
New Approaches to the Role of Thrombin in Acute Coronary Syndromes: Quo Vadis Bivalirudin, a Direct Thrombin Inhibitor?

Abstract
:1. Introduction
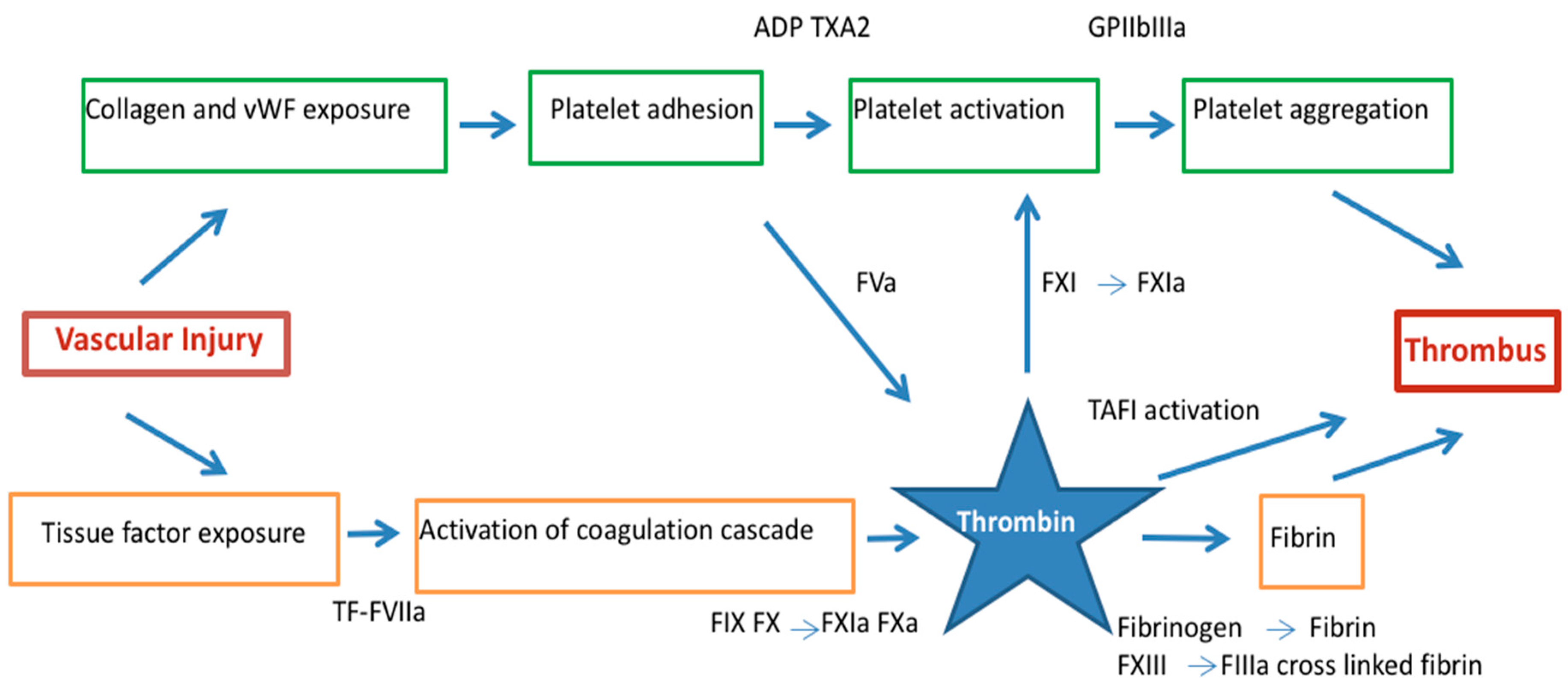
2. Pathophysiology of ACS and the Role of Thrombin
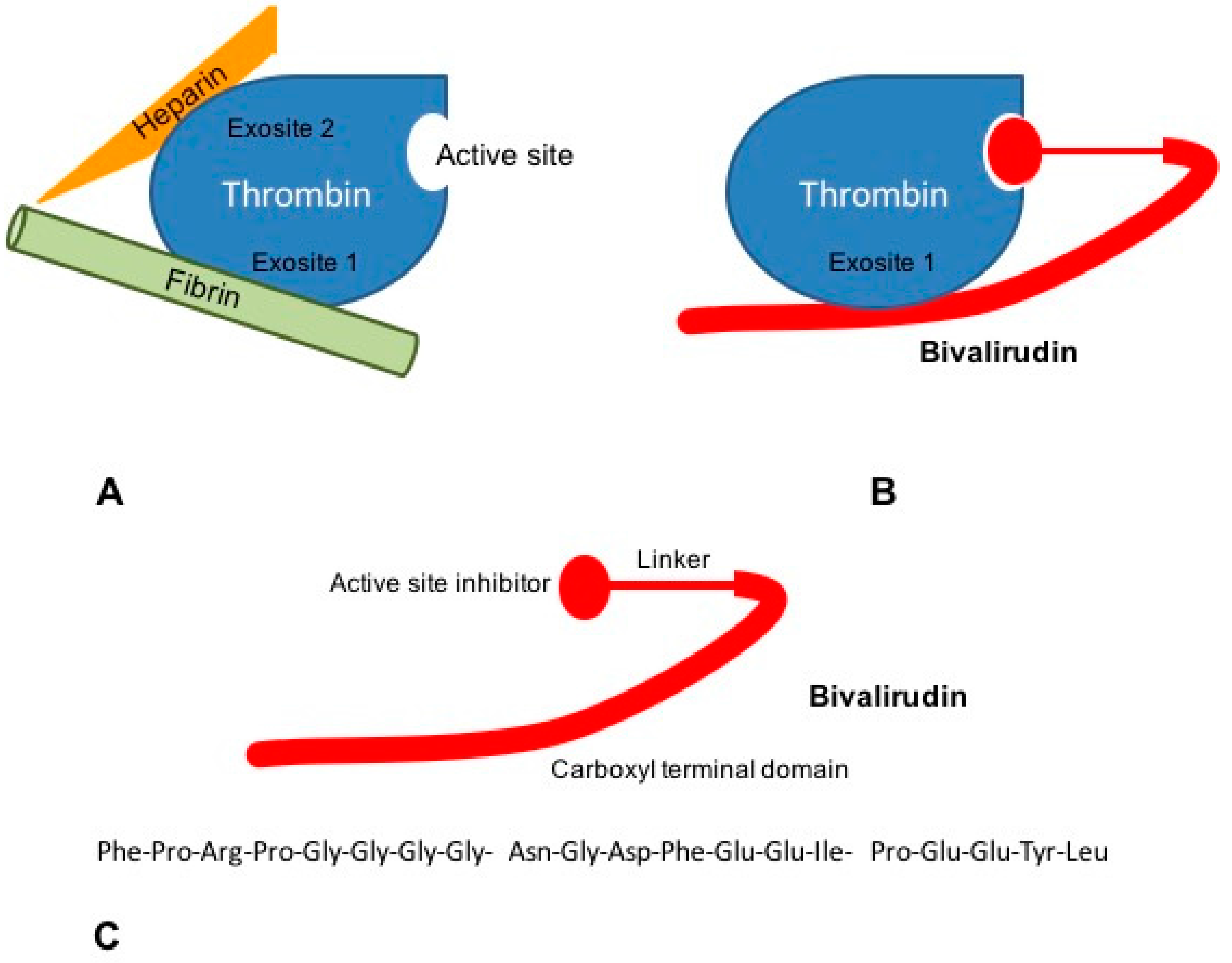
3. Bivalirudin Mechanism of Action
4. Bivalirudin Pharmacokinetics and Pharmacodynamics
5. Bivalirudin in Acute Coronary Syndromes
6. Clinical Guidelines Management of Antithrombotic Treatment in STEMI and Non-STEMI Patients
7. New Evidence–New Approaches
8. Questions to Be Answered
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Capodanno, D.; de Caterina, R. Bivalirudin for acute coronary syndromes: Premises, promises and doubts. Thromb. Haemost. 2015, 113, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, D.; Stavrou, K.; Koniari, I.; Gkizas, V.; Perperis, A.; Kontoprias, K.; Vogiatzi, C.; Bampouri, T.; Xanthopoulou, I. Ticagrelor vs. prasugrel one-month maintenance therapy: Impact on platelet reactivity and bleeding events. Thromb. Haemost. 2014, 112, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Shen, H.; Wang, Z.J.; Yang, S.W.; Liu, X.L.; Zhou, Y.J. Risk and benefit of direct oral anticoagulants or PAR-1 antagonists in addition to antiplatelet therapy in patients with acute coronary syndrome. Thromb. Res. 2015, 136, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Fuster, V.; Badimon, J.; Chesebro, J.H.; Fallon, J.T. Plaque rupture, thrombosis, and therapeutic implications. Haemostasis 1996, 26 (Suppl. 4), 269–284. [Google Scholar] [PubMed]
- He, L.W.; Dai, W.C.; Li, N.G. Development of Orally Active Thrombin Inhibitors for the Treatment of Thrombotic Disorder Diseases. Molecules 2015, 20, 11046–11062. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.L.; Goswami, L.N.; Dikshit, D.K. Progress in the design of low molecular weight thrombin inhibitors. Med. Res. Rev. 2005, 25, 66–92. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.G.; Brummel, K.; Butenas, S. What is all that thrombin for? J. Thromb. Haemost. 2003, 1, 1504–1514. [Google Scholar] [CrossRef] [PubMed]
- Ansell, J. Factor Xa or thrombin: Is factor Xa a better target? J. Thromb. Haemost. 2007, 5 (Suppl. 1), 60–64. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.; Bates, E.R.; Valgimigli, M.; Wallentin, L.; Kristensen, S.D.; Anderson, J.L.; Lopez Sendon, J.L.; Tubaro, M.; Granger, C.B.; Bode, C.; et al. Antiplatelet and anticoagulation agents in acute coronary syndromes: What is the current status and what does the future hold? Am. Heart J. 2014, 168, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Cavender, M.A.; Sabatine, M.S. Bivalirudin versus heparin in patients planned for percutaneous coronary intervention: a meta-analysis of randomised controlled trials. Lancet 2014, 384, 599–606. [Google Scholar] [CrossRef]
- Straub, A.L.; Roehrig, S.; Hillisch, A. Oral, direct thrombin and factor Xa inhibitors: the replacement for warfarin, leeches, and pig intestines? Angew. Chem. Int. Ed. Engl. 2011, 50, 4574–4590. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, G.; Valgimigli, M.; Pagnotta, P.; Presbitero, P. Bivalirudin versus heparin in patients with acute myocardial infarction: A meta-analysis of randomized trials. Catheter. Cardiovasc. Interv. 2015, 86, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Showkathali, R.; Natarajan, A. Antiplatelet and antithrombin strategies in acute coronary syndrome: State-of-the-art review. Curr. Cardiol. Rev. 2012, 8, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N. Triggers, targets and treatments for thrombosis. Nature 2008, 451, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Fuster, V.; Badimon, L.; Badimon, J.J.; Chesebro, J.H. The pathogenesis of coronary artery disease and the acute coronary syndromes. Part I. N. Engl. J. Med. 1992, 326, 242–250. [Google Scholar] [PubMed]
- Epstein, F.; Fuster, V.; Badimon, L.; Badimon, J.J.; Chesebro, J.H. The pathogenesis of coronary artery disease and the acute coronary syndromes. Part II. N. Engl. J. Med. 1992, 326, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Falk, E.; Shah, P.K.; Fuster, V. Coronary plaque disruption. Circulation 1995, 92, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Furie, B.; Furie, B.C. Molecular and cellular biology of blood coagulation. N. Engl. J. Med. 1992, 326, 800–806. [Google Scholar] [PubMed]
- Goel, M.S.; Diamond, S.L. Factor VIIa-mediated tenase function on activated platelets under flow. J. Thromb. Haemost. 2004, 2, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- De Cristofaro, R.; de Candia, E. Thrombin domains: structure, function and interaction with platelet receptors. J. Thromb. Thrombolysis 2003, 15, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.R.; Camerer, E.; Hamilton, J.R. Protease-activated receptors in hemostasis, thrombosis, and vascular biology. In Protease Activated Receptors in Hemostasis, Thrombosis, and Vascular Biology, Hemostasis and Thrombosis: Basic Principles and Clinical Practice, 5th ed.; Colman, R.W., Mardeer, V.J., Clowes, A.W., George, J.N., Goldhaber, S.Z., Eds.; Lippincott, Williams, & Wilkins: Philadelphia, PA, USA, 2006; pp. 555–566. [Google Scholar]
- Angiolillo, D.J.; Capodanno, D.; Goto, S. Platelet thrombin receptor antagonism and atherothrombosis. Eur. Heart J. 2010, 31, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.R. Thrombin signalling and protease-activated receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Monroe, D.M.; Hoffman, M.; Roberts, H.R. Platelets and thrombin generation. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Weitz, J.I.; Hudoba, M.; Massel, D.; Maraganore, J.; Hirsh, J. Clot-bound thrombin is protected from inhibition by heparin-antithrombin III but is susceptible to inactivation by antithrombin III-independent inhibitors. J. Clin. Unvest. 1990, 86, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Béguin, S.; Hemker, H.C. The influence of fibrinogen and fibrin on thrombin generation evidence for feedback activation o f the clotting system by clot bound thrombin. Thromb. Haemost. 1994, 72, 713–721. [Google Scholar] [PubMed]
- Davie, E.W.; Kulman, J.D. An overview of the structure and function of thrombin. Semin. Thromb. Hemost. 2006, 32, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Weitz, J.I. Insights into the role of thrombin in the pathogenesis of recurrent ischaemia after acute coronary syndrome. Thromb. Haemost. 2014, 112, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Bajzar, L.; Manuel, R.; Nesheim, M.E. Purification and characterization of TAFI, a thrombin-activable fibrinolysis inhibitor. J. Biol. Chem. 1995, 270, 14777–14784. [Google Scholar]
- Sakharov, D.V.; Plow, E.F.; Rijken, D.C. In the mechanism of the antifibrinolytic activity of plasma carboxypeptidase B. J. Biol. Chem. 1997, 272, 14477–14482. [Google Scholar] [CrossRef] [PubMed]
- Ewald, G.A.; Eisenberg, P.R. Plasmin-mediated activation of contact system in response to pharmacological thrombolysis. Circulation 1995, 91, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, P.K.; Miletich, J.R.; Sobel, B.E.; Jaffe, A.S. Differential effects of activation of prothrombin by streptokinase compared with urokinase and tissue-type plasminogen activation (t-PA). Thromb. Res. 1998, 50, 707–717. [Google Scholar] [CrossRef]
- Seitz, R.; Pelzer, H.; Immel, A.; Egbring, R. Prothrombin activation by thrombolytic agents. Fibrinolysis 1993, 7, 109–115. [Google Scholar] [CrossRef]
- Glover, C.J.; McIntire, L.V.; Leverett, L.B.; Hellums, J.D.; Brown, C.H.; Natelson, E.A. Effect of shear stress on clot structure formation. Trans. Am. Soc. Artif. Intern. Organs 1974, 20B, 463–468. [Google Scholar]
- Fenton, F.W., II; Ofosu, F.A.; Brezniak, D.V.; Hassouna, H.I. Thrombin and antithrombotics. Semin. Thormb. Hemost. 1998, 24, 89–91. [Google Scholar]
- Tricoci, P.; Huang, Z.; Held, C.; Moliterno, D.J.; Armstrong, P.W.; van de Werf, F.; White, H.D.; Aylward, P.E.; Wallentin, L.; Chen, E.; et al. TRACER Investigators. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N. Engl. J. Med. 2012, 366, 20–33. [Google Scholar] [PubMed]
- Burke, D.A.; Warraich, H.J.; Pinto, D.S. Which antithrombin for whom? Identifying the patient population that benefits most from novel antithrombin agents. Curr. Cardiol. Rep. 2012, 14, 493–501. [Google Scholar] [CrossRef] [PubMed]
- De Caterina, R.; Husted, S.; Wallentin, L.; Andreotti, F.; Arnesen, H.; Bachmann, F.; Baigent, C.; Huber, K.; Jespersen, J.; Kristensen, S.D.; et al. Parenteral anticoagulants in heart disease: Current status and perspectives (Section II). Position paper of the ESC Working Group on Thrombosis-Task Force on Anticoagulants in Heart Disease. Thromb. Haemost. 2013, 109, 769–786. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.V.; Ohman, E.M. Anticoagulant therapy for percutaneous coronary intervention. Circ. Cardiovasc. Interv. 2010, 3, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Chew, D. Promise of factor Xa inhibition in acute coronary syndromes. Curr. Cardiol. Rep. 2012, 14, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.M.; Murphy, S.A.; Montalescot, G.; Morrow, D.A.; Ardissino, D.; Cohen, M.; Gulba, D.C.; Kracoff, O.H.; Lewis, B.S.; Roguin, N.; et al. Percutaneous coronary intervention in patients receiving enoxaparin or unfractionated heparin after fibrinolytic therapy for ST-segment elevation myocardial infarction in the ExTRACT-TIMI 25 trial. J. Am. Coll. Cardiol. 2007, 49, 2238–2246. [Google Scholar] [CrossRef] [PubMed]
- Montalescot, G.; White, H.D.; Gallo, R.; Cohen, M.; Steg, P.G.; Aylward, P.E.; Bode, C.; Chiariello, M.; King, S.B., 3rd; Harrington, R.A.; et al. Enoxaparin versus unfractionated heparin in elective percutaneous coronary intervention. N. Engl. J. Med. 2006, 355, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, S.; Mehta, S.R.; Chrolavicius, S.; Afzal, R.; Pogue, J.; Granger, C.B.; Budaj, A.; Peters, R.J.; Bassand, J.P.; Wallentin, L.; Joyner, C.; Fox, K.A. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N. Engl. J. Med. 2006, 354, 1464–1476. [Google Scholar] [PubMed]
- Yusuf, S.; Mehta, S.R.; Chrolavicius, S.; Afzal, R.; Pogue, J.; Granger, C.B.; Budaj, A.; Peters, R.J.; Bassand, J.P.; Wallentin, L.; et al. Effects of fondaparinux on mortality and reinfarction in patients with acute ST-segment elevation myocardial infarction: The OASIS-6 randomized trial. JAMA 2006, 295, 1519–1530. [Google Scholar] [PubMed]
- Bates, S.M.; Weitz, J. Direct thrombin inhibitors for treatment of arterial thrombosis: potential differences between bivalirudin and hirudin. Am. J. Cardiol. 1998, 82, 12P–18P. [Google Scholar] [CrossRef]
- Ansell, J.E. Universal, class-specific and drug-specific reversal agents for the new oral anticoagulants. J. Thromb. Thrombolysis 2016, 41, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Outes, A.; Suárez-Gea, M.L.; Lecumberri, R.; Terleira-Fernández, A.I.; Vargas-Castrillón, E. Direct-acting oral anticoagulants: pharmacology, indications, management, and future perspectives. Eur. J. Haematol. 2015, 95, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Heidbuchel, H.; Verhamme, P.; Alings, M.; Antz, M.; Hacke, W.; Oldgren, J.; Sinnaeve, P.; Camm, A.J.; Kirchhof, P. European Heart Rhythm Association Practical Guide on the use of new oral anticoagulants in patients with non-valvular atrial fibrillation. Europace 2013, 15, 625–651. [Google Scholar]
- Eikelboom, J.W.; Weitz, J.I. “Realworld” use of non-vitamin K antagonist oral anticoagulants (NOACs): Lessons from the Dresden NOAC Registry. Thromb. Haemost. 2015, 113, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Markwardt, F. Past, present, and future of hirudin. Haemostasis 1991, 21 (Suppl. I), 11–26. [Google Scholar] [CrossRef] [PubMed]
- Maragonore, J.M.; Bourdon, P.; Jablonsky, J.; Ramachandran, K.L.; Fenton, J.W. Design and characterization of hirulogs: A novel class of bivalent peptide inhibitors of thrombin. Biochemistry 1990, 29, 7095–7101. [Google Scholar] [CrossRef]
- De Caterina, R.; Husted, S.; Wallentin, L.; Andreotti, F.; Arnesen, H.; Bachmann, F.; Baigent, C.; Huber, K.; Jespersen, J.; Kristensen, S.D.; et al. General mechanisms of coagulation and targets of anticoagulants (Section I). Position Paper of the ESC Working Group on Thrombosis-Task Force on Anticoagulants in Heart Disease. Thromb. Haemost. 2013, 109, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Witting, J.L.; Bourdon, P.; Brenziak, D.V.; Fenton, J.W. Thrombin-specific inhibition by an slow cleavage of hirulog-1. Biochem. J. 1992, 283, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Parry, M.A.; Maraganore, J.M.; Stone, S.R. Kinetic mechanism for the interaction of Hirulog with thrombin. Biochemistry 1994, 33, 14807–14814. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.M.; Weitz, J.I. The mechanism of action of thrombin inhibitors. J. Invasive Cardiol. 2000, 12 (Suppl. F), 27F–32F. [Google Scholar] [PubMed]
- Warkentin, T.E.; Greinacher, A.; Koster, A. Bivalirudin. Thromb Haemost. 2008, 99, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Gladwell, T.D. Bivalirudin: a direct thrombin inhibitor. Clin. Ther. 2002, 24, 38–58. [Google Scholar] [CrossRef]
- Angiomax™ (Package insert) Parsippany; The Medicines Company: Parsippany, NJ, USA, 2005.
- Robson, R.; White, H.; Aylward, P.; Frampton, C. Bivalirudin pharmacokinetics and pharmacodynamics: Effect of renal function, dose, and gender. Clin. Pharmacol. Ther. 2002, 71, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Lidón, R.M.; Théroux, P.; Juneau, M.; Adelman, B.; Maraganore, J. Initial experience with a direct antithrombin, Hirulog, in ustable angina. Anticoagulant, antithrombotic and clinical effects. Circulation 1993, 88, 1495–1501. [Google Scholar] [CrossRef] [PubMed]
- Topol, E.J.; Bonan, R.; Jewitt, D.; Sigwar, U.; Kakkar, V.V.; Rothman, M.; de Bono, D.; Ferguson, J.; Willerson, J.T.; Strony, J.; et al. Use of a direct antithrombin, Hirulog, in place of heparin during coronary angioplasty. Circulation 1993, 87, 1622–1629. [Google Scholar] [CrossRef] [PubMed]
- Lui, H.K. Dosage, pharmacological effects and clinical outcomes for bivalirudin in percutaneous coronary intervention. J. Invasive Cardiol. 2000, 12 (Suppl. F), 41F–52F. [Google Scholar] [PubMed]
- Bittl, J.A.; Strony, J.; Brinker, J.A.; Ahmed, W.H.; Meckel, C.R.; Chaitman, B.R.; Maraganore, J.; Deutsch, E. Adelman B, for the Hirulog Angioplasty Study Investigatiors. Treatment with bivalirudin (Hirulog) as compared with heparin during coronary angioplasty for unstable or postinfarction angina. N. Engl. K. Med. 1995, 333, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Pötzch, B.; Hund, S.; Madlener, K.; Seelig, C.; Riess, C.F.; Greinacher, A.; Müller-Berghaus, G. Monitoring of r-hirudin anticoagulation during cardiopulmonary bypass: Assessment of the whole blood ecarin clotting time. Thromb. Haemost. 1997, 77, 920–925. [Google Scholar]
- Koster, A.; Chew, D.; Gründel, M.; Bauer, M.; Kuppe, H.; Spiess, B.D. Bivalirudin monitored with the ecarin clotting time for anticoagulation during cardiopulmonary bypass. Anesth. Analg. 2003, 96, 383–386. [Google Scholar] [PubMed]
- Zhang, D.; Wang, Z.; Zhao, X.; Lu, W.; Gu, J.; Cui, Y. Pharmacokinetics, pharmacodynamics, tolerability and safety of single doses of bivalirudin in healthy chinese subjects. Biol. Pharm. Bull. 2011, 34, 1841–1848. [Google Scholar] [CrossRef] [PubMed]
- Fox, I.; Dawson, A.; Loynds, P.; Eisner, J.; Findlen, K.; Levin, E.; Hanson, D.; Mant, T.; Wagner, J.; Maraganore, J. Anticoagulant activity of Hirulog, a direct thrombin inhibitor, in humans. Thromb. Haemost. 1993, 69, 157–163. [Google Scholar] [PubMed]
- Stone, G.W.; Witzenbichler, B.; Guagliumi, G.; Peruga, J.Z.; Brodie, B.R.; Dudek, D.; Kornowski, R.; Hartmann, F.; Gersh, B.J.; Pocock, S.J.; et al. Bivalirudin during primary PCI in acute myocardial infarction. N. Engl. J. Med. 2008, 358, 2218–2230. [Google Scholar] [CrossRef] [PubMed]
- Steg, P.G.; van’t Hof, A.; Hamm, C.W.; Clemmensen, P.; Lapostolle, F.; Coste, P.; Ten Berg, J.; van Grunsven, P.; Jan Eggink, G.; Nibbe, L.; et al. Bivalirudin started during emergency transport for primary PCI. N. Engl. J. Med. 2013, 369, 2207–2217. [Google Scholar] [CrossRef] [PubMed]
- Clemmensen, P.; Wiberg, S.; van’t Hof, A.; Deliargyris, E.N.; Coste, P.; Ten Berg, J.; Cavallini, C.; Hamon, M.; Dudek, D.; Zeymer, U.; et al. Acute stent thrombosis after primary percutaneous coronary intervention: insights from the EUROMAX trial (European Ambulance Acute Coronary Syndrome Angiography). JACC Cardiovasc. Interv. 2015, 8 (1 Pt B), 214–220. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, A.; Kemp, I.; Mars, C.; Wilson, K.; Roome, C.; Cooper, R.; Andron, M.; Appleby, C.; Fisher, M.; Khand, A.; et al. Unfractionated heparin versus bivalirudin in primary percutaneous coronary intervention (HEAT-PPCI): An open-label, single centre, randomised controlled trial. Lancet 2014, 384, 1849–1858. [Google Scholar] [CrossRef]
- Han, Y.; Guo, J.; Zheng, Y.; Zang, H.; Su, X.; Wang, Y.; Chen, S.; Jiang, T.; Yang, P.; Chen, J.; et al. Bivalirudin vs. heparin with or without tirofiban during primary percutaneous coronary intervention in acute myocardial infarction: The BRIGHT randomized clinical trial. JAMA 2015, 313, 1336–1346. [Google Scholar] [CrossRef] [PubMed]
- Schulz, S.; Richardt, G.; Laugwitz, K.-L.; Morath, T.; Neudecker, J.; Hoppmann, P.; Mehran, R.; Gershlick, A.; Tolg, R.; Anette Fiedler, K.; et al. Prasugrel plus bivalirudin vs. clopidogrel plus heparin in patients with ST-segment elevation myocardial infarction. Eur. Heart J. 2014, 35, 2285–2294. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.M.; Morrow, D.A.; Murphy, S.A.; Palabrica, T.M.; Jennings, L.K.; Stone, P.H.; Lui, H.; Bulle, T.; Lakkis, N.; Kovach, R.; et al. A randomized trial to evaluate the relative protection against post-percutaneous coronary intervention microvascular dysfunction, ischemia, and inflammation among antiplatelet and antithrombotic agents: The PROTECT-TIMI-30 trial. J. Am. Coll. Cardiol. 2006, 47, 2364–2373. [Google Scholar] [CrossRef] [PubMed]
- White, H.D.; Chew, D.P.; Hoekstra, J.W.; Miller, C.D.; Pollack, C.V.; Feit, F.; Lincoff, A.; Bertrand, M.; Pocock, S.; Ware, J.; et al. Safety and efficacy of switching from either unfractionated heparin or enoxaparin to bivalirudin in patients with non-ST-segment elevation acute coronary syndromes managed with an invasive strategy: Results from the ACUITY (Acute Catheterization and Urgent Intervention Triage strategY) trial. J. Am. Coll. Cardiol. 2008, 51, 1734–1741. [Google Scholar] [PubMed]
- Sibbing, D.; Bernlochner, I.; Schulz, S.; Massberg, S.; Schömig, A.; Mehilli, J.; Kastrati, A. Prognostic value of a high on-clopidogrel treatment platelet reactivity in bivalirudin versus abciximab treated non-ST-segment elevation myocardial infarction patients. ISAR-REACT 4 (Intracoronary Stenting and Antithrombotic Regimen: Rapid Early Action for Coronary Treatment-4) platelet substudy. J. Am. Coll. Cardiol. 2012, 60, 369–377. [Google Scholar] [PubMed]
- Steg, P.G.; James, S.K.; Atar, D.; Badano, L.P.; Blömstrom-Lundqvist, C.; Borger, M.; Di Mario, C.; Dickstein, K.; Ducrocq, G.; Fernandez-Aviles, F.; et al. ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur. Heart J. 2012, 33, 2569–2619. [Google Scholar] [CrossRef] [PubMed]
- O’Gara, P.T.; Kushner, F.G.; Ascheim, D.D.; Casey, D.E.; Chung, M.K.; de Lemos, J.A.; Ettinger, S.; Fang, J.; Fesmire, F.; Franklin, B.; et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: Developed in collaboration with the American College of Emergency Physicians and Society for Cardiovascular Angiography and Interventions. Catheter. Cardiovasc. Interv. 2013, 82, E1–E27. [Google Scholar] [PubMed]
- National Clinical Guideline Centre (UK). Myocardial Infarction with ST-Segment Elevation: The Acute Management of Myocardial Infarction with ST-Segment Elevation [Internet]; Royal College of Physicians (UK): London, UK, 2013. [Google Scholar]
- Windecker, S.; Kolh, P.; Alfonso, F.; Collet, J.-P.; Cremer, J.; Falk, V.; Filippatos, G.; Hamm, C.; Head, S.; Juni, P.; et al. 2014 ESC/EACTS Guidelines on myocardial revascularization: The Task Force on Myocardial Revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS)Developed with the special contribution of the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur. Heart J. 2014, 35, 2541–2619. [Google Scholar] [PubMed]
- National Clinical Guideline Centre (UK). Unstable Angina and NSTEMI: The Early Management of Unstable Angina and Non-ST-Segment-Elevation Myocardial Infarction; Royal College of Physicians (UK): London, UK, 2010. [Google Scholar]
- Roffi, M.; Patrono, C.; Collet, J-P.; Mueller, C.; Valgimigli, M.; Andreotti, F.; Bax, J.J.; Borger, M.A.; Brotons, C.; Chew, D.P.; et al. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: Task Force for the Management of Acute Coronary Syndromes in Patients Presenting without Persistent ST-Segment Elevation of the European Society of Cardiology (ESC). Eur. Heart J. 2016, 37, 267–315. [Google Scholar] [PubMed]
- Amsterdam, E.A.; Wenger, N.K.; Brindis, R.G.; Casey, D.E.; Ganiats, T.G.; Holmes, D.R.; Jaffe, A.; Jneid, H.; Kelly, R.; Kontos, M.; Levine, G.; et al. 2014 AHA/ACC Guideline for the Management of Patients with Non-ST-Elevation Acute Coronary Syndromes: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J. Am. Coll Cardiol. 2014, 64, e139–e228. [Google Scholar] [CrossRef] [PubMed]
- Valgimigli, M.; Frigoli, E.; Leonardi, S.; Rothenbühler, M.; Gagnor, A.; Calabrò, P.; Garducci, S.; Rubartelli, P.; Briguori, C.; Andò, G.; et al. Bivalirudin or Unfractionated Heparin in Acute Coronary Syndromes. N. Engl. J. Med. 2015, 373, 997–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farag, M.; Gorog, D.A.; Prasad, A.; Srinivasan, M. Bivalirudin versus unfractionated heparin: A meta-analysis of patients receiving percutaneous coronary intervention for acute coronary syndromes. Open Heart 2015, 2, e000258. [Google Scholar] [CrossRef] [PubMed]
- Bittl, J.A.; He, Y.; Lang, C.D.; Dangas, G.D. Factors Affecting Bleeding and Stent Thrombosis in Clinical Trials Comparing Bivalirudin With Heparin During Percutaneous Coronary Intervention. Circ. Cardiovasc Interv. 2015, 8, e002789. [Google Scholar] [CrossRef] [PubMed]
- Navarese, E.P.; Schulze, V.; Andreotti, F.; Kowalewski, M.; Kołodziejczak, M.; Kandzari, D.E.; Rassaf, T.; Gorny, B.; Brockmeyer, M.; Meyer, C.; et al. Comprehensive meta-analysis of safety and efficacy of bivalirudin versus heparin with or without routine glycoprotein IIb/IIIa inhibitors in patients with acute coronary syndrome. JACC Cardiovasc. Interv. 2015, 8, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, R.; de Biase, C.; D’Anna, C.; Trimarco, B.; Piscione, F.; Galasso, G. Early stent thrombosis with bivalirudin in patients undergoing percutaneous coronary intervention. A meta-analysis of randomised clinical trials. Thromb. Haemost. 2015, 113, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Alexander, W. Bivalirudin Versus Heparin: A Fight Far From Finished?: Efficacy, Safety, and Cost Remain Battlegrounds for the Treatment Of ST-Segment Elevation Myocardial Infarction. Pharm. Ther. 2015, 40, 209–217. [Google Scholar]



{kind=link}
{kind=link}
| Main Trials | HORIZONS-AMI [68] | EUROMAX [69] | HEAT-PPCI [71] | BRIGHT [72] | BRAVE 4 [73] | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Year | 2008 | 2013 | 2014 | 2015 | 2014 | ||||||
| Enrolment | 2005–2007 | 2010–2013 | 2012–2013 | 2012–2013 | 2009-2013 | ||||||
| Centres | 123 | 65 | 1 | 82 | 3 | ||||||
| Treatment | Bivalirudin: 0.75 mg/kg bolus followed by an infusion of 1.75 mg/kg/h for the duration of PCI. Heparin: UFH 60 UI/kg bolus with additional bolus to target ACT 200–250 s. | Bivalirudin: 0.75 mg/kg bolus followed by an infusion of 1.75 mg/kg/h for at least 4 h after (0.25–1.75 mg/kg/h). Heparin: UFH 100 UI/kg bolus without GPI or 60 UI/kg with GPI; or enoxaparin 0.5 mg/kg bolus. | Bivalirudin: 0.75 mg/kg bolus followed by an infusion of 1.75 mg/kg/h for the duration of PCI. Heparin: UFH 70 UI/kg bolus with additional doses if ACT < 200 s. | Bivalirudin: 0.75 mg/kg bolus followed by an infusion of 1.75 mg/kg/h for at least 4 h. After reduced infusion 0.2 mg/kg/h for up 20 h. Heparin: UFH 60 UI/kg bolus with additional doses if ACT < 200 s. Heparin + GPI: UFH 60 UI/kg bolus + tirofiban 10 µg/kg bolus followed by infusion 0.15 µg/kg/min for 18 to 36 h. | Bivalirudin + Prasugrel: Prasugrel loading dose of 60 mg + 10 mg daily. Bivalirudin: 0.75 mg/kg bolus followed by an infusion of 1.75 mg/kg/h for the duration of PCI. Heparin + Clopidogrel: Clopidogrel loading dose 600 mg + 75 mg daily. UFH 70–100 UI/kg | ||||||
| HORIZONS-AMI [68] | EUROMAX [69] | HEAT-PPCI [71] | BRIGHT [72] | BRAVE 4 [73] | |||||||
| Bivalirudin | Heparin | Bivalirudin | Heparin | Bivalirudin | Heparin | Bivalirudin | Heparin | Heparin + GPI | Bivalirudin | Heparin | |
| Patients | 1800 | 1802 | 1089 | 1109 | 905 | 907 | 735 | 729 | 730 | 271 | 277 |
| Age | 59.8 (26.0–92.3) | 60.7 (21.6–91.6) | 61 (52–71) | 62 (52–72) | 62.9 (53.7–74.0) | 63.6 (54.0–73.8) | 57.3 ± 11.6 | 58.1 ± 11.7 | 58.2 ± 11.8 | 61.4 (51.9–71.7) | 61.4 (52.9–71.5) |
| Male | 77.1% | 76.1% | 74.7% | 77.6% | 71.0% | 73.0% | 82.7% | 81.6% | 82.1% | 76% | 79% |
| Clopidogrel | 95.7% | 95.1% | 50.0% | 51.5% | 11.8% | 10.0% | 100% | 100% | 99.9% | 3.7% | 90.2% |
| Prasugrel | 0% | 0% | 30.8% | 28.9% | 27.3% | 27.6% | 0% | 0% | 0% | 94.6% | 7.1% |
| Ticagrelor | 0% | 0% | 19.2% | 19.4% | 61.2% | 62.7% | 0% | 0% | 0% | 0% | 0% |
| GPI use | 7.2% | 94.5% | 11.5% | 69.1% | 13.5% | 15.5% | 4.4% | 5.6% | 100% | 3.0% | 6.1% |
| Radial access | 5.7% | 5.4% | 47.7% | 46.3% | 80.3% | 82.0% | 78.4% | 79.0% | 78.2% | <1% | <1% |
| Drug-eluting stent | NA | NA | 57.1% | 55.9% | 79.7% | 79.8% | 99.3% | 99.3% | 99.6% | 82.3% | 82.3% |
| PCI perform | 93.2% | 92.2% | 87.1% | 85.4% | 83.0% | 81.6% | 98.4% | 98.6% | 98.9% | 88.6% | 86.6% |
| NACE | 9.2% | 12.1% | 5.1% | 8.5% | 8.7% | 5.7% | 8.8% | 13.2% | 17.0% | 15.6% | 14.5% |
| Bleeding events | 4.9% | 8.3% | 2.6% | 6.0% | 3.5% | 3.1% | 4.1% | 7.5% | 12.3% | 14.1% | 12.0% |
| Main Trials | PROTECT-30 [74] | ACUITY [75] | ISAR-REACT [76] | ||||
|---|---|---|---|---|---|---|---|
| Year | 2006 | 2007 | 2011 | ||||
| Enrolment | 2003–2004 | 2003–2007 | 2006–2011 | ||||
| Centres | 100 | 450 | 8 | ||||
| Treatment | Bivalirudin: 0.75 mg/kg bolus followed by an infusion of 1.75 mg/kg/h for the duration of PCI and 4 h after at physician’s discretion. Heparin: UFH 50 UI/kg bolus with additional bolus to target ACT 200–250 s + epifibatide. LMWH: Bolus of 0.5 mg/Kg + epifibatide. | Bivalirudin alone: 0.75 mg/kg bolus followed by an infusion of 1.75 mg/kg/h. Bivalirudin + GPI: 0.75 mg/kg bolus followed by an infusion of 1.75 mg/kg/h GPI: Epifibatide or Tirofiban. Heparin + GPI: UFH 60 UI/kg bolus plus infusion of 12 UI/kg/h to target ACT 200–250 s or enoxaparin 1 mg/kg twice daily plus bolus 0.3-0.75 mg/kg i.v. during PIC. GPI: epifibatide or tirofiban. | Bivalirudin: 0.75 mg/kg bolus followed by an infusion of 1.75 mg/kg/h for the duration of PCI. Heparin: UFH 70 UI/kg bolus with additional doses if ACT < 200 s + GPI (abciximab 0.25 mg/kg bolus + 0.125 µg/kg/min for 12 h). | ||||
| Bivalirudin | Heparin | Bivalirudin | Bivalirudin + GPI | Heparin + GPI | Bivalirudin | Heparin | |
| Patients | 284 | 573 | 2619 | 2609 | 2561 | 860 | 862 |
| Age | 59.7 ± 9.8 | 60.0 ± 11.1 | 63 (30–92) | 62 (21–95) | 63 (25–91) | 67.5 ± 11.2 | 67.5 ± 10.8 |
| Male | 68.3% | 66.0% | 73.0% | 74.0% | 73.0% | 76.8% | 76.9% |
| Clopidogrel | NA | 100% | 100% | 100% | 100% | 100% | |
| Prasugrel | NA | 0% | 0% | 0% | 0% | 0% | |
| Ticagrelor | NA | 0% | 0% | 0% | 0% | 0% | |
| GPI use | 3.0% | 99% | 9.0% | 97.0% | 97.0% | 0% | 100% |
| Radial access | NA | 6% | <1% (6 patients) | ||||
| Drug-eluting stent | 79% | 60.0% | 60.0% | 61.0% | 88.2% | 88.9% | |
| PCI performed | 100% | 100% | 56.8% | 56.7% | 56% | 99.8% | 99.8% |
| NACE | * 1.43% | 1.33% | 12.0% | 15.0% | 13.0% | 11.0% | 10.9% |
| Bleeding events | 0.4% | 2.5% | 4.0% | 8.0% | 7.0% | 2.6% | 4.6% |
| ACS | 2012 ESC GUIDELINES [77] | 2013 AHA/ACC GUIDELINES [78] | 2014 REVASCULARIZATION GUIDELINES [80] | NICE Guidelines July 2013 [79] |
| Stemi Patients | Bivalirudin with use of GP IIb/IIIa blocker restricted to bailout) is recommended over UFH and a GP IIb/IIIa blocker. | Bivalirudin with or without prior treatment with UFH is recommended. | Bivalirudin 0.75 mg/kg i.v. bolus followed by i.v. infusion of 1.75 mg/kg/h for up 4 h after the procedure. | Bivalirudin in combination with aspirin and clopidogrel is recommended for the treatment of adults with STEMI undergoing primary PCI. |
| Class I, Level B | Class I, Level B | Class IIa, Level A | ||
| ACS | 2015 ESC GUIDELINES [82] | 2014 AHA/ACC GUIDELINES [83] | 2014 REVASCULARIZATION GUIDELINES [80] | NICE Guidelines March 2010 [81] |
| Non-Stemi Patients | Bivalirudin (0.75 mg/kg bolus, followed by 1.75 mg/kg/h for up to 4 h after the procedure) is recommended as alternative to UFH plus GP IIb/IIIa receptor inhibitor during PCI. | Bivalirudin 0.10 mg/kg loading dose followed by 0.25 mg/kg per hour (only in patients managed with an early invasive strategy) continued until diagnostic angiography or PCI, with only provisional use of GP IIb/IIIa inhibitor, provided the patient is also treated with DAPT. | Bivalirudin (0.75 mg/kg bolus, followed by 1.75 mg/kg/h for up to 4 h after the procedure) is recommended as alternative to UFH plus GP IIb/IIIa receptor inhibitor during PCI. | As an alternative to the combination of UFH plus GPI, consider bivalirudin for patients who: Are at intermediate or higher risk of adverse cardiovascular events (predicted 6 moth mortality above 3%) Are not already receiving GPI or fondaparinux Are scheduled to undergo angiography within 24 h of admission. |
| Class I, Level A | Class I, Level B | Class I, Level A |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esteve-Pastor, M.A.; Hernández-Romero, D.; Valdés, M.; Marín, F. New Approaches to the Role of Thrombin in Acute Coronary Syndromes: Quo Vadis Bivalirudin, a Direct Thrombin Inhibitor? Molecules 2016, 21, 284. https://doi.org/10.3390/molecules21030284
Esteve-Pastor MA, Hernández-Romero D, Valdés M, Marín F. New Approaches to the Role of Thrombin in Acute Coronary Syndromes: Quo Vadis Bivalirudin, a Direct Thrombin Inhibitor? Molecules. 2016; 21(3):284. https://doi.org/10.3390/molecules21030284
Chicago/Turabian StyleEsteve-Pastor, María Asunción, Diana Hernández-Romero, Mariano Valdés, and Francisco Marín. 2016. "New Approaches to the Role of Thrombin in Acute Coronary Syndromes: Quo Vadis Bivalirudin, a Direct Thrombin Inhibitor?" Molecules 21, no. 3: 284. https://doi.org/10.3390/molecules21030284
APA StyleEsteve-Pastor, M. A., Hernández-Romero, D., Valdés, M., & Marín, F. (2016). New Approaches to the Role of Thrombin in Acute Coronary Syndromes: Quo Vadis Bivalirudin, a Direct Thrombin Inhibitor? Molecules, 21(3), 284. https://doi.org/10.3390/molecules21030284