
New 3-Cyano-2-Substituted Pyridines Induce Apoptosis in MCF 7 Breast Cancer Cells

 ,
,
Abstract
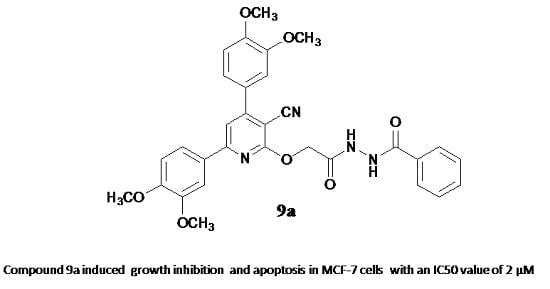
:
1. Introduction
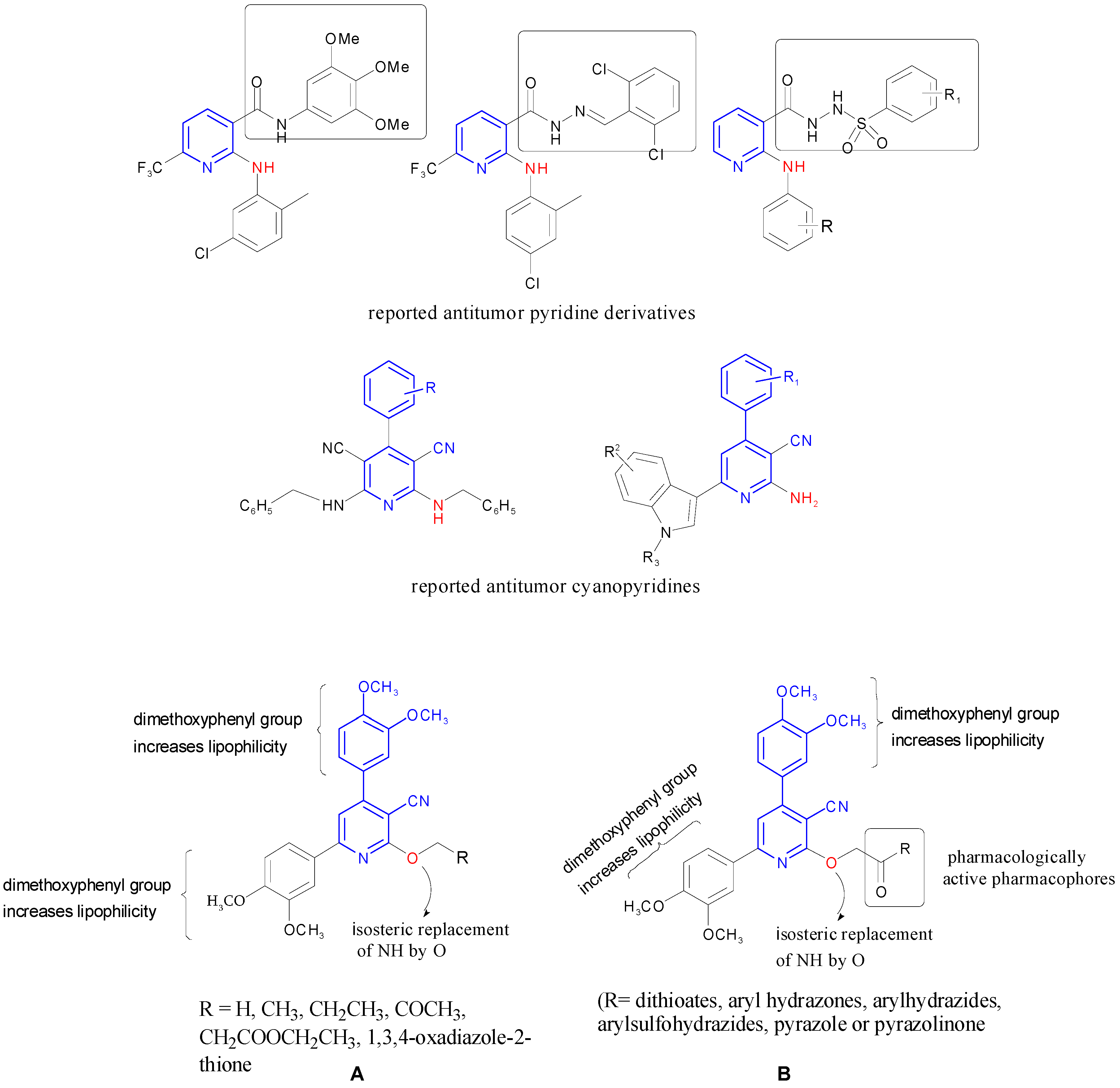
2. Results and Discussion
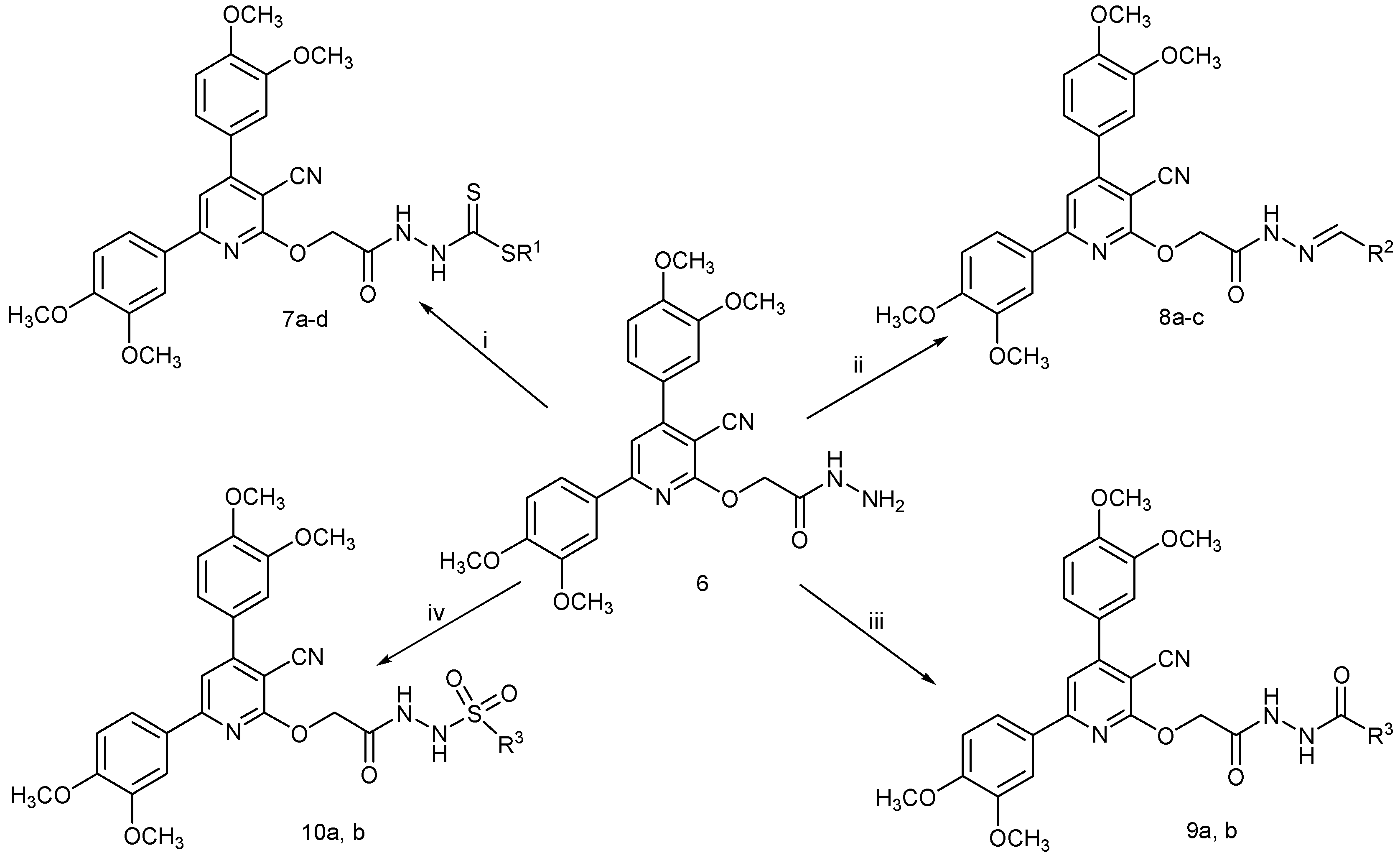
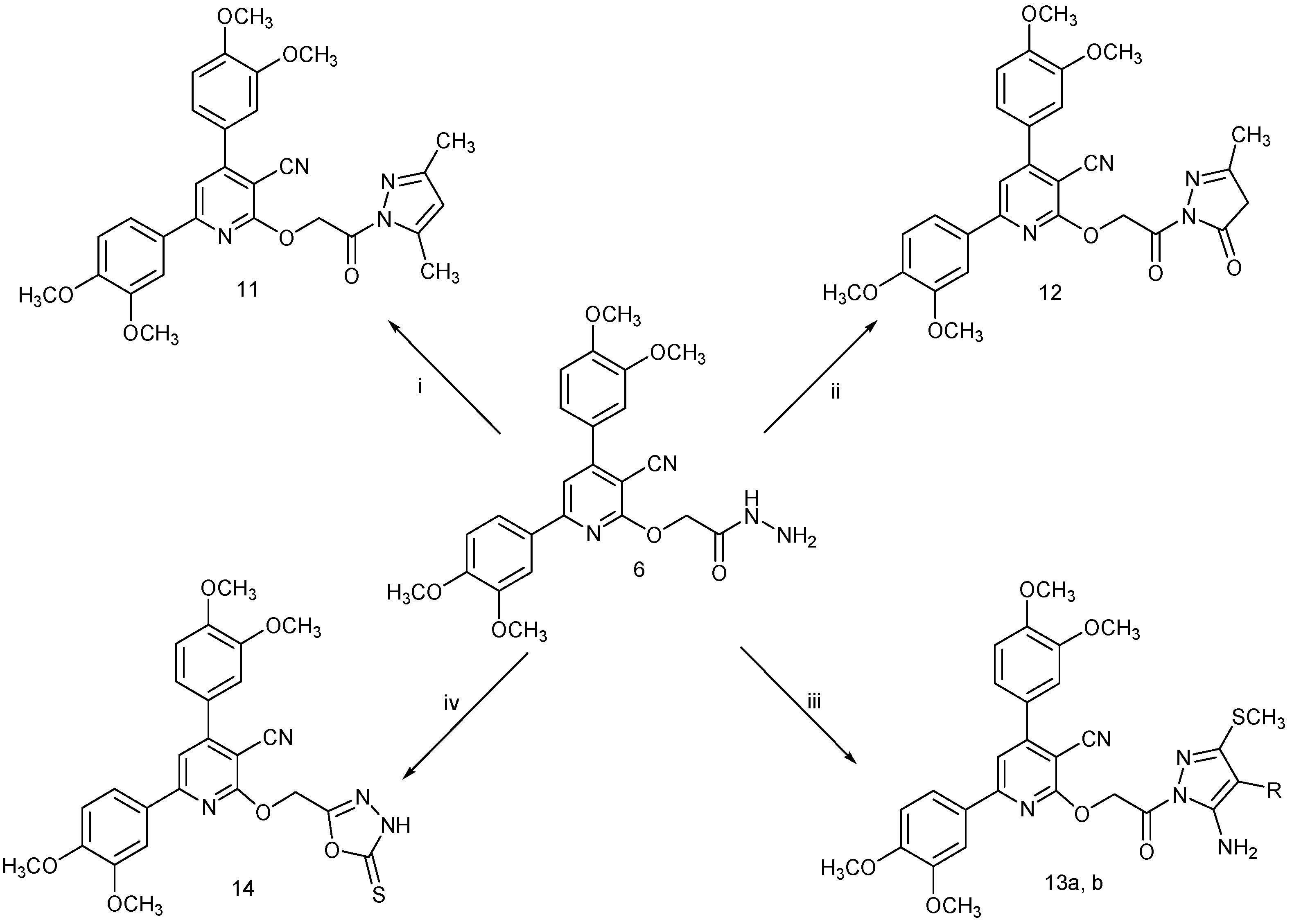
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Effect of the Synthesized Compounds on Cell Viability of Different Cancer Cell Lines
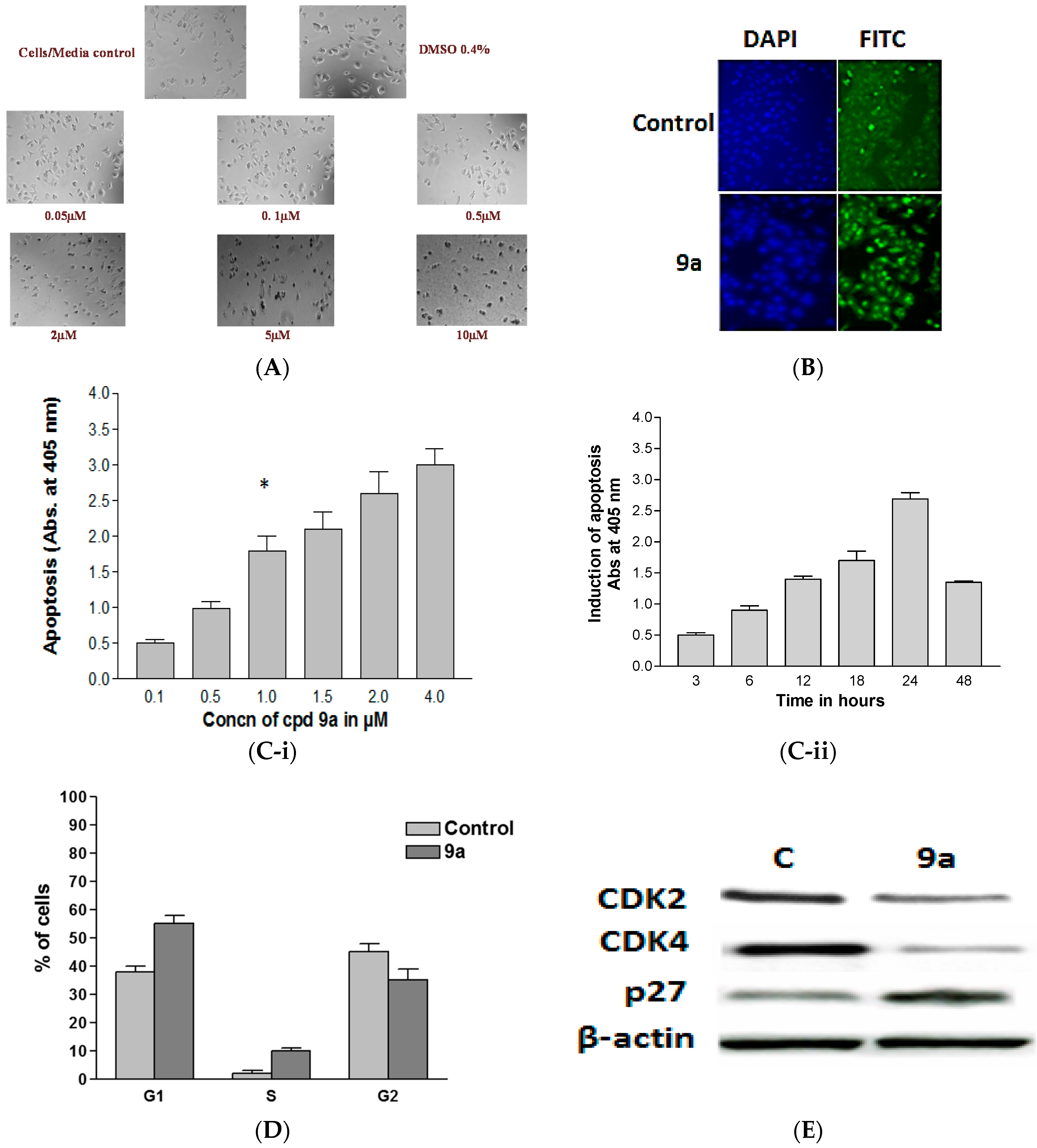
2.2.2. Compound 9a induces apoptosis in MCF-7 cells
Morphological Changes
TUNEL Assay
Enzyme Linked Immunosorbent Apoptosis Assay
Flow Cytometric Analysis
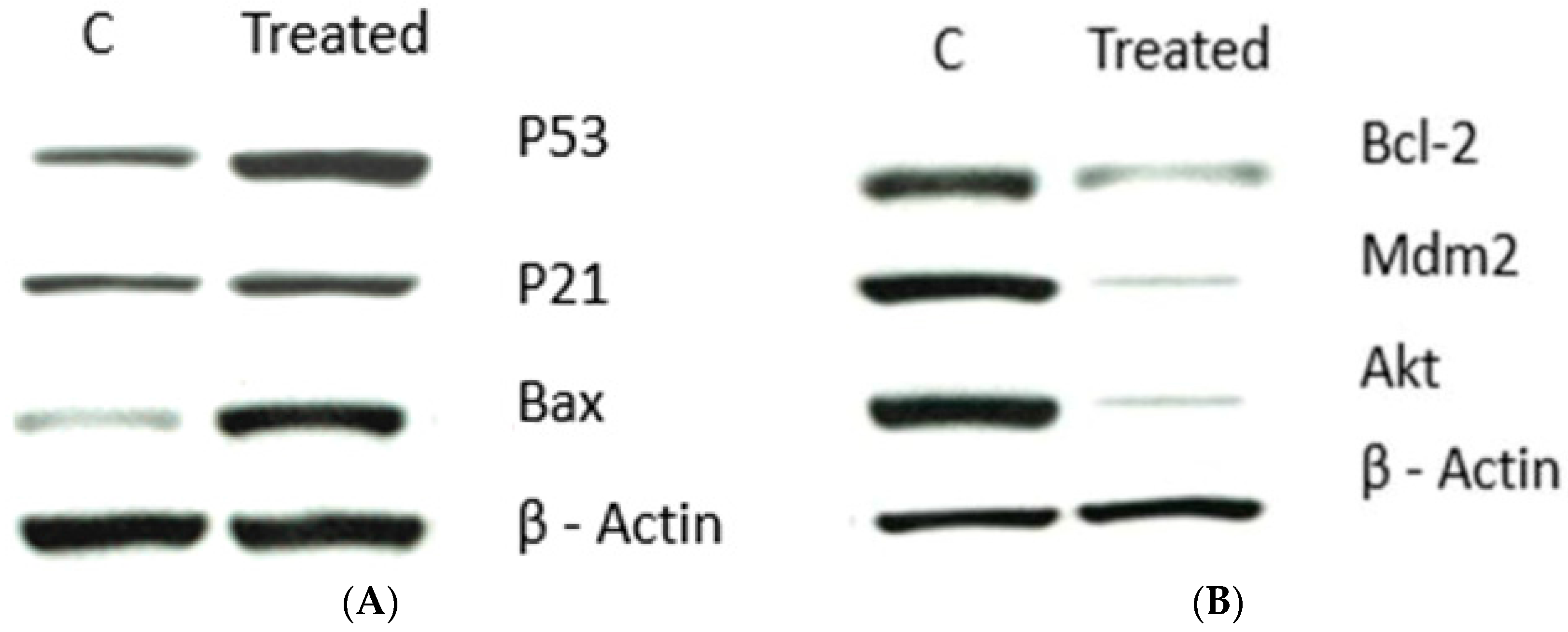
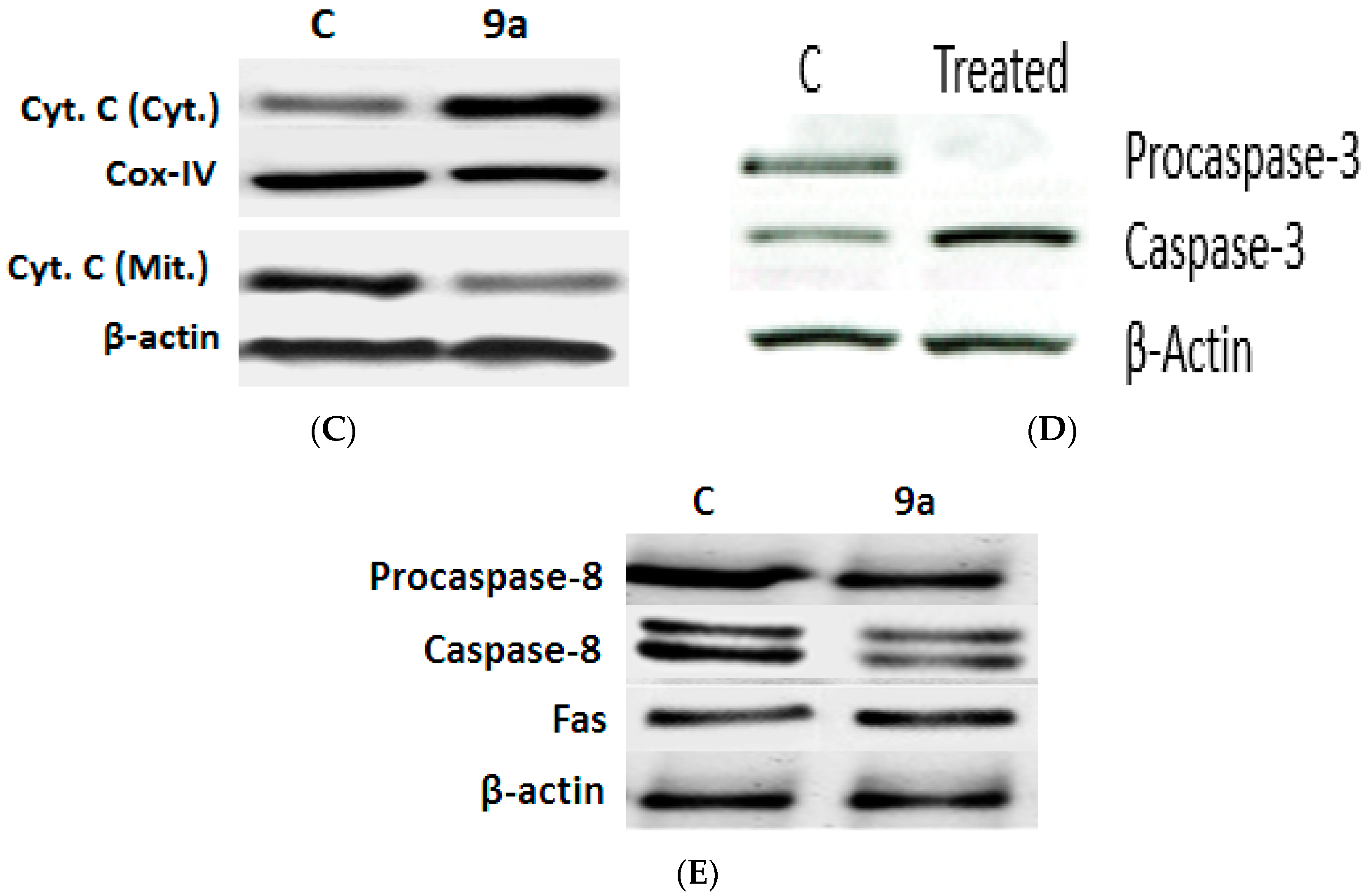
Impact of 9a on Apoptotic Signaling Pathways
The Effect of 9a on the Expression of Angiogenesis-Related Genes
3. Experimental Section
3.1. General Informstion
3.2. Chemistry
3.2.1. 4,6-bis(3,4-Dimethoxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (3)
3.2.2. General Procedure for the Synthesis of Compounds 4a–d
3.2.3. Ethyl 2-(3-Cyano-4,6-bis(3,4-dimethoxyphenyl)pyridin-2-yloxy)acetate (5)
3.2.4. 2-{[3-Cyano-4,6-bis(3,4-dimethoxyphenyl)pyridin-2-yl]oxy}acetohydrazide (6)
3.2.5. General Procedure for the Synthesis of Compounds 7a–d
3.2.6. General Procedure for the Synthesis of Compounds 8a–c
3.2.7. General Procedure for the Synthesis of Compounds 9a,b and 10a,b
3.2.8. General Procedure for the Synthesis of Compounds 11 and 12
3.2.9. General Procedure for the Synthesis of Compounds 13a,b
3.3. Biological Evaluation Methodology
3.3.1. Cell Lines and Cell Culture
3.3.2. Drug Stock Preparation
3.3.3. Cell Viability Assay (WST-1 Assay)
3.3.4. Morphological Examination
3.3.5. TUNEL Assay
3.3.6. Enzyme Linked Immunosorbent Apoptosis Assay
3.3.7. Flow Cytometry
Cell Cycle Analysis
3.3.8. Western Blot
3.3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Weigelt, B.; Reis-Filho, J.S. Histological and molecular types of breast cancer: Is there a unifying taxonomy? Nat. Rev. Clin. Oncol. 2009, 6, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Forouzanfar, M.H.; Foreman, K.J.; Delossantos, A.M.; Lozano, R.; Lopez, A.D.; Murray, C.J.L.; Naghavi, M. Breast and cervical cancer in 187 countries between 1980 and 2010: A systematic analysis. Lancet 2011, 378, 1461–1484. [Google Scholar] [CrossRef]
- O’Shaughnessy, J. Extending survival with chemotherapy in metastatic breast cancer. Oncologist 2005, 10 (Suppl. S3), 20–29. [Google Scholar] [CrossRef] [PubMed]
- Vandooren, J.; Van den Steen, P.E.; Opdenakker, G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): The next decade. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 222–272. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, G.; Warsch, S.; Glück, S.; Avancha, K.; Montero, A.J. Treating breast cancer in the 21st century: Emerging biological therapies. J. Cancer 2013, 4, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Wind, N.S.; Holen, I. Multidrug resistance in breast cancer: From in vitro models to clinical studies. Int. J. Breast Cancer 2011, 2011, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Inhibitor of apoptosis (IAP) proteins as therapeutic targets for radiosensitization of human cancers. Cancer Treat. Rev. 2012, 38, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Perez-Rebolledo, A.; Ayala, J.D.; de Lima, G.M.; Marchini, N.; Bombieri, G.; Zani, C.L.; Souza-Fagundes, E.M.; Beraldo, H. Structural studies and cytotoxic activity of N(4)-phenyl-2-benzoylpyridine thiosemicarbazone Sn(IV) complexes. Eur. J. Med. Chem. 2005, 40, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Khan, M.N.A.; Reddy, K.S.; Rohini, K. Synthesis of a new class of 2-anilino substituted nicotinyl arylsulfonylhydrazides as potential anticancer and antibacterial agents. Bioorg. Med. Chem. 2007, 15, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- Onnis, V.; Cocco, M.T.; Lilliu, V.; Congiu, C. Synthesis and evaluation of antitumoral activity of ester and amide derivatives of 2-arylamino-6-trifluoromethyl-3-pyridinecarboxylic acids. Bioorg. Med. Chem. 2008, 16, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Abadi, A.H.; Ibrahim, T.M.; Abouzid, K.M.; Lehmann, J.; Tinsley, H.N.; Gary, B.D.; Piazza, G.A. Design, synthesis and biological evaluation of novel pyridine derivatives as anticancer agents and phosphodiesterase 3 inhibitors. Bioorg. Med. Chem. 2009, 17, 5974–5982. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, H.A.; Moustafa, A.H.; Haikal, A.E.-F.Z.; Abu-El-Halawa, R.; el Ashry, E.-S.H. Synthesis, antitumor and antimicrobial activities of 4-(4-chlorophenyl)-3-cyano-2-(β-O-glycosyloxy)-6-(thien-2-yl)-nicotinonitrile. Eur. J. Med. Chem. 2011, 46, 2948–2954. [Google Scholar] [CrossRef] [PubMed]
- Onnis, V.; Cocco, M.T.; Fadda, R.; Congiu, C. Synthesis and evaluation of anticancer activity of 2-arylamino-6-trifluoromethyl-3-(hydrazonocarbonyl)pyridines. Bioorg. Med. Chem. 2009, 17, 6158–6165. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Pennati, M.; Barraja, P.; Montalbano, A.; Parrino, B.; Spano, V.; Lopergolo, A.; Sbarra, S.; Doldi, V.; Zaffaroni, N.; et al. Synthesis and antiproliferative activity of substituted 3[2-(1H-indol-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[3,2-b]pyridines, marine alkaloid Nortopsentin analogues. Curr. Med. Chem. 2014, 21, 1654–1666. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Pennati, M.; Parrino, B.; Lopergolo, A.; Barraja, P.; Montalbano, A.; Spanò, V.; Sbarra, S.; Doldi, V.; de Cesare, M.; et al. Novel 1H-Pyrrolo[2,3-b]pyridine Derivative Nortopsentin Analogues: Synthesis and Antitumor Activity in Peritoneal Mesothelioma Experimental Models. J. Med. Chem. 2013, 56, 7060–7072. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Parrino, B.; di Vita, G.; Attanzio, A.; Spanò, V.; Montalbano, A.; Barraja, P.; Tesoriere, L.; Antonia Livrea, M.; Diana, P.; et al. Synthesis and antiproliferative activity of thiazolyl-bis-pyrrolo[2,3-b]pyridines and indolyl-thiazolyl-pyrrolo[2,3-c]pyridines, Nortopsentin analogues. Mar. Drugs 2015, 13, 460–492. [Google Scholar] [CrossRef] [PubMed]
- Parrino, B.; Carbone, A.; Di Vita, G.; Ciancimino, C.; Attanzio, A.; Spanò, V.; Montalbano, A.; Barraja, P.; Tesoriere, L.; Antonia Livrea, M.; et al. 3-[4-(1H-Indol-3-yl)-1,3-thiazol-2-yl]-1H-pyrrolo[2,3-b]pyridines, Nortopsentin analogues with antiproliferative activity. Mar. Drugs 2015, 13, 1901–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thapa, P.; Karki, R.; Thapa, U.; Jahng, Y.; Jung, M.-J.; Nam, J.M.; Na, Y.; Kwon, Y.; Lee, E.-S. 2-Thienyl-4-furyl-6-aryl pyridine derivatives: Synthesis, topoisomerase I and II inhibitory activity, cytotoxicity, and structure–activity relationship study. Bioorg. Med. Chem. 2010, 18, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Jun, K.-Y.; Kwon, H.; Park, S.-E.; Lee, E.; Karki, R.; Thapa, P.; Lee, J.-H.; Lee, E.-S.; Kwon, Y. Discovery of dihydroxylated 2,4-diphenyl-6-thiophen-2-yl pyridine as a non-intercalative DNA-binding topoisomerase II-specific catalytic inhibitor. Eur. J. Med. Chem. 2014, 80, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Song, C.; Kadayat, T.M.; Magar, T.B.T.; Bist, G.; Shrestha, A.; Na, Y.; Kwon, Y.; Lee, E.-S. Topoisomerase I and II inhibitory activity, cytotoxicity, and structure-activity relationship study of dihydroxylated 2,6-diphenyl-4-aryl pyridines. Bioorg.Med.Chem. 2015, 23, 3638–3654. [Google Scholar] [CrossRef] [PubMed]
- Kraker, A.J.; Hartl, B.G.; Amar, A.M.; Barvian, M.R.; Showalter, H.D.; Moore, C.W. Biochemical and cellular effects of c-Src-kinase-selective pyrido[2,3-d]pyrimidine tyrosine kinase inhibitors. Biochem. Pharmacol. 2000, 60, 885–898. [Google Scholar] [CrossRef]
- Moasser, M.M.; Srethapakdi, M.; Sachar, K.S.; Kraker, A.J.; Rosen, N. Inhibition of Src-kinases by a selective tyrosine kinase inhibitor causes mitotic arrest. Cancer Res. 1999, 59, 6145–6152. [Google Scholar] [PubMed]
- Chand, K.; Prasad, S.; Tiwari, R.K.; Shirazi, A.N.; Kumar, S.; Parang, K.; Sharma, S.K. Synthesis and evaluation of c-Src-kinase inhibitory activity of pyridine 2-(1H)-one derivatives. Bioorg. Chem. 2014, 53, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Waud, W.R.; Struck, R.F. Determination of the phamacophore of penclomedine, a clinically evaluated antitumor pyridine derivative. Bioorg. Med. Chem. 2002, 10, 3593–3598. [Google Scholar] [CrossRef]
- Finch, R.A.; Liu, M.; Grill, S.P.; Rose, W.C.; Loomis, R.; Vasquez, K.M.; Cheng, Y.; Sartorelli, A.C. Triapine (3-aminopyridine-2-carboxaldehyde-thiosemicarbazone): A potent inhibitor of ribonucleotide reductase activitywith broad spectrum antitumor activity. Biochem. Pharmacol. 2000, 59, 983–991. [Google Scholar] [CrossRef]
- Wang, G.T.; Wang, X.; Wang, W.; Hasvold, L.A.; Sullivan, G.; Hutchins, C.W.; O’Conner, S.; Gentiles, R.; Sowin, T.; Cohen, J.; et al. Design and synthesis of O-trifluoromethylbiphenyl substituted 2-amino-nicotinonitriles as inhibitors of farnesyltransferase. Bioorg. Med. Chem. Lett. 2005, 15, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Cocco, M.T.; Congiu, C.; Lilliu, V.; Onnis, V. Synthesis and antiproliferative activity of 2,6-dibenzylamino-3,5-dicyanopyridines on human cancer cell lines. Eur. J. Med. Chem. 2005, 40, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Cocco, M.T.; Congiu, C.; Lilliu, V.; Onnis, V. Synthesis and in vitro antitumoral activity of new 3,5-dicyanopyridine derivatives. Bioorg. Med. Chem. 2007, 15, 1859–1867. [Google Scholar] [CrossRef] [PubMed]
- Rostom, S.A.F.; Faidallah, H.M.; Al-Saadi, M.S. A facile synthesis of some 3-cyano-1,4,6-trisubstituted-2(1H)pyridinones and their biological evaluation as anticancer agents. Med. Chem. Res. 2011, 20, 1260–1272. [Google Scholar] [CrossRef]
- Rostom, S.A.F.; Ashour, H.M.A.; Abd ElRazik, H.A. Synthesis and biological evaluation of some novel polysubstituted pyrimidine derivatives as potential antimicrobial and anticancer agents. Arch. Pharm. Chem. Life Sci. 2009, 342, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Ashour, H.M.A.; Abdel Wahab, A.E. Synthesis and biological evaluation of novel pyrazoles and pyrazolo[3,4-d]pyrimidines incorporating a benzenesulfonamide moiety. Arch. Pharm. Chem. Life Sci. 2009, 342, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Rostom, S.A.F.; Badr, M.H.; Abd ElRazik, H.A.; Ashour, H.M.A.; Abdel Wahab, A.E. Synthesis of some pyrazolines and pyrimidines derived from polymethoxy chalcones as anticancer and antimicrobial agents. Arch. Pharm. Chem. Life Sci. 2011, 344, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Malki, A.; Elbayaa, R.Y.; Ashour, H.M.A.; Loffredo, C.A.; Youssef, A.M. Novel thiosemicarbazides induced apoptosis in human MCF-7 breast cancer cells via JNK signaling. J. Enzyme Inhib. Med. Chem. 2015, 30, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-F.; Yang, J.-S.; Chang, C.-Y.; Kuo, S.-C.; Lee, M.-R.; Huang, L.-J. Synthesis and anticancer activity of benzyloxybenzaldehyde derivatives against HL-60 cells. Bioorg. Med. Chem. 2005, 13, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Barboni, L.; Giarlo, G.; Ballini, R.; Fontana, G. 14β-hydroxy-10-deacetylbaccatin III as a convenient, alternative substrate for the improved synthesis of methoxylated second-generation taxanes. Bioorg. Med. Chem. Lett. 2006, 16, 5389–5391. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, Y.; Wang, W.-J.; Xiang, P.; Luo, X.-M.; Yang, L.; Yang, S.-Y.; Zhao, Y.-L. Synthesis and biological evaluation of novel (E)-N′-(2,3-dihydro-1H-inden-1-ylidene) benzohydrazides as potent LSD1 inhibitors. Bioorg. Med. Chem. Lett. 2016, in press. [Google Scholar]
- Saini, M.; Kumar, P.; Kumar, M.; Ramasamy, K.; Mani, V.; Mishra, R.K.; Abdul Majeed, A.B.; Narasimhan, B. Synthesis, in vitro antimicrobial, anticancer evaluationand QSAR studies of N-(substituted)-4-(butan-2-ylideneamino)benzohydrazides. Arab. J. Chem. 2014, 7, 448–460. [Google Scholar] [CrossRef]
- Li, R.-D.; Wang, H.-L.; Li, Y.-B.; Wang, Z.-Q.; Wang, X.; Wang, Y.-T.; Ge, Z.-M.; Li, R.-T. Discovery and optimization of novel dual dithiocarbamates as potent anticancer agents. Eur. J. Med. Chem. 2015, 93, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.-C.; Ma, Y.-C.; Zhang, E.; Shi, X.-J.; Wang, M.-M.; Ye, X.-W.; Liu, H.-M. Design and synthesis of novel 1,2,3-triazole-dithiocarbamate hybrids as potential anticancer agents. Eur. J. Med. Chem. 2013, 62, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Nasr, T.; Bondock, S.; Youns, M. Anticancer activity of new coumarin substituted hydrazide-hydrazonederivatives. Eur. J. Med. Chem. 2014, 76, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Baviskar, A.T.; Banerjee, U.C.; Gupta, M.; Singh, R.; Kumar, S.; Gupta, M.K.; Kumar, S.; Raut, S.K.; Khullar, M.; Singh, S.; et al. Synthesis of imine-pyrazolopyrimidinones and their mechanistic interventions on anti-cancer activity. Bioorg. Med. Chem. 2013, 21, 5782–5793. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Shi, L.; Ke, S. Acylhydrazone derivatives as potential anticancer agents: Synthesis, bioevaluation and mechanism of action. Bioorg. Med. Chem. Lett. 2015, 25, 5772–5776. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.W.; Li, Y.; Ge, D.; Zhao, B.X.; Liu, Y.R.; Lv, H.S.; Ding, J.; Miao, J.Y. Synthesis of novel oxime-containing pyrazole derivatives and discovery of regulators for apoptosis and autophagy in A549 lung cancer cells. Bioorg. Med. Chem. Lett. 2010, 20, 4766–4770. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.-W.; Zhu, J.; Zhao, B.-X.; Huang, Y.-H.; Ding, J.; Miao, J.-Y. Synthesis, crystal structure and biological evaluation of novel 2-[(5-hydroxymethyl)-3-phenyl-1H-pyrazol-1-yl)-1-phenylethanol derivatives. Eur. J. Med. Chem. 2010, 45, 5792–5799. [Google Scholar] [CrossRef] [PubMed]
- Puthiyapurayil, P.; Poojary, B.; Chikkanna, C.; Buridipad, S.K. Design, synthesis and biological evaluation of a novel series of 1,3,4-oxadiazole bearing N-methyl-4-(trifluoromethyl)phenyl pyrazole moiety as cytotoxic agents. Eur. J. Med. Chem. 2012, 53, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, S.A.; Yar, M.; Bajda, M.; Jadoon, B.; Khan, Z.A.; Naqvi, S.A.R.; Shaikh, A.J.; Hayat, K.; Mahmmod, A.; Mahmood, N.; et al. Synthesis and biological evaluation of novel oxadiazole derivatives: A new class of thymidine phosphorylase inhibitors as potential anti-tumor agents. Bioorg. Med. Chem. 2014, 22, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.C.; Bhatt, N.; Somani, H.; Trivedi, A. Synthesis, antimicrobial and cytotoxic activities of some novel thiazole-clubbed 1,3,4-oxadiazoles. Eur. J. Med. Chem. 2013, 67, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Al-Saadi, M.S.; Rostom, S.A.F.; Faidallah, H.M. Synthesis and biological evaluation of some 3-cyano-4-(1-methyl-1H-pyrrol-2-yl)-6-substituted-2(1H)-pyridinones and their 2-imino isosters. Alex. J. Pharm. Sci. 2005, 19, 15–21. [Google Scholar]
- Kornblum, N.; Berrigan, P.J.; le Noble, W.J. Solvation as a factor in alkylation of ambident anions: The importance of hydrogen bonding capacity of the solvent. J. Am. Chem. Soc. 1963, 85, 1141–1147. [Google Scholar] [CrossRef]
- Galic, N.; Peric, B.; Joji-Prodic, B.; Cimeriman, Z. Structural and spectroscopic characteristics of aroylhydrazones derived from nicotinic acid hydrazide. J. Mol. Struct. 2001, 559, 187–194. [Google Scholar] [CrossRef]
- Wyrzykiewicz, E.; Prukah, D. New isomeric N-substituted hydrazones of 2-, 3- and 4-pyridinecarboxaldehydes. J. Heterocycl. Chem. 1998, 35, 381–387. [Google Scholar] [CrossRef]
- Rando, D.G.; Sats, D.N.; Siqueira, L.; Malvezzi, A.; Leite, C.O.F.; Amaral, A.T.; Ferreira, F.I.; Tavares, L.C. Potential tuberculostatic agents. Topliss application on benzoic acid [(5-Nitrothiophen-2-yl)methylene]hydrazide series. Bioorg. Med. Chem. 2002, 10, 557–560. [Google Scholar] [CrossRef]
- Mohan, S.; Ananthan, S. A new approach for the synthesis of some novel sulphur bridged pyrazoles and their characterization. J. Chem. Pharm. Res. 2011, 3, 402–413. [Google Scholar]
- Shrivastava, H.Y.; Ravikumar, T.; Shanmugasundaram, N.; Babu, M.; Nair, B.U. Cytotoxicity studies of chromium (III) complexes on human dermal fibroblasts. Free Radic. Biol. Med. 2005, 38, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Chu, I.M.; Hengst, L.; Slingerland, J.M. The Cdk inhibitor p27 in human cancer: Prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 2008, 8, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Berger, M.; Goldberg, Z.; Haupt, Y. Apoptosis–the p 53 network. J. Cell Sci. 2003, 116, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Kato, Y.; Baba, Y.; Nishimura, G.; Tanigaki, Y.; Horiuchi, C.; Mochimatsu, I.; Tsukuda, M. Protein levels of p21, p27, cyclin E and Bax predict sensitivity to cisplatin and paclitaxel in head and neck squamous cell carcinomas. Oncol. Rep. 2004, 11, 421–446. [Google Scholar]
- Riedl, S.J.; Shi, Y.G. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004, 5, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Pellecchia, M.; Reed, J.C. Inhibition of anti-apoptotic Bcl-2 family proteins by natural polyphenols new avenues for cancer chemoprevention and chemotherapy. Curr. Pharm. Des. 2004, 10, 1387–1398. [Google Scholar] [CrossRef] [PubMed]
- Shiojima, I.; Walsh, K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ. Res. 2002, 90, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Dakeng, S.; Duangmano, S.; Jiratchariyahul, W.; U-Pratya, Y.; Bogler, O.; Patmasriwat, P. Inhibition of Wnt signaling by cucurbitacin B in breast cancer cells: Reduction of Wnt-associated proteins and reduced translocation of galectin-3 mediated β-catenin to the nucleus. J. Cell Biochem. 2012, 113, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Morini, M.; Mottolese, M.; Ferrari, N.; Ghiorzo, F.; Buglioni, S.; Mortarini, R.; Noonan, D.M.; Natal, P.G.; Albini, A. The α3β1 integrin is associated with mammary carcinoma cell metastasis, invasion and gelatinase B (mmp-9) activity. Int. J. Cancer 2000, 87, 336–342. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–14 are available from the authors.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | SK-OV-3 b | MCF-7 c | MDA-MB-231 d | HCT-116 e | RKO f |
|---|---|---|---|---|---|
| 4a | 11.37 ± 0.72 | 9.08 ± 0.37 | 32.10 ± 0.84 | 20.30 ± 0.85 | 13.21 ± 0.77 |
| 4b | 18.16 ± 0.98 | 15.52 ± 0.46 | 40.04 ± 1.36 | 26.25 ± 1.22 | 19.80 ± 0.37 |
| 4c | 22.12 ± 1.32 | 12.28 ± 0.77 | 44.93 ± 1.74 | 10.06 ± 0.76 | 18.14 ± 1.03 |
| 4d | 25.19 ± 0.76 | 20.30 ± 0.98 | 35.80 ± 0.96 | 13.14 ± 0.84 | 17.39 ± 0.44 |
| 5 | 10.15 ± 0.44 | 15.42 ± 0.94 | 25.12 ± 0.87 | 14.88 ± 0.98 | 22.13 ± 0.97 |
| 7a | 9.01 ± 0.42 | 11.23 ± 0.22 | 30.10 ± 0.88 | 23.20 ± 0.95 | 11.25 ± 0.91 |
| 7b | 12.33 ± 0.88 | 18.14 ± 1.76 | 37.87 ± 1.25 | 9.10 ± 0.97 | 15.21 ± 1.13 |
| 7c | 5.12 ± 0.11 | 7.10 ± 0.44 | 31.73 ± 0.89 | 26.90 ± 0.97 | 20.30 ± 0.76 |
| 7d | 10.10 ± 0.98 | 14.30 ± 0.95 | 35.53 ± 1.03 | 11.16 ± 1.26 | 24.98 ± 1.33 |
| 8a | 14.13 ± 0.95 | 12.01 ± 0.96 | 25.34 ± 1.24 | 28.82 ± 0.31 | 12.32 ± 0.02 |
| 8b | 10.21 ± 0.92 | 9.31 ± 0.98 | 32.26 ± 0.87 | 25.20 ± 0.89 | 14.16 ± 0.91 |
| 8c | 12.14 ± 0.98 | 15.27 ± 0.74 | 20.31 ± 1.06 | 25.87 ± 0.83 | 16.32 ± 0.37 |
| 9a | 9.06 ± 0.24 | 2.04 ± 0.13 | 22.34 ± 0.88 | 7.12 ± 0.37 | 8.22 ± 0.99 |
| 9b | 7.08 ± 0.16 | 10.18 ± 0.92 | 19.27 ± 0.86 | 17.90 ± 0.74 | 9.95 ± 1.06 |
| 10a | 11.34 ± 0.02 | 13.39 ± 0.98 | 26.82 ± 0.33 | 21.82 ± 0.85 | 12.10 ± 1.26 |
| 10b | 9.17 ± 0.38 | 8.12 ± 0.47 | 22.44 ± 1.71 | 16.09 ± 1.07 | 15.65 ± 0.86 |
| 11 | 26.19 ± 0.95 | >50 | >50 | 33.90 ± 0.87 | >50 |
| 12 | 20.27 ± 1.72 | >50 | >50 | 40.35 ± 1.76 | >50 |
| 13a | >50 | >50 | >50 | 36.30 ± 1.71 | >50 |
| 13b | >50 | >50 | >50 | 30.14 ± 0.76 | >50 |
| 14 | 30.30 ± 1.12 | >50 | >50 | >50 | >50 |
| 5-FU | 32.19 ± 1.03 | 7.06 ± 0.91 | 15.13 ± 0.94 | 12.19 ± 1.10 | 20.18 ± 1.16 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malki, A.; Mohsen, M.; Aziz, H.; Rizk, O.; Shaban, O.; El-Sayed, M.; Sherif, Z.A.; Ashour, H. New 3-Cyano-2-Substituted Pyridines Induce Apoptosis in MCF 7 Breast Cancer Cells. Molecules 2016, 21, 230. https://doi.org/10.3390/molecules21020230
Malki A, Mohsen M, Aziz H, Rizk O, Shaban O, El-Sayed M, Sherif ZA, Ashour H. New 3-Cyano-2-Substituted Pyridines Induce Apoptosis in MCF 7 Breast Cancer Cells. Molecules. 2016; 21(2):230. https://doi.org/10.3390/molecules21020230
Chicago/Turabian StyleMalki, Ahmed, Mona Mohsen, Hassan Aziz, Ola Rizk, Omima Shaban, Mohamed El-Sayed, Zaki A. Sherif, and Hayam Ashour. 2016. "New 3-Cyano-2-Substituted Pyridines Induce Apoptosis in MCF 7 Breast Cancer Cells" Molecules 21, no. 2: 230. https://doi.org/10.3390/molecules21020230
APA StyleMalki, A., Mohsen, M., Aziz, H., Rizk, O., Shaban, O., El-Sayed, M., Sherif, Z. A., & Ashour, H. (2016). New 3-Cyano-2-Substituted Pyridines Induce Apoptosis in MCF 7 Breast Cancer Cells. Molecules, 21(2), 230. https://doi.org/10.3390/molecules21020230