
Water Will Be the Coal of the Future—The Untamed Dream of Jules Verne for a Solar Fuel



 and
and
Abstract
:
1. Introduction
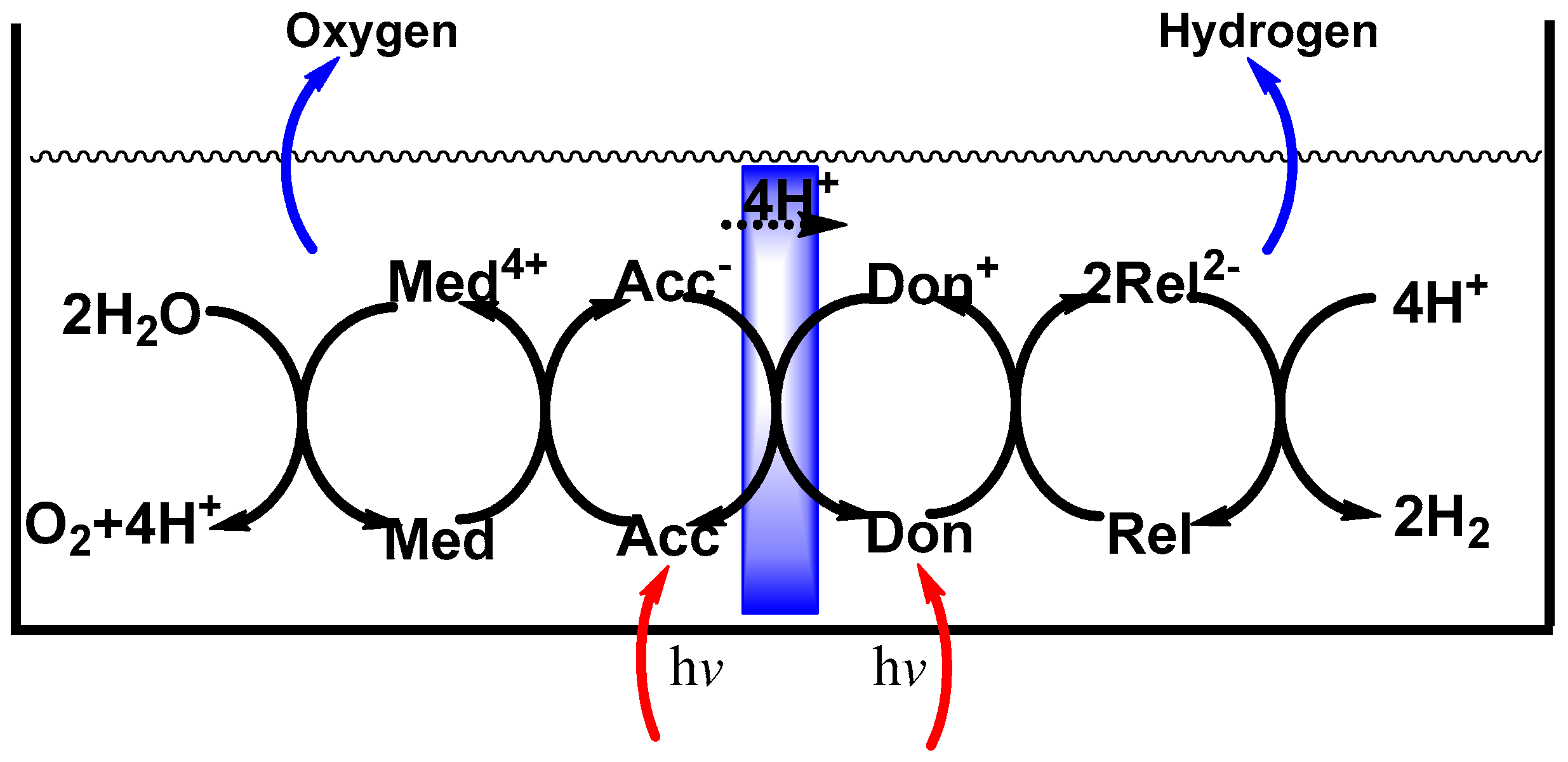
2. Approaches to the Photosplitting of Water
- (1)
- Among the active photocatalysts are most of the alkali halides [29], the non-reducible metal oxides (e.g., BeO, MgO, CaO, γ-Al2O3, and SiO2) and the reducible metal oxides (e.g., La2O3 and ZrO2). These oxides differ with regard to dissociative and molecular adsorption of water molecules at regular and defective surface sites [30].
- (2)
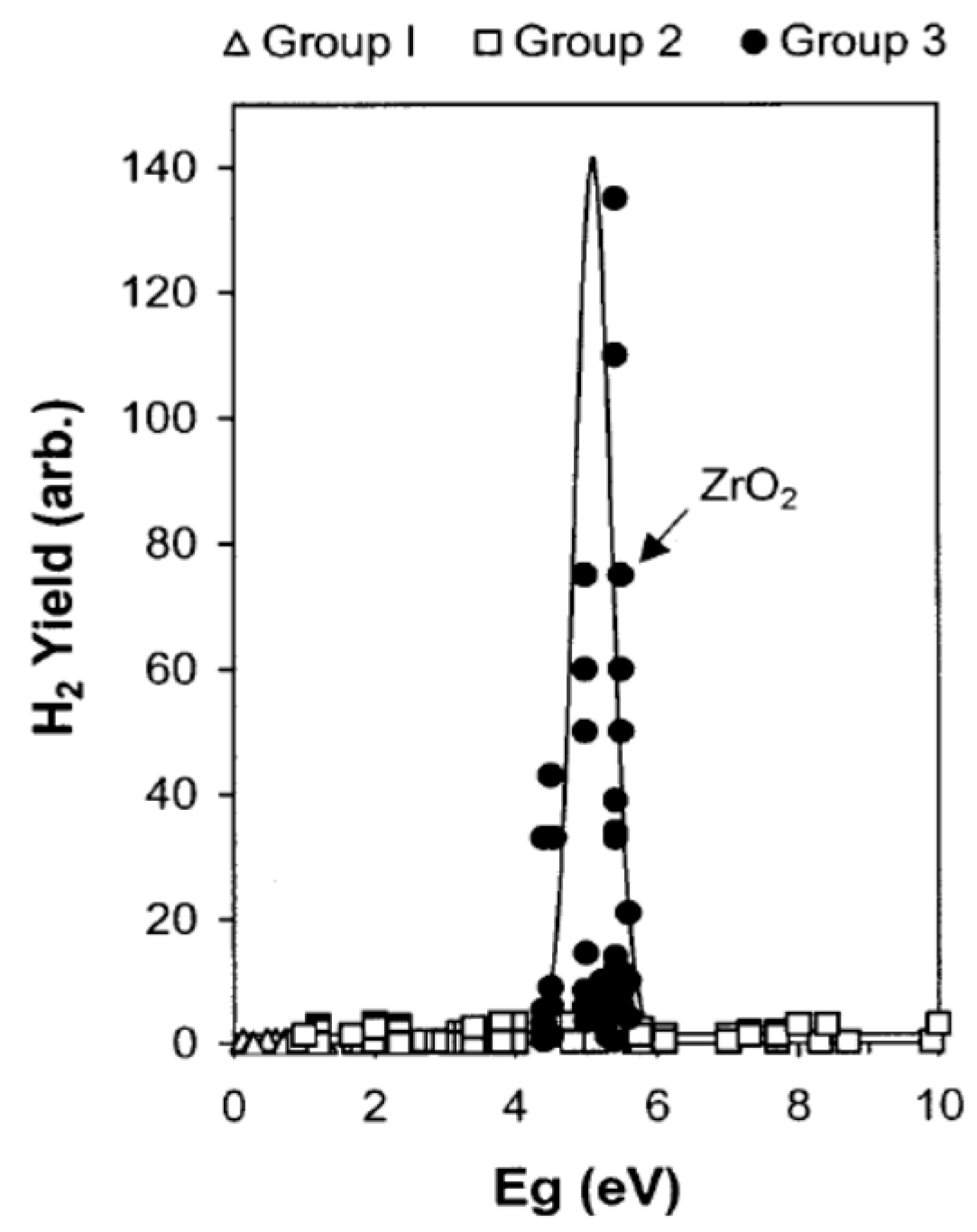
- A noticeable activity is shown by the oxides (BeO, γ-Al2O3 MgO, and SiO2) and alkali halides (except KI, RbI, and CsI) with bandgap energies, Ebg, or energy of the exciton bands for alkali halides [29], greater than 6.2 eV. Accordingly, photoexcitation of such solids by (non-vacuum) UV/visible light into their fundamental absorption bands was not possible.
- (3)
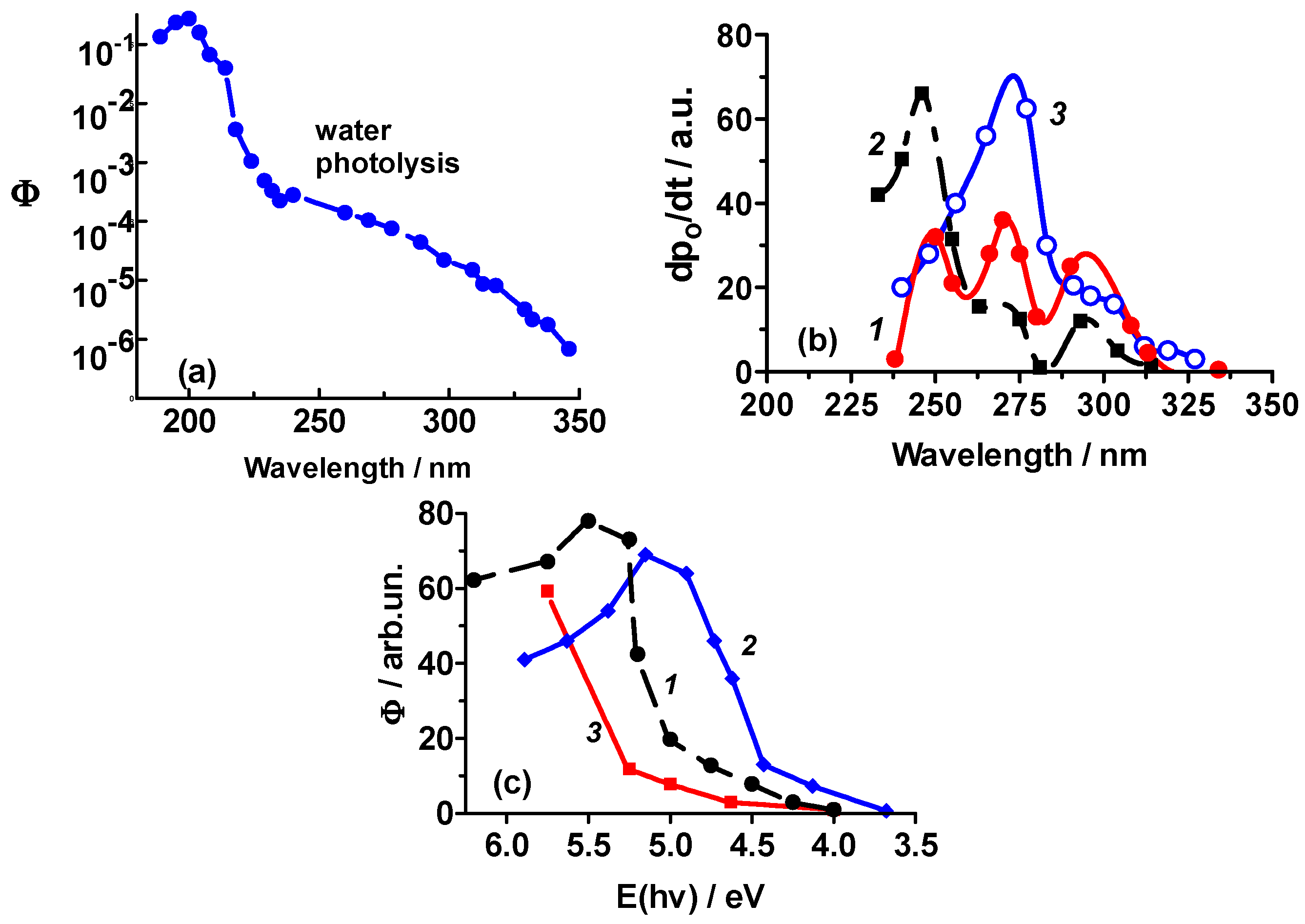
- The red limit of the photosplitting of adsorbed water lies in the rather narrow energy range of 5.4–4.75 eV (230–260 nm) for all the samples examined. The shift of the red limit for the photosplitting of water adsorbed on metal oxides and alkali halides is about 1.0–1.75 eV. A red limit (240 nm or 5.15 eV) within this interval was also reported for the photolysis of water adsorbed on the Pt(111) surface at 85 K [31].
- (4)
- Illumination of samples with adsorbed water evolves H2 into the gas phase, while oxygen tends to remain adsorbed at the surface. Photolytic oxygen can be detected in some cases in the gas phase on heating the samples after irradiation (Table 1). At times, formation of oxygen in the reaction manifested itself via formation of secondary products (e.g., CO) through interaction of oxygen with some surface organic contaminants. In the case of a few catalysts (BeO and ZrO2 are good examples [5,6]) oxygen began to evolve into the gas phase in the expected stoichiometric ratio for water splitting (at a definite time delay) after prolonged illumination at ambient temperature.
- (5)
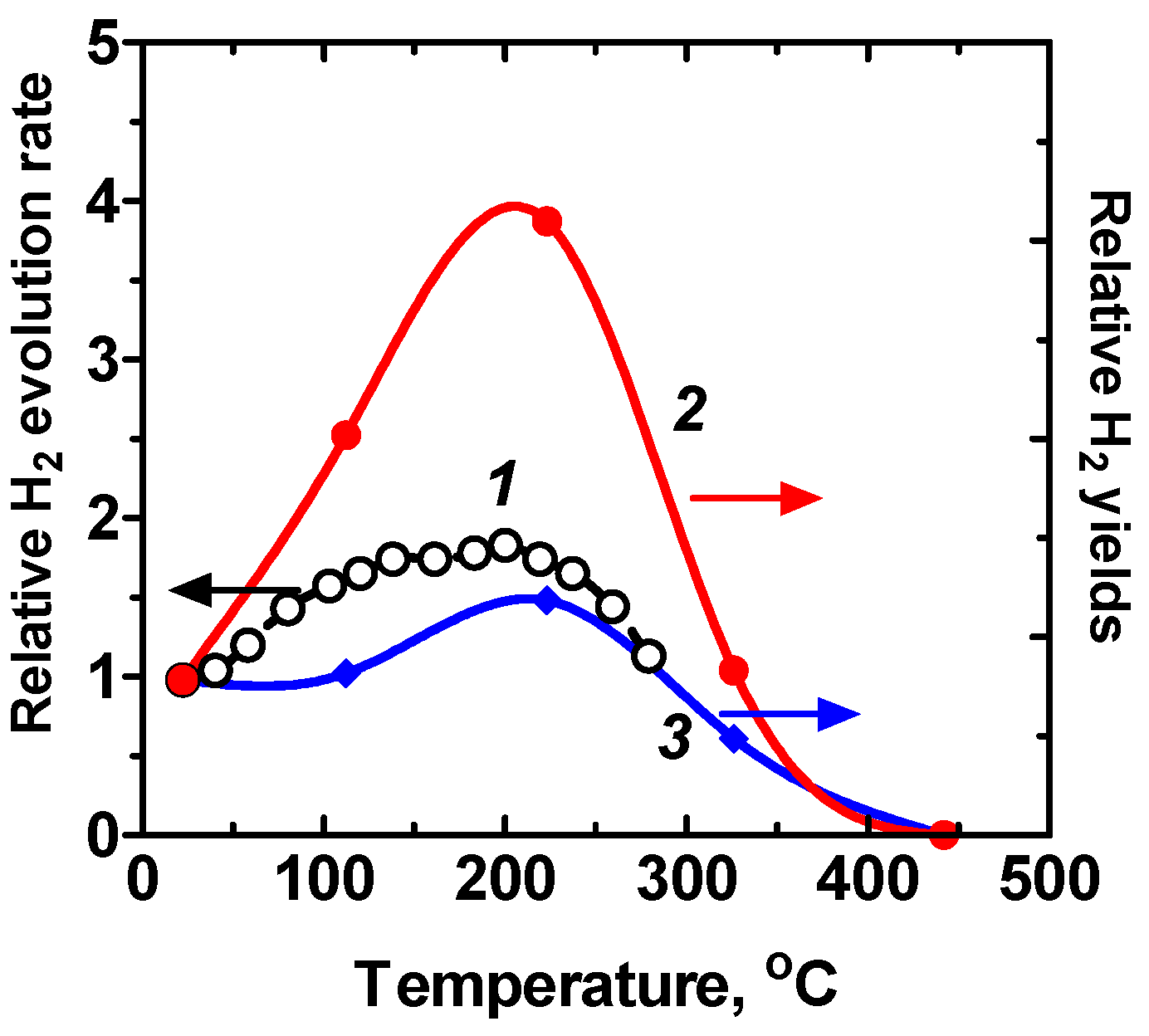
- The rate of photodecomposition of water on metal oxides can usually be enhanced by moderate heating of the samples in the presence of adsorbed water. This infers some selective activity of chemisorbed water species.
- (6)
- In the case of BeO, for which experiments were carried out using a water vapor flow photoreactor, molecular hydrogen and molecular oxygen evolved in the gas phase in stoichiometric amounts at elevated temperature (T = 200 °C) under UV illumination for a few hours without decrease in activity [6]. A noticeable decrease of the reaction rate occurred, however, under prolonged illumination at ambient temperature for this oxide and other solids.
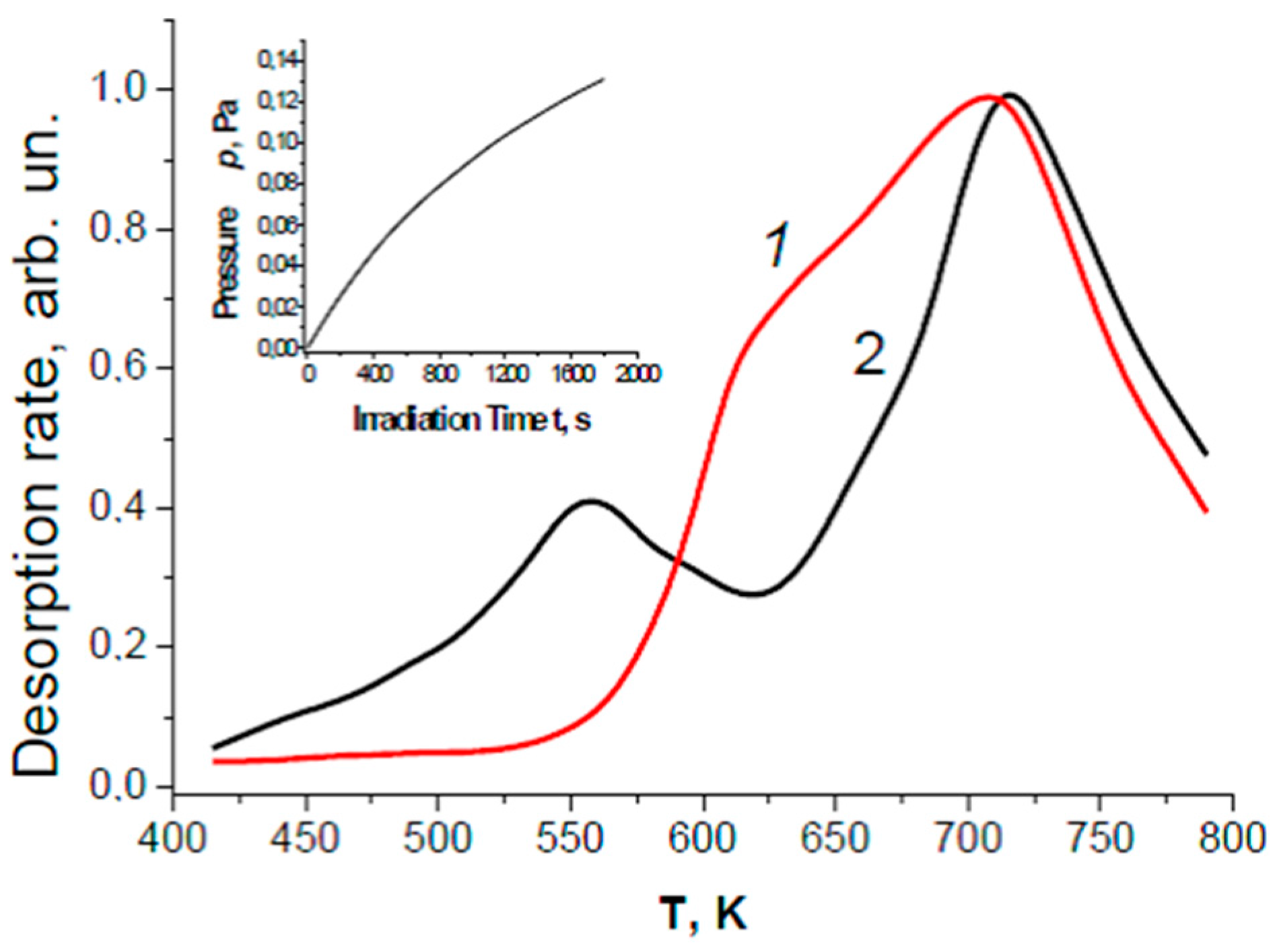
2.1. Adsorbed Active Water Species Subsequent to UV-Induced Dissociation
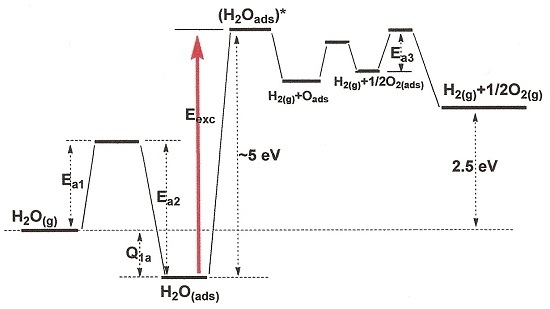
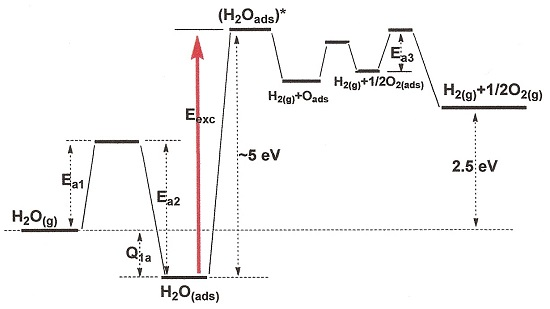
3. Photoexcitation of Adsorbed Water
3.1. Direct Photolysis of Adsorbed Water Molecules
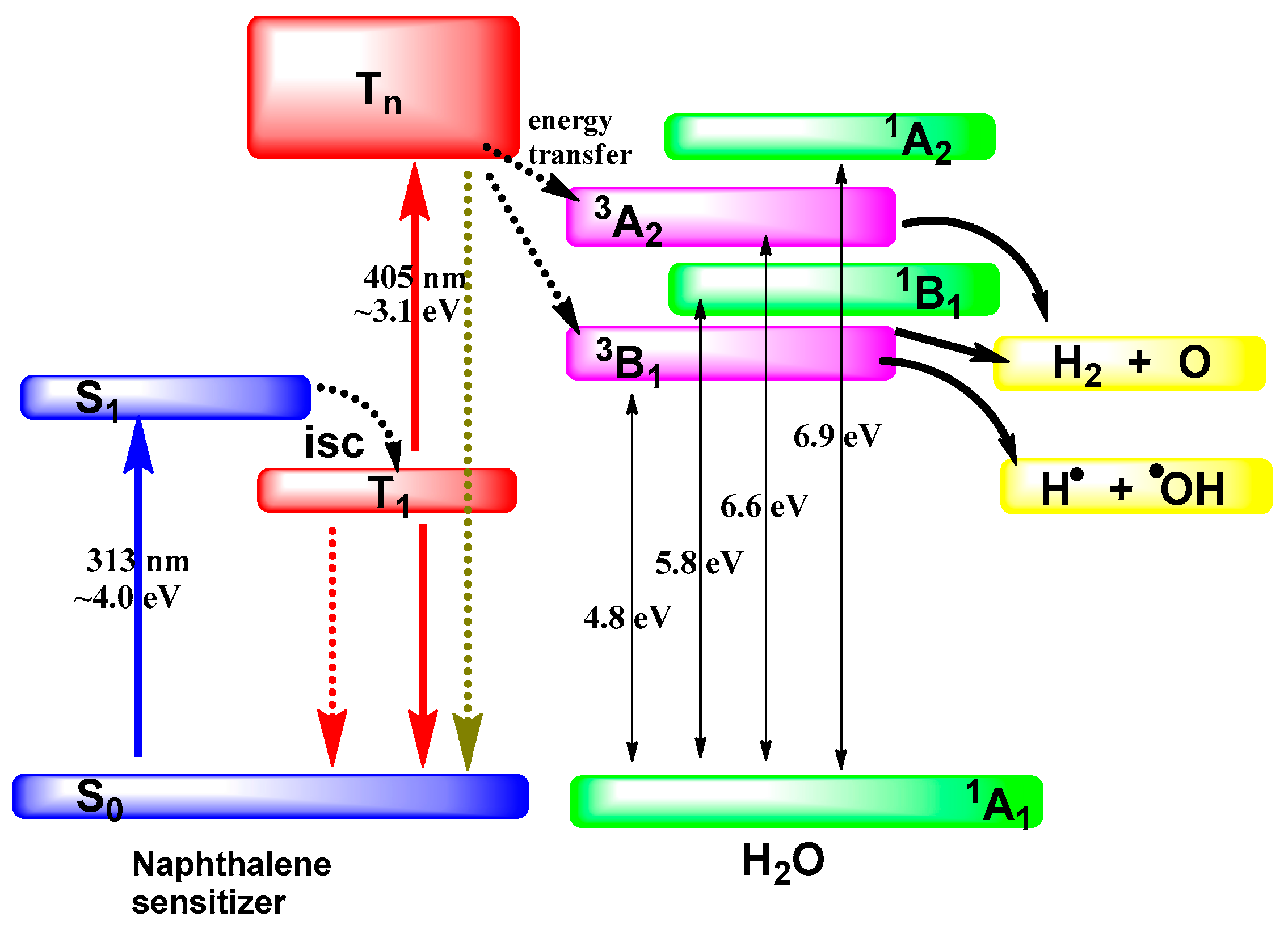
3.2. Photolysis of Adsorbed Water Molecules via Photoexcitation of the Solid Photocatalyst
3.3. Quantum Yields of the Photosplitting of Adsorbed Water and the Photoadsorption of O2 and H2 on Metal Oxides
4. Mechanistic Considerations of the Photosplitting of Adsorbed Water
5. Final Remarks on Photosplitting of Water and Future Outlook
Acknowledgments
Conflicts of Interest
References
- Jules, V. The Mysterious Island; Pierre-Jules Hetzel Publisher: Paris, France, 1874. [Google Scholar]
- Giacomo, C. The Photochemistry of the Future. Science 1912, 36, 385–394. [Google Scholar]
- Venturi, M.; Balzani, V.; Gandolfi, M.T. Fuels from Solar Energy. A dream of Giacomo Ciamician, the father of photochemistry. In Proceedings of the 2005 Solar World Congress, Orlando, FL, USA, 6–12 August 2005.
- Kotel’nikov, V.A.; Terenin, A.N. Photochemical processes on aluminum oxide surface. Dokl. Akad. Nauk SSSR 1967, 174, 1366–1369. [Google Scholar]
- Basov, L.L.; Efimov, Y.P.; Solonitzyn, Y.P. The studies of water photolysis in adsorption state. Uspekhi Fotoniki Adv. Photonics 1974, 4, 12–18. [Google Scholar]
- Basov, L.L.; Kotel’nikov, V.A.; Solonitzyn, Y.P. Photodissociation of simple molecules on oxide adsorbents. In Spectroscopy of Molecular Photo Transformations; Krasnovsky, A.A., Ed.; Nauka: Leningrad, Russia, 1977; pp. 228–239. [Google Scholar]
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Balzani, V.; Moggi, L.; Manfrin, M.F.; Bolletta, F.; Gleria, M. Solar Energy Conversion by Water Photodissociation. Science 1975, 189, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.R. Solar Fuels. Science 1978, 202, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, V.N.; Lisachenko, A.A.; Vilesov, T.I. Photocatalytic decomposition of water adsorbed on BeO. High Energy Chem. 1973, 7, 230–233. [Google Scholar]
- Jaeger, C.D.; Bard, A.J. Spin trapping and electron spin resonance detection of radical intermediates in the photodecomposition of water at TiO2 particulate systems. J. Phys. Chem. 1979, 83, 3146–3152. [Google Scholar] [CrossRef]
- Ueno, A.; Kakuta, N.; Park, K.H.; Finlayson, M.F.; Bard, A.J.; Campion, A.; Fox, M.A.; Webber, S.E.; White, J.M. Silica-Supported ZnS–CdS Mixed Semiconductor Catalysts for Photo-generation of Hydrogen. J. Phys. Chem. 1985, 89, 3828–3833. [Google Scholar] [CrossRef]
- Bard, A.J.; Fox, M.A. Artificial Photosynthesis: Solar Splitting of Water to Hydrogen and Oxygen. Acc. Chem. Res. 1995, 28, 141–145. [Google Scholar] [CrossRef]
- Gonzalez, M.G.; Oliveros, E.; Worner, M.; Braun, A.M. Vacuum-ultraviolet photolysis of aqueous reaction systems. J. Photochem. Photobiol. C Photochem. Rev. 2004, 5, 225–246. [Google Scholar] [CrossRef]
- Ismail, A.A.; Bahnemann, D.W. Photochemical splitting of water for hydrogen production by photo- catalysis: A review. Sol. Energy Mater. Sol. Cells 2014, 128, 85–101. [Google Scholar] [CrossRef]
- Shi, Z.; Wen, X.; Guan, Z.; Cao, D.; Luo, W.; Zou, Z. Recent progress in photoelectrochemical water splitting for solar hydrogen production. Ann. Phys. 2015, 358, 236–247. [Google Scholar] [CrossRef]
- Currao, A. Photoelectrochemical Water Splitting. Chimia 2007, 61, 815–819. [Google Scholar] [CrossRef]
- Chen, Z.; Dinh, H.N.; Miller, E. Photoelectrochemical Water Splitting—Standards, Experimental Methods, and Protocols; Springer: New York, NY, USA, 2013. [Google Scholar]
- Cho, S.; Jang, J.-W.; Lee, K.-H.; Lee, J.S. Research Update: Strategies for efficient photoelectrochemical water splitting using metal oxide photoanodes. APL Mater. 2014, 2, 010703. [Google Scholar] [CrossRef]
- Protti, S.; Albini, A.; Serpone, N. Photocatalytic generation of solar fuels from the reduction of H2O and CO2: A look at the patent literature. Phys. Chem. Chem. Phys. 2014, 16, 19790–19827. [Google Scholar] [CrossRef] [PubMed]
- Serpone, N.; Emeline, A.V.; Ryabchuk, V.K.; Kuznetsov, V.N.; Artem’ev, Y.M.; Horikoshi, S. Why do Hydrogen and Oxygen Yields from Semiconductor-based Photocatalyzed Water Splitting Remain Disappointingly Low? Intrinsic and extrinsic factors impacting surface redox reactions. ACS Energy Lett. 2016, 1, 931–948. [Google Scholar] [CrossRef]
- Jafari, T.; Moharreri, E.; Amin, A.S.; Miao, R.; Song, W.; Suib, S.L. Photocatalytic Water Splitting—The Untamed Dream: A Review of Recent Advances. Molecules 2016, 21, 900. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Chen, Z.; Gao, T.; Niu, F.; Qin, L.; Huang, Y. Hydrogen Generation from Water Splitting on TiO2 Nanotube-Array-Based Photocatalysts. Energy Technol. 2015, 3, 888–895. [Google Scholar] [CrossRef]
- Li, R.; Weng, Y.; Zhou, X.; Wang, X.; Mi, Y.; Chong, R.; Han, H.; Li, C. Achieving overall water splitting using titanium dioxide-based photocatalysts of different phases. Energy Environ. Sci. 2015, 8, 2377–2382. [Google Scholar] [CrossRef]
- Rodríguez-Hernández, F.; Tranca, D.C.; Szyja, B.M.; van Santen, R.A.; Martínez-Mesa, A.; Uranga-Piña, L.; Seifert, G. Water Splitting on TiO2-Based Electrochemical Cells: A Small Cluster Study. J. Phys. Chem. C 2016, 120, 437–449. [Google Scholar] [CrossRef]
- Salas, S.E. Photocatalytic Water Splitting Using a Modified PtTiO2. Kinetic Modeling and Hydrogen Production Efficiency. Ph.D. Thesis, The University of Western Ontario, London, ON, Canada, August 2013. [Google Scholar]
- Henderson, M.A. Fundamental Investigations of Water Splitting on Model TiO2 Photocatalysts Doped for Visible Light Absorption. Available online: https://www.hydrogen.energy.gov/pdfs/review08/bes_10_henderson.pdf (accessed 8 November 2016).
- Oudenhoven, J.; Scheijen, F.; Wolffs, M. Fundamentals of Photocatalytic Water splitting by Visible light. Chemistry of Catalytic System 2: Photocatalysis. 2004. Available online: http://large.stanford.edu/courses/2011/ph240/waisberg2/docs/oudenhoven.pdf (accessed 8 November 2016).
- Basov, L.L.; Solonitzin, Y.P.; Ryabchuk, V.K. Photoadsorption, photooxidation and photodecomposition of simple molecules on alkali halides surfaces. React. Kinet. Catal. Lett. 1988, 32, 119–124. [Google Scholar]
- Freund, H.-J.; Dillmann, B.; Seiferth, O.; Klivenyi, G.; Bender, M.; Ehrlich, D.; Hemmerich, I.; Cappus, D. Molecules on oxide surface. Catal. Today 1996, 32, l–10. [Google Scholar] [CrossRef]
- Gilarowsky, G.; Erley, V.; Ibach, H. Photochemical reactions of water adsorbed on Pt(111). Surf. Sci. 1996, 351, 156–164. [Google Scholar] [CrossRef]
- Serpone, N.; Emeline, A.V.; Horikoshi, S.; Kuznetsov, V.N.; Ryabchuk, V.K. On the genesis of hetero-geneous photocatalysis: A brief historical perspective in the period 1910 to the mid 1980s. Photochem. Photobiol. Sci. 2012, 11, 1121–1150. [Google Scholar] [CrossRef] [PubMed]
- De Boer, J.H. Electron Emission and Adsorption Phenomena; Cambridge University Press: Cambridge, UK, 1935. [Google Scholar]
- De Boer, J.H.; Houben, G. Misleading color reactions. Proc. Konickl. Nederland. Akad. Wet. 1951, 54, 421–429. [Google Scholar]
- Terenin, A.N. Izbrannie Trudi; Nauka: Leningrad, Russia, 1975; Volume 3, p. 346. [Google Scholar]
- Roth, O.; Dahlgren, B.; LaVerne, J.A. Radiolysis of Water on ZrO2 Nanoparticles. J. Phys. Chem. C 2012, 116, 17619–17624. [Google Scholar] [CrossRef]
- Basov, L.L.; Drobinin, A.N.; Filimonov, V.N. Studies of the effect of hydration of surface at water photolysis and photoadsorption of oxygen and hydrogen on beryllium oxide. Kinetika i Kataliz 1982, 23, 108–112. [Google Scholar]
- Readhead, P.A. Thermal desorption of gases. Vacuum 1962, 12, 203–211. [Google Scholar] [CrossRef]
- Poston, M.J.; Aleksandrov, A.B.; Sabo, D.E.; Zhang, Z.J.; Orlando, T.M. UV Photon-Induced Water Decomposition on Zirconia Nanoparticles. J. Phys. Chem. C 2014, 118, 12789–12795. [Google Scholar] [CrossRef]
- Petrik, N.G.; Alexandrov, A.B.; Vall, A.I. Interfacial Energy Transfer during Gamma Radiolysis of Water on the Surface of ZrO2 and Some Other Oxides. J. Phys. Chem. B 2001, 105, 5935–5944. [Google Scholar] [CrossRef]
- Okabe, H. Photochemistry of Small Molecules; John Wiley & Sons: New York, NY, USA, 1978. [Google Scholar]
- Yi, W.; Park, J.; Lee, J. Photodissociation dynamics of water at Lyman alpha (121.6 nm). Chem. Phys. Lett. 2007, 439, 46–49. [Google Scholar] [CrossRef]
- Bachler, V.; Gartne, W. Electric Field-Assisted Photochemical Water Splitting Should Operate with 287 nm Light. Photochem. Photobiol. 2016, 92, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Strehlow, W.H.; Cook, E.L. Compilation of energy band-gaps in elemental and binary compound semiconductors and insulators. J. Phys. Chem. Ref. Data 1973, 2, 163–193. [Google Scholar] [CrossRef]
- Sterrer, M.; Diwald, O.; Knozinger, E.; Sushko, P.V.; Shluger, A.L. Energies and dynamics of photoinduced electron and hole processes on MgO powders. J. Phys. Chem. B 2002, 106, 12478–12482. [Google Scholar] [CrossRef]
- Diwald, O.; Sterrer, M.; Knozinger, E.; Sushko, P.V.; Shluger, A.L. Wavelength selective excitation of surface oxygen anions on highly dispersed MgO. J. Chem. Phys. 2002, 116, 1707–1712. [Google Scholar] [CrossRef]
- Volodin, A.M. Photoinduced phenomena on the surface of wide band-gap oxide catalysts. J. Catal. 2000, 58, 103–114. [Google Scholar] [CrossRef]
- Crawford, H., Jr. Defects and Defect Processes in Ionic Oxides: Where Do We Stand Today? Nucl. Instrum. Methods Phys. Res. 1984, B1, 159–165. [Google Scholar] [CrossRef]
- Kuznetsov, V.N.; Lisachenko, L.L. Spectral evidence of intrinsic defects participation in surface photoreactions on wide band gap oxides. Russ. J. Phys. Chem. 1991, 6, 1589–1593. [Google Scholar]
- Kuznetsov, V.N.; Lisachenko, A.A.; Vilesov, T.I. Photodissociation of water sensitized by hafnium oxide. Kinetika i Kataliz 1972, 13, 1082–1084. [Google Scholar]
- Emeline, A.V.; Kuzmin, G.N.; Purevdorj, D.; Ryabchuk, V.K.; Serpone, N. Spectral Dependencies of the Quantum Yield of Photochemical Processes on the Surface of Wide Band Gap Solids. 3. Gas/Solid Systems. J. Phys. Chem. B 2000, 104, 2989–2999. [Google Scholar] [CrossRef]
- Emeline, A.V.; Ryabchuk, V.K.; Serpone, N. Spectral Dependencies of the Quantum Yield of Photochemical Processes on the Surface of Nano-/Micro-Particulates of Wide Band Gap Metal Oxides: I. Theoretical Approach. J. Phys. Chem. B. 1999, 103, 1316–1324. [Google Scholar] [CrossRef]
- Garibov, A.A.; Parmon, V.N.; Agaev, T.N.; Kasumov, R.D. Influence of the polymorphous forms of the oxide and the temperature on the transfer of energy during radiation-induced heterogeneous processes in Al2O3 + H2O system. High Energy Chem. 1991, 25, 86–90. [Google Scholar]
- Claydon, C.R.; Segal, G.A.; Taylor, H.S. Theoretical interpretation of the optical and electron scattering spectra of H2O. J. Chem. Phys. 1971, 54, 3799–3815. [Google Scholar] [CrossRef]
- Korotkov, V.I.; Basov, L.L.; Kholmogorov, V.E. Two-quantum reactions of adsorbed molecules—Sensitized water photolysis. Dokl. Akad. Sci. USSR 1973, 209, 392–395. [Google Scholar]
- Sharma, D.; Paterson, M.J. Ground and excited states of naphthalene–water (naphtha–W6) clusters: A computational study. RSC Adv. 2015, 5, 28281–28291. [Google Scholar] [CrossRef]
- Sony, P.; Shukla, A. Large-scale correlated study of excited state absorptions in naphthalene and anthracene. J. Chem. Phys. 2009, 131, 014302. [Google Scholar] [CrossRef] [PubMed]
- Rubio, M.; Serrano-Andres, L.; Merchan, M. Excited states of the water molecules: Analysis of the valence and Rydberg character. J. Chem. Phys. 2008, 128, 104305. [Google Scholar] [CrossRef] [PubMed]
- Winter, N.W.; Goddard, W.A., III; Bobrowitz, F.W. Configuration interaction studies of the excited states of water. J. Chem. Phys. 1975, 62, 4325–4331. [Google Scholar] [CrossRef]
- Birks, J.B.; Christophorou, L.G.; Huebner, R.H. Excited electronic states of benzene and naphthalene. Nature 1968, 217, 809–812. [Google Scholar] [CrossRef]
- Schrauzer, G.N.; Guth, T.D. Photocatalytic reactions. 1. Photolysis of water and photoreduction of nitrogen on titanium dioxide. J. Am. Chem. Soc. 1977, 99, 7189–7193. [Google Scholar] [CrossRef]
- Kawai, T.; Sakata, T. Photocatalytic decomposition of gaseous water over TiO2 and TiO2-RuO2 surfaces. Chem. Phys. Lett. 1980, 72, 87–89. [Google Scholar] [CrossRef]
- Smart, J.C.; Pinsky, B.L.; Fredrich, M.F.; Day, V.W. On the Photoassisted Decomposition of Water at the Gas-Solid Interface on TiO2. J. Am. Chem. Soc. 1979, 101, 4371–4373. [Google Scholar] [CrossRef]
- Reddy, V.R.; Huang, D.W.; Lee, J.S. Photocatalytic Water Splitting over ZrO2 Prepared by Precipitation Method. Korean J. Chem. Eng. 2003, 20, 1026–1029. [Google Scholar] [CrossRef]
- Pan, Z.; Hisatomi, T.; Wang, Q.; Chen, S.; Nakabayashi, M.; Shibata, N.; Pan, C.; Takata, T.; Katayama, M.; Minegishi, T.; et al. Photocatalyst Sheets Composed of Particulate LaMg1/3Ta2/3O2N and Mo-Doped BiVO4 for Z-Scheme Water Splitting under Visible Light. ACS Catal. 2016, 6, 7188–7196. [Google Scholar] [CrossRef]
- Emeline, A.V.; Panasuk, A.V.; Sheremetyeva, N.; Serpone, N. Mechanistic studies of the formation of different states of oxygen on irradiated ZrO2 and the photocatalytic nature of photoprocesses from determination of turnover numbers. J. Phys. Chem. B 2005, 109, 2785–2792. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Lu, D.; Domen, K. Solar-Driven Z-scheme Water Splitting Using Modified BaZrO3−BaTaO2N Solid Solutions as Photocatalysts. ACS Catal. 2013, 3, 1026–1033. [Google Scholar] [CrossRef]
- Etacheri, V.; Di Valentin, C.; Schneider, J.; Bahnemann, D.; Pillai, S.C. Visible-light activation of TiO2 photocatalysts: Advances in theory and experiments. J. Photochem. Photobiol. C Photochem. Rev. 2015, 25, 1–29. [Google Scholar] [CrossRef]
- Zangeneh, H.; Zinatizadeh, A.A.L.; Habibi, M.; Akia, M.; Isa, M.H. Photocatalytic oxidation of organic dyes and pollutants in wastewater using different modified titanium dioxides: A comparative review. J. Ind. Eng. Chem. 2015, 26, 1–36. [Google Scholar] [CrossRef]
- Wang, M.; Ye, M.; Iocozzia, J.; Lin, C.; Lin, Z. Plasmon-Mediated Solar Energy Conversion via Photocatalysis in Noble Metal/Semiconductor Composites. Adv. Sci. 2016, 3, 1600024. [Google Scholar] [CrossRef] [PubMed]
- Cates, E.L.; Chinnapongse, S.L.; Kim, J.-H.; Kim, J.-H. Engineering Light: Advances in Wavelength Conversion Materials for Energy and Environmental Technologies. Environ. Sci. Technol. 2012, 46, 12316–12328. [Google Scholar] [CrossRef] [PubMed]
- Serpone, N.; Emeline, A.V. Semiconductor Photocatalysis—Past, Present, and Future Outlook. J. Phys. Chem. Lett. 2012, 3, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, Y.; Lee, S.-T.; Yang, S.; Kang, Z. Coupling surface plasmon resonance of gold nanoparticles with slow-photon-effect of TiO2 photonic crystals for synergistically enhanced photo- electrochemical water splitting. Energy Environ. Sci. 2014, 7, 1409–1419. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Gong, Y.; Bai, S.; Ju, H.; Wang, C.; Xu, Q.; Zhu, J.; Jiang, J.; Xiong, Y. Towards full-spectrum photocatalysis: Achieving a Z-scheme between Ag2S and TiO2 by engineering energy band alignment with interfacial Ag. Nano Res. 2015, 8, 3621–3629. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
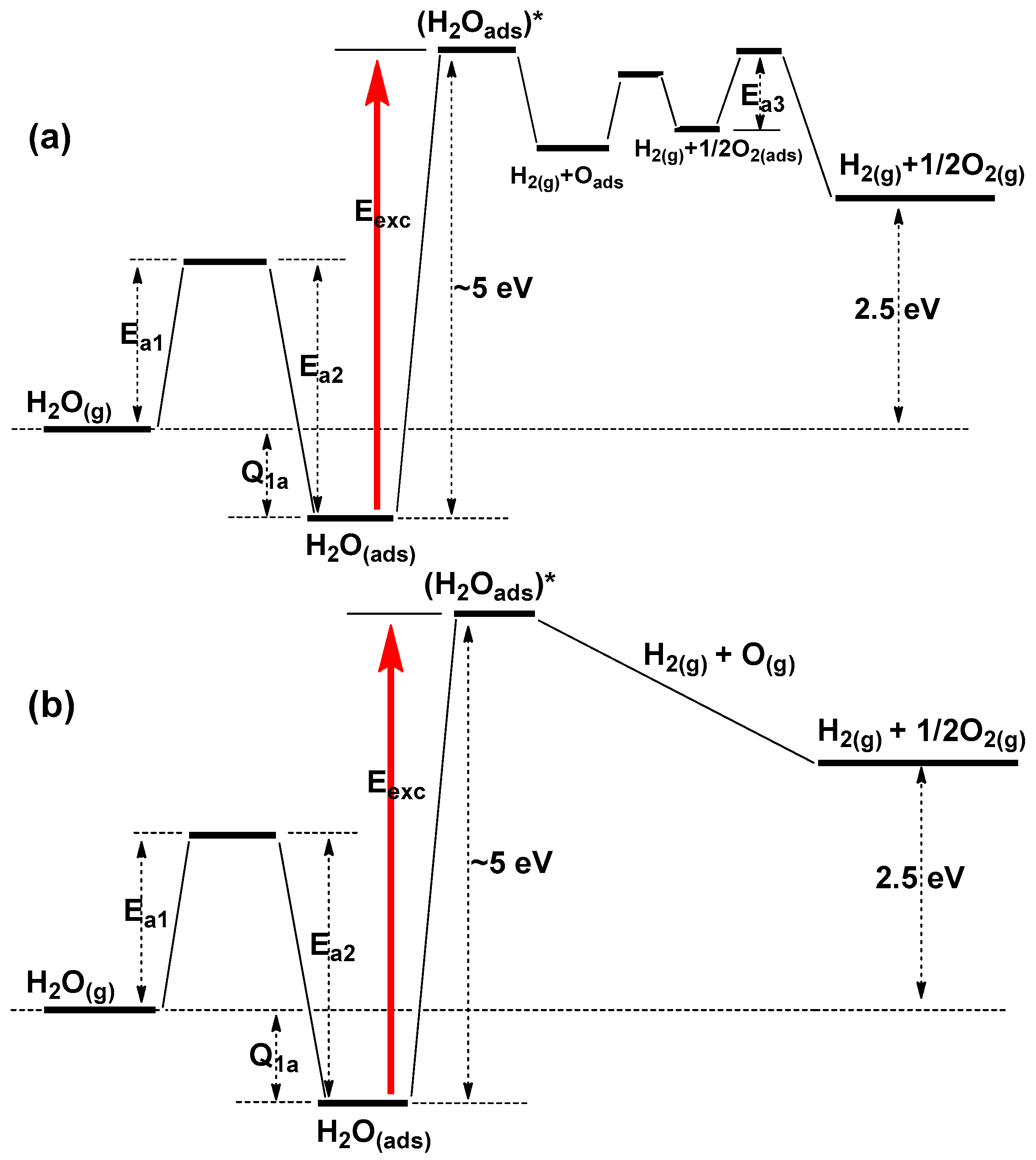{kind=link}
| Solid | Bandgaps (eV) | Relative Activity (Arbitrary Units) | Products Detected in the Gas Phase from the Photosplitting of H2O | |
|---|---|---|---|---|
| BeO | 10.5 | 100 | H2 | O2 |
| Be(OH)2 | - | 30 | H2 | O2 |
| γ-Al2O3 | 9.5 | 30 | H2 | O2 |
| ZrO2 | 5.0 | 10 | H2 | O2 |
| La2O3 | 5.5 | 9 | H2 | O2 |
| ThO2 | 5.7 | 8 | H2 | O2 |
| Na2(AlF6) | - | 7 | H2 | O2 |
| H3BO4 | - | 6 | H2 | O2 |
| HfO2 | 5.6 | 6 | H2 | O2 |
| SrO | 5.8 | 6 | H2 | O2 |
| SiO2 | 8.2 | 3 | H2 | O2 |
| Ho2O3 | 5.4 | 1 | H2 | O2 |
| Gd2O3 | 5.3 | <1 | H2 | O2 |
| Sc2O3 | 6.3 | <1 | H2 | O2 |
| MgO | 7.6 | <1 | H2 | O2 |
| TiO2 | 3.2 | <<1 | H2 | - |
| ZnO | 3.4 | <<1 | H2 | - |
| GeO2 | 5.6 | <<1 | H2 | - |
| Yb2O3 | 3.0 | <<1 | H2 | - |
| Dy2O3 | 4.9 | <<1 | H2 | - |
| Nb2O5 | (3.5) | <<1 | H2 | - |
| Zeolite NaX | - | <<1 | H2 | O2 |
| KBr | 7.5 | 7.0 | H2 | - |
| KCl | 8.7 | 0.4 | H2 | - |
| NaCl | 8.5 | 0.5 | H2 | - |
| H2O (“snow”) | - | 1.0 | H2 | O2 |
| Solids | Ebg (eV) | Red Limits of Photoreactions | ||
|---|---|---|---|---|
| Photoreduction of O2 (eV) | Photooxidation of H2 (eV) | Photosplitting of H2O (eV) | ||
| TiO2 | 3.2 | 2.2 | ~2.2 | ~3.1 |
| ZnO | 3.2 (3.4) * | 1.7 | - | - |
| MgO | 8.7 (7.6) * | 4.0 | 2.7 | 4.9 |
| γ-Al2O3 | 9.5 | 3.75 | ~4.0 | 3.75 |
| MgAlO4 | 9.0 | 4.75 | 4.75 | - |
| MgAlO4 (Cr) | 9.0 | 3.0 | 3.0 | - |
| ZrO2 | 5.4 (5.0) * | 3.1 | 3.1 | - |
| LiF | 13.6 | 3.9 | 4.9 | 4.9 |
| NaF | 11.6 | 3.9 | 4.9 | 4.9 |
| KBr | 7.5 | 3.7, 1.6 | - | 4.9 |
| SiO2 | >10 (8.2) * | 4.95 | - | 4.95 |
| SiO2 (V) | >10 | - | 3.7 | - |
| SiO2 naphthalene | >10 | - | - | 3.9 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryabchuk, V.K.; Kuznetsov, V.N.; Emeline, A.V.; Artem’ev, Y.M.; Kataeva, G.V.; Horikoshi, S.; Serpone, N. Water Will Be the Coal of the Future—The Untamed Dream of Jules Verne for a Solar Fuel. Molecules 2016, 21, 1638. https://doi.org/10.3390/molecules21121638
Ryabchuk VK, Kuznetsov VN, Emeline AV, Artem’ev YM, Kataeva GV, Horikoshi S, Serpone N. Water Will Be the Coal of the Future—The Untamed Dream of Jules Verne for a Solar Fuel. Molecules. 2016; 21(12):1638. https://doi.org/10.3390/molecules21121638
Chicago/Turabian StyleRyabchuk, Vladimir K., Vyacheslav N. Kuznetsov, Alexei V. Emeline, Yurii M. Artem’ev, Galina V. Kataeva, Satoshi Horikoshi, and Nick Serpone. 2016. "Water Will Be the Coal of the Future—The Untamed Dream of Jules Verne for a Solar Fuel" Molecules 21, no. 12: 1638. https://doi.org/10.3390/molecules21121638
APA StyleRyabchuk, V. K., Kuznetsov, V. N., Emeline, A. V., Artem’ev, Y. M., Kataeva, G. V., Horikoshi, S., & Serpone, N. (2016). Water Will Be the Coal of the Future—The Untamed Dream of Jules Verne for a Solar Fuel. Molecules, 21(12), 1638. https://doi.org/10.3390/molecules21121638