lomonossoff, G.P.; Rejzek, M.; Fairhurst, S.; Olsen, C.E.; Motawia, M.S.; Melton, R.E.; Hemmings, A.M.; et al. Biochemical analysis of a multifunctional cytochrome P450 (CYP51) enzyme required for synthesis of antimicrobial triterpenes in plants. Proc. Natl. Acad. Sci. USA 2013, 110, E3360–E3367. [Google Scholar] [CrossRef] [PubMed]
- Madl, T.; Sterk, H.; Mittelbach, M.; Rechberger, G.N. Tandem Mass Spectrometric Analysis of a Complex Triterpene Saponin Mixture of Chenopodium quinoa. J. Am. Soc. Mass Spectrom. 2006, 17, 795–806. [Google Scholar] [PubMed]
- Khakimov, B.; Motawia, M.S.; Bak, S.; Engelsen, S.B. The use of trimethylsilyl cyanide derivatization for robust and broad-spectrum high-throughput gas chromatography-mass spectrometry based metabolomics. Anal. Bioanal. Chem. 2013, 405, 9193–9205. [Google Scholar] [CrossRef] [PubMed]
- Khakimov, B. Metabolomics and Bioactive Substances in Plants. Ph.D. Thesis, University of Copenhagen, Frederiksberg, Denmark, 2013. [Google Scholar]
- Harshman, R.A. PARAFAC2: Mathematical and technical notes. UCLA Work. Pap. Phon. 1972, 22, 30–47. [Google Scholar]
- Bro, R.; Andersson, C.A.; Kiers, H.A.L. PARAFAC2—Part II. Modeling chromatographic data with retention time shifts. J. Chemometr. 1999, 13, 295–309. [Google Scholar] [CrossRef]
- Khakimov, B.; Amigo, J.M.; Bak, S.; Engelsen, S.B. Plant metabolomics: Resolution and quantification of elusive peaks in liquid chromatography-mass spectrometry profiles of complex plant extracts using multi-way decomposition methods. J. Chromatogr. A 2012, 1266, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Nørgaard, L.; Bro, R.; Westad, F.; Engelsen, S.B. A modification of canonical variates analysis to handle highly collinear multivariate data. J. Chemometr. 2006, 20, 425–435. [Google Scholar] [CrossRef]
- Nielsen, N.J.; Nielsen, J.; Staerk, D. New Resistance-Correlated Saponins from the Insect-Resistant Crucifer Barbarea vulgaris. J. Agric. Food Chem. 2010, 58, 5509–5514. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.P.; Yu, Y.Y.; Zhou, S.R.; Tian, S.G.; Cao, S.W. Isolation and identification of phenolic compounds from Gynura divaricata leaves. Pharmacogn. Mag. 2011, 7, 101–108. [Google Scholar] [PubMed]
- Agrawal, P.K. Assigning stereodiversity of the 27-Me group of furostane-type steroidal saponins via NMR chemical shifts. Steroids 2005, 70, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.; Amberg, A.; Barrett, D.; Beale, M.; Beger, R.; Daykin, C.; Fan, T.; Fiehn, O.; Goodacre, R.; Griffin, J.; et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Khakimov, B.; Mongi, R.J.; Sørensen, K.M.; Ndabikunze, B.K.; Chove, B.E.; Engelsen, S.B. A comprehensive and comparative GC-MS metabolomics study of non-volatiles in Tanzanian grown mango, pineapple, jackfruit, baobab and tamarind fruits. Food Chem. 2016, 213, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not Avaiable.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | RT (min) | Aglycone (Da) A | Sugar Moieties B | Mass Spectra Fragmentation C | Anomeric Protons (ppm) D |
|---|---|---|---|---|---|
| 1 | 27.3 | 448 | 2 × Methylpentose 3 × Hexose | [M1225 − H − 162 − 146 − 162 − 146 − 162]− = 447 | 4.13 (d, J = 7.8) 4.17 (d, J = 7.8) 4.29 (d, J = 7.8) |
| 2 | 27.7 | 478 | 2 × Methylpentose 1 × Pentose 2 × Hexose | [M1225 − H − 162 − 132 – 162 − 146 − 146]− = 477 | |
| 3 | 49.5 | 504 | 1 × Methylpentose 2 × Hexose | [M973 − H − 146 − 162 − 162]− = 503 | 4.21 (d, J = 7.8) 4.41 (d, J = 7.8) 4.42 (d, J = 7.8) 5.25 * (t, J = 3.4) |
| 4 | 50.5 | 504 | 1 × Methylpentose 2 × Hexose | [M973 − H − 146 − 162 − 162]− = 503 | 4.21 (d, J = 7.8) 4.41 (d, J = 7.8) 4.42 (d, J = 7.8) 5.25 * (t, J = 3.4) |
| 5 | 51.2 | 504 | 1 × Methylpentose 2 × Hexose | [M973 − H − 162 − 146 − 162]− = 503 | 4.21 (d, J = 7.8) 4.39 (d, J = 7.8) 5.25 * (t, J = 3.4) |
| 6 | 52.5 | 649 ** | 3 × Hexose | [M1135 − H − 162 − 162 − 162]− = 649 | 4.41 (d, J = 8.0) 4.43 (d, J = 8.0) 5.55 (d, J = 8.2) |
| 7 | 56.2 | 649 ** | 2 × Hexose | [M973 − H − 162 − 162]− = 649 | 4.41 (d, J = 8.0) 5.52 (d, J = 8.2) |
| 8 | 59.5 | 649 ** | 2 × Hexose | [M973 − H − 162 − 162]− = 649 | 4.41 (d, J = 8.0) 5.52 (d, J = 8.2) |
| 9 | 60.6 | 472 | 4 × Hexose | [M1119 − H − 162 − 162 − 162 − 162]− = 471 | 4.34 (d, J = 7.8) 4.41 (d, J = 8.0) 4.43 (d, J = 7.8) 5.55 (d, J = 8.0) |
| 10 | 61.2 | 633 | 3 × Hexose | [M1119 − H − 162 − 162 − 162]− = 633 | 4.34 (d, J = 7.8) 4.41 (d, J = 8.0) 4.43 (d, J = 7.8) 5.55 (d, J = 8.0) |
| 11 | 63.4 | 472 | 3 × Hexose | [M957 − H − 162 − 162 − 162]− = 471 | 4.27 (d, J = 7.8) 4.34 (d, J = 7.8) 4.41 (d, J = 7.8) 5.24 * (t, J = 3.4) 5.53 (d, J = 8.0) |
| 12 | 64.0 | 458 | 3 × Hexose | [M943 − H − 162 − 162 − 162]− = 457 | 4.27 (d, J = 7.8) 4.34 (d, J = 7.8) 4.41 (d, J = 7.8) |
| 13 | 66.2 | 619 ** | 2 × Hexose | [M943 − H − 162 − 162]− = 457 | 4.17 (d, J = 7.8) 4.27 (d, J = 7.8) 4.34 (d, J = 7.8) 4.35 (d, J = 7.8) 4.41 (d, J = 7.8) 5.20 * (t, J = 3.4) |
| 14 | 66.6 | 472 | 3 × Hexose | [M957 − H − 162 − 162 − 162]− = 471 | |
| 15 | 67.2 | 458 | 3 × Hexose | [M943 – H − 162 − 162 − 162]− = 457 | 4.18 (d, J = 7.8) 4.26 (d, J = 7.8) 4.35 (d, J = 7.8) 4.41 (d, J = 7.8) 5.25 * (t, J = 3.4) 5.38 (d, J = 8.3) |
| 16 | 67.6 | 472 | 4 × Hexose | [M1119 − H − 162 − 162 − 162 − 162]− = 471 | |
| 17 | 68.2 | 458 | 3 × Hexose | [M943 − H − 162 − 162 − 162]− = 457 | 4.18 (d, J = 7.8) 4.22 (d, J = 7.8) 4.27 (d, J = 6.0) 4.30 (d, J = 6.0) 4.35 (d, J = 6.9) 4.41 (d, J = 7.8) 5.20 * (t, J = 3.4) 5.25 * (t, J = 3.4) |
| 18 | 68.3 | 472 | 3 × Hexose | [M957 − H − 162 – 162 − 162]− = 471 | |
| 19 | 68.5 | 472 | 2 × Methylpentose 1 × Hexose | [M925 − H − 146 − 146 − 162]− = 471 | |
| 20 | 69.1 | 472 | 1 × Methylpentose 2 × Hexose | [M941 − H − 146 − 162 − 162]− = 471 | 4.16 (d, J = 6.6) 4.35 (d, J = 7.8) 4.41 (d, J = 7.8) 5.20 * (t, J = 3.4) 5.25 * (t, J = 3.4) |
| 21 | 69.5 | 458 | 3 × Hexose | [M943 − H − 162 − 162 − 162]− = 457 | |
| 22 | 69.7 | 456 | 3 × Hexose | [M941 − H − 162 − 162 − 162]− = 455 | |
| 23 | 70.2 | 472 | 1 × Methylpentose 2 × Hexose | [M941 − H − 146 − 162 − 162]− = 471 | 4.35 (d, J = 7.8) 4.41 (d, J = 7.8) 5.25 * (t, J = 3.4) 5.30 * (t, J = 3.4) |
| 24 | 70.4 | 456 | 3 × Hexose | [M941 − H − 162 − 162 − 162]− = 455 | |
| 25 | 70.4 | 472 | 2 × Methylpentose 1 × Hexose | [M925 − H − 146 − 162 − 146]− = 471 | |
| 26 | 72.8 | 472 Hederagenin cellobioside | 2 × Hexose | [M795 – H − 162 − 162]− = 471 | 4.41 (d, J = 7.8) 4.42 (d, J = 7.8) 5.24 * (t, J = 3.4) |
| 27 | 75.8 | 456 Oleanolic ac. cellobioside | 2 × Hexose | [M779 − H − 162 − 162]− = 455 | 4.34 (d, J = 7.8) 4.41 (d, J = 7.8) 5.23 * (t, J = 3.4) |
| No. | RT (min) | Aglycone (Da) A | Sugar Moieties B | Fragmentation Pattern in Mass Spectra C | Anomeric Protons (ppm) D |
|---|---|---|---|---|---|
| 1 | 45.2 | 448 | 5 × Hexose | [M1257 − H − 162 − 162 − 162 − 162 − 162]− = 447 | 4.13 (d, J = 7.8) 5.20 (d, J = 7.8) 5.28 (d, J = 7.8) |
| 2 | 45.7 | 464 | 3 × Methylpentose 2 × Hexose | [M1225 − H − 162 − 146 − 162 − 146 − 146]− = 463 | |
| 3 | 46.4 | 478 | 1 × Pentose 1 × Hexose 1 × Methylpentose | [M917 − H − 132 − 162 − 146]− = 477 | 4.13 (d, J = 7.8) 4.63 (d, J = 7.8) 5.17 (d, J = 7.8) |
| 4 | 54.1 | 448 | 2 × Methylpentose 1 × Hexose | [M901 − H − 146 − 146 − 162]− = 447 | 4.35 (d, J = 8.0) 4.39 (d, J = 8.0) 4.58 (d, J = 8.0) 4.60 (d, J = 8.0) 5.12 (d, J = 7.8) 5.23 (d, J = 7.8) |
| 5 | 54.8 | 464 | 3 × Methylpentose | [M901 − H − 146 − 146 − 146]− = 463 | |
| 6 | 56.8 | 448 | 2 × Methylpentose 1 × Hexose | [M901 − H − 146 − 146 − 162]− = 447 | 4.04 (d, J = 8.0) 4.42 (d, J = 8.0) 4.75 (d, J = 7.8) |
| 7 | 57.7 | 448 | 2 × Methylpentose 1 × Hexose | [M901 − H − 146 − 146 − 162]− = 447 | 4.29 (d, J = 7.8) 4.43 (d, J = 7.8) 4.66 (d, J = 7.8) |
| 8 | 58.4 | 609* | 1 × Methylpentose 1 × Hexose | [M917 − H − 146 − 162]− = 609 | 4.19 (d, J = 7.8) 4.31 (d, J = 7.8) 5.14 (d, J = 7.8) |
| 9 | 59.6 | 448 | 1 × Methylpentose 2 × Hexose | [M917 − H − 162 − 162 − 146]− = 447 | 4.19 (d, J = 7.8) 4.39 (d, J = 7.8) 4.53 (d, J = 7.8) |
| 10 | 61.2 | 448 | 2 × Methylpentose 1 × Hexose | [M901 − H − 146 − 146 − 162]− = 447 | 4.37 (d, J = 7.8) 4.42 (d, J = 8.0) 4.60 (d, J = 8.0) 4.62 (d, J = 8.0) 5.09 (d, J = 7.8) 5.20 (d, J = 7.8) |
| 11 | 61.8 | 448 | 2 × Methylpentose 1 × Hexose | [M901 − H − 146 − 146 − 162]− = 447 | |
| 12 | 62.7 | 472 | 2 × Hexose | [M795 − H − 162 − 162]− = 471 | 4.19 (d, J = 7.8) 5.14 (d, J = 7.8) |
| 13 | 63.1 | 448 | 2 × Methylpentose 1 × Hexose | [M901 − H − 146 − 146 − 162]− = 447 | 4.42 (d, J = 8.0) 4.60 (d, J = 8.0) 4.62 (d, J = 8.0) |
| 14 | 64.1 | 623* | 1 × Methylpentose 1 × Hexose | [M931 − H − 146 − 162]− = 623 | 4.15 (d, J = 8.0) 4.19 (d, J = 7.8) 4.62 (d, J = 8.0) 4.68 (d, J = 8.0) 5.13 (d, J = 7.8) 5.28 (d, J = 7.8) 5.57 (d, J = 8.0) |
| 15 | 64.6 | 448 | 2 × Methylpentose 1 × Hexose | [M901 − H − 146 − 146 − 162]− = 447 | |
| 16 | 77.4 | 474 | 4 × Hexose | [M1121 − H − 162 − 162 − 162 − 162 ]− = 473 | 4.30 (d, J = 7.8) 4.40 (d, J = 7.8) 4.41 (d, J = 7.8) 4.71 (d, J = 8.0) |
| 17 | 78.4 | 474 | 3 × Hexose | [M959 − H − 162 − 162 − 162 ]− = 473 | 4.27 (d, J = 7.8) 4.41 (d, J = 7.8) 4.41 (d, J = 7.8) |
| 18 | 79.1 | 458 | 4 × Hexose | [M1105 − H − 162 − 162 − 162 − 162]− = 457 | 4.27 (d, J = 7.8) 4.34 (d, J = 7.8) 4.38 (d, J = 7.8) 4.39 (d, J = 7.8) 4.41(d, J = 7.8) 4.62 (d, J = 7.8) 5.53 (d, J = 8.2) 5.60 (d, J = 8.2) |
| 19 | 79.4 | 472 | 4 × Hexose | [M1119 − H − 162 − 162 − 162 − 162 ]− = 471 | |
| 20 | 79.8 | 474 | 1 × Methylpentose 3 × Hexose | [M1105 − H − 146 − 162 − 162 − 162 ]− = 473 | |
| 21 | 80.4 | 458 | 4 × Hexose | [M943 − H − 162 − 162 − 162 ]− = 457 | 4.27 (d, J = 7.8) 4.31 (d, J = 7.8) 4.34 (d, J = 7.8) 4.38 (d, J = 7.8) 4.39 (d, J = 7.8) 4.41 (d, J = 7.8) 4.61 (d, J = 7.8) |
| 22 | 80.8 | 472 | 1 × Methylpentose 3 × Hexose | [M1103 − H − 162 − 162 − 146 − 162 ]− = 471 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khakimov, B.; Tseng, L.H.; Godejohann, M.; Bak, S.; Engelsen, S.B. Screening for Triterpenoid Saponins in Plants Using Hyphenated Analytical Platforms. Molecules 2016, 21, 1614. https://doi.org/10.3390/molecules21121614
Khakimov B, Tseng LH, Godejohann M, Bak S, Engelsen SB. Screening for Triterpenoid Saponins in Plants Using Hyphenated Analytical Platforms. Molecules. 2016; 21(12):1614. https://doi.org/10.3390/molecules21121614
Chicago/Turabian StyleKhakimov, Bekzod, Li Hong Tseng, Markus Godejohann, Søren Bak, and Søren Balling Engelsen. 2016. "Screening for Triterpenoid Saponins in Plants Using Hyphenated Analytical Platforms" Molecules 21, no. 12: 1614. https://doi.org/10.3390/molecules21121614
APA StyleKhakimov, B., Tseng, L. H., Godejohann, M., Bak, S., & Engelsen, S. B. (2016). Screening for Triterpenoid Saponins in Plants Using Hyphenated Analytical Platforms. Molecules, 21(12), 1614. https://doi.org/10.3390/molecules21121614