
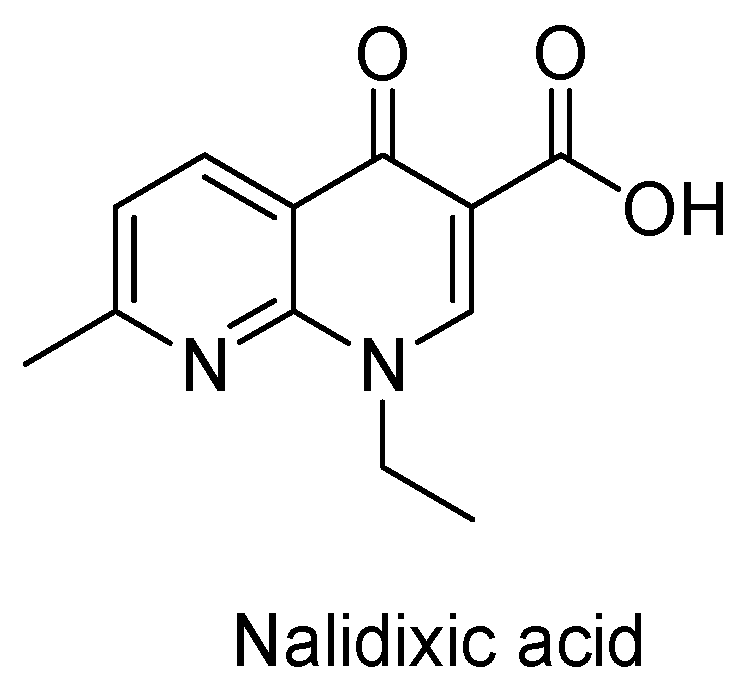
The Current Case of Quinolones: Synthetic Approaches and Antibacterial Activity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
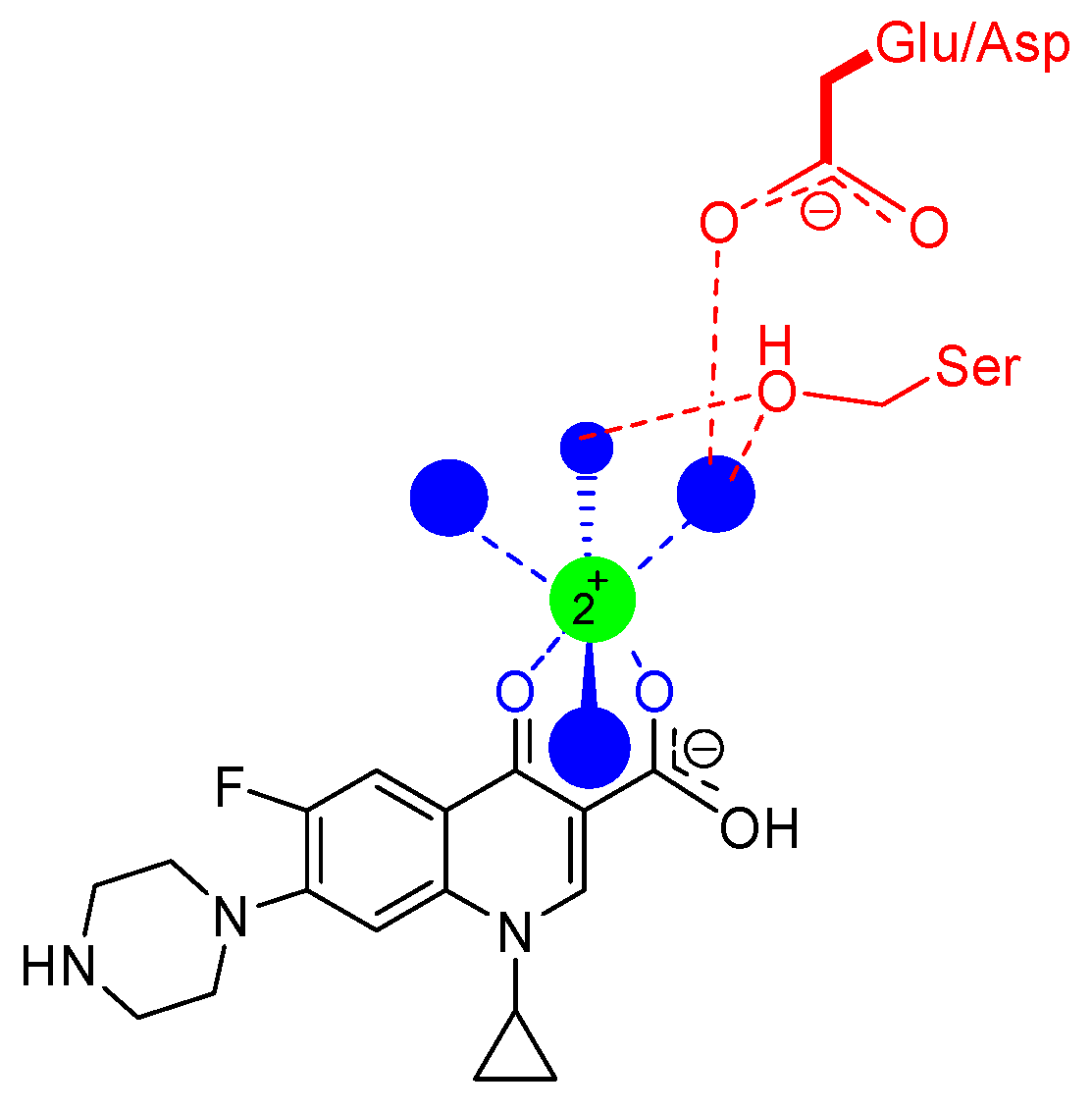{kind=link}
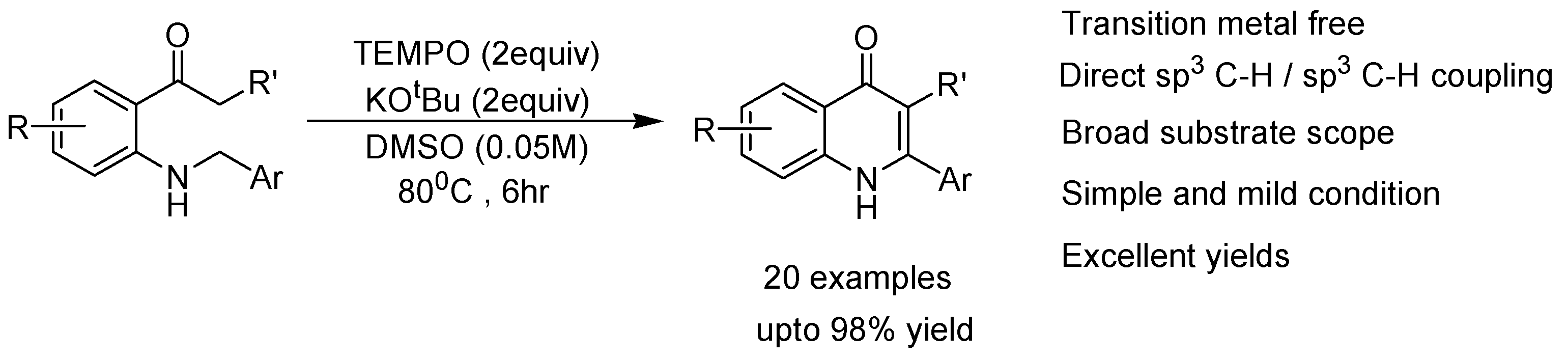{kind=link}
{kind=link}
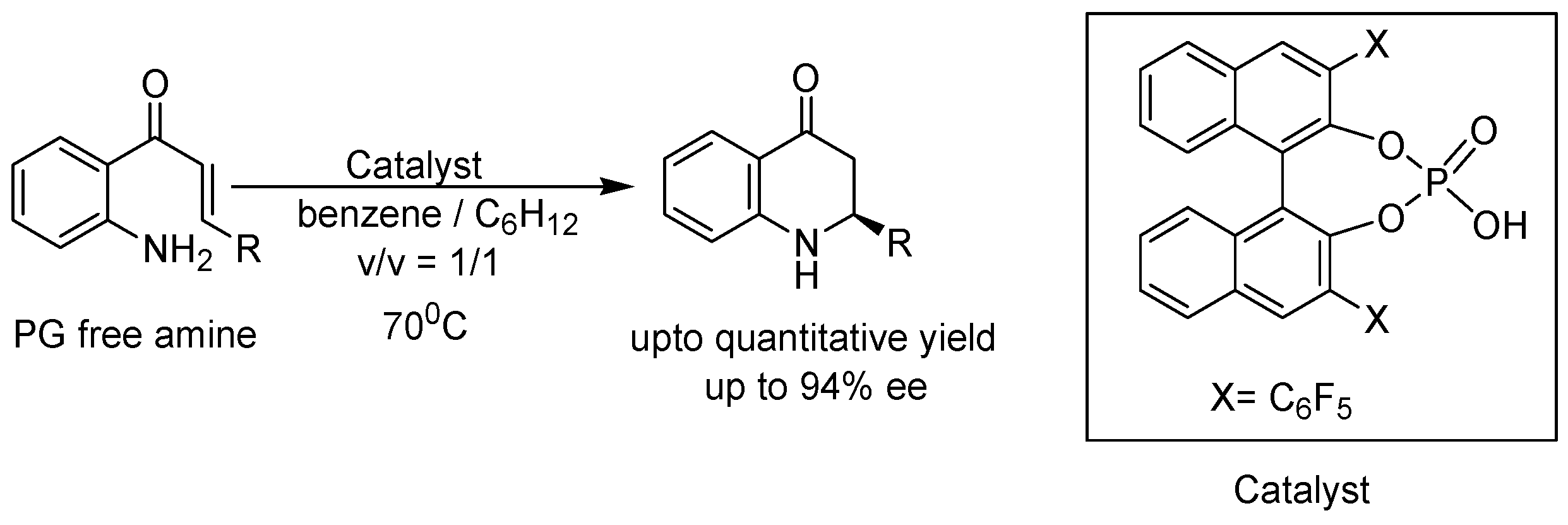{kind=link}
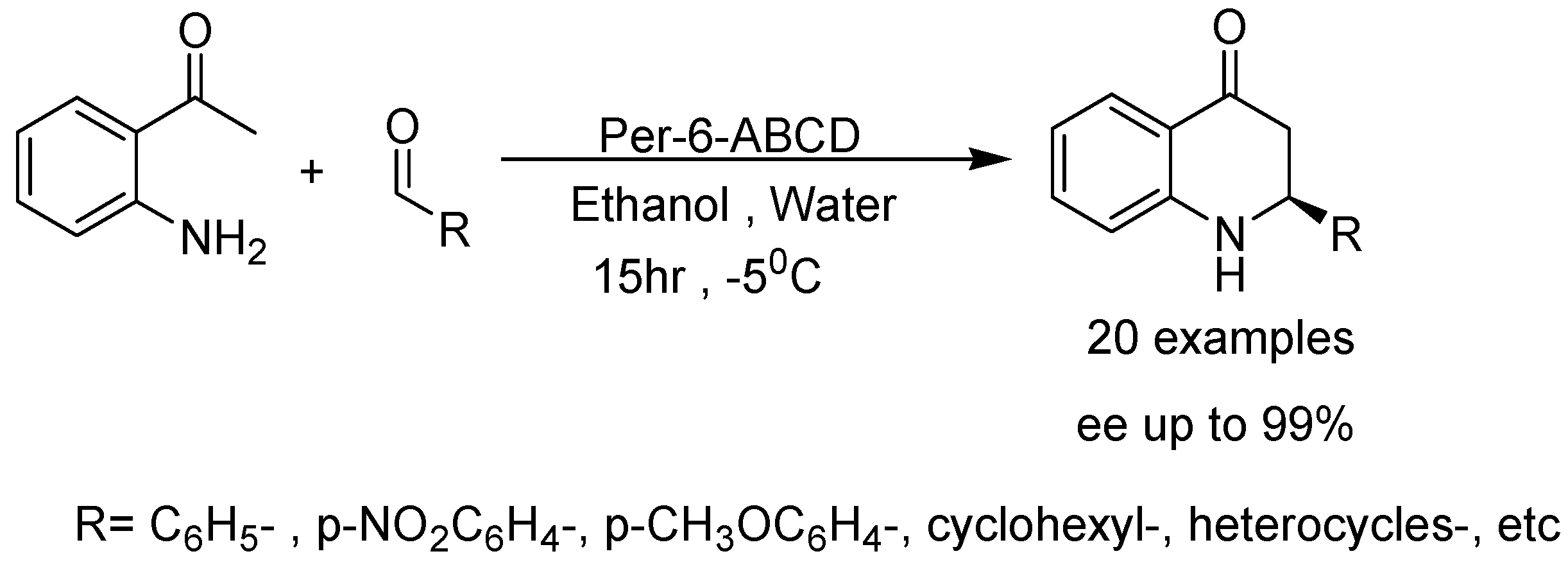{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
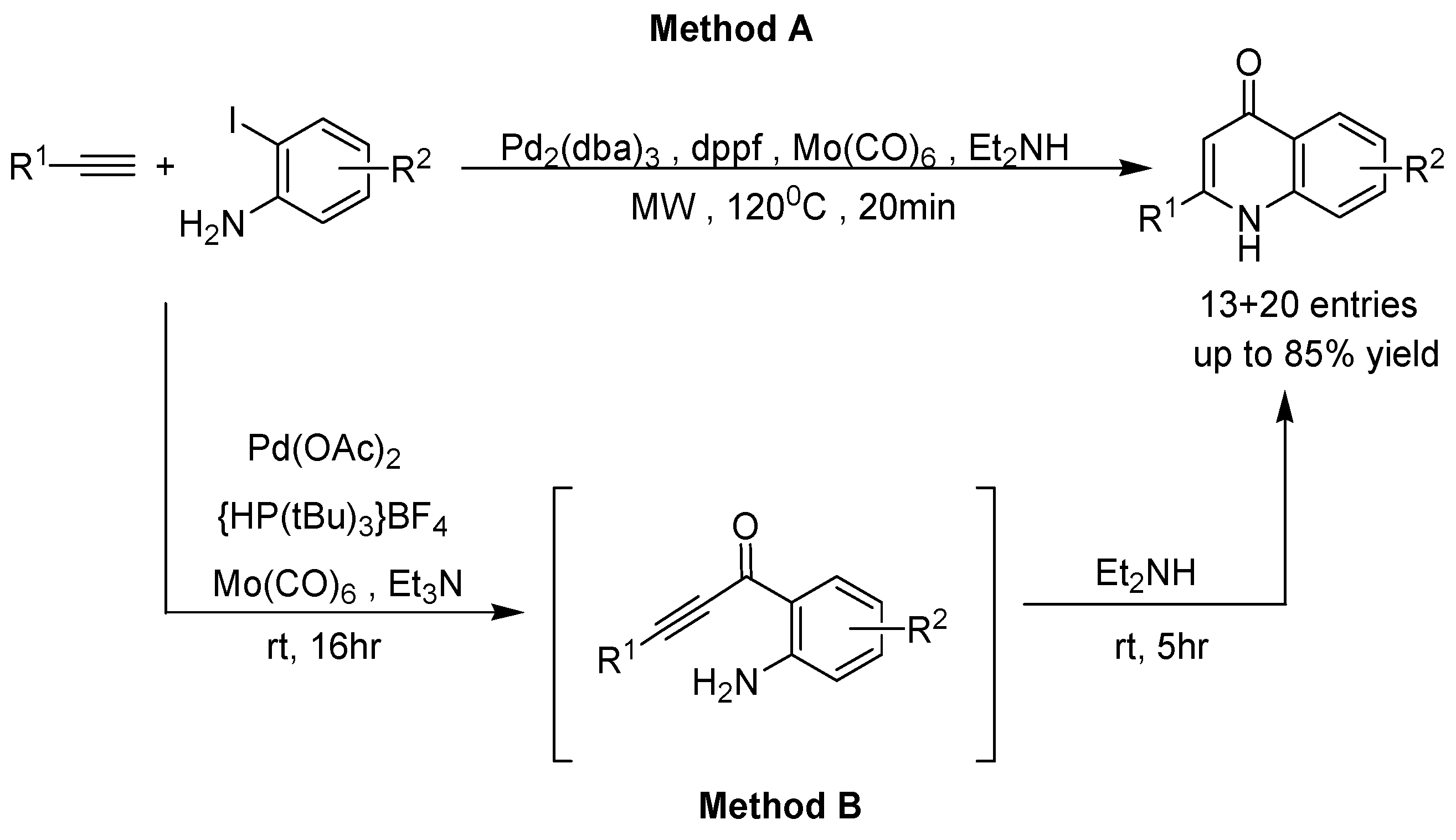
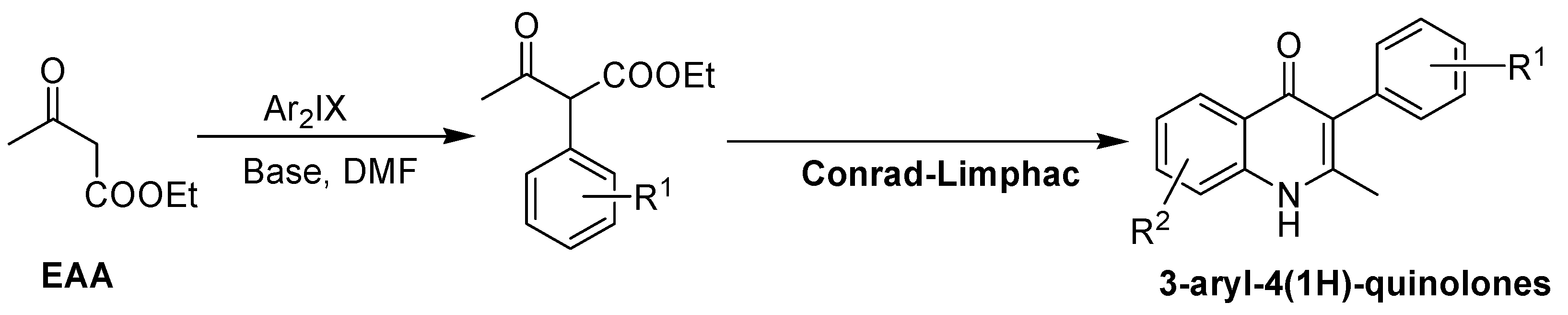
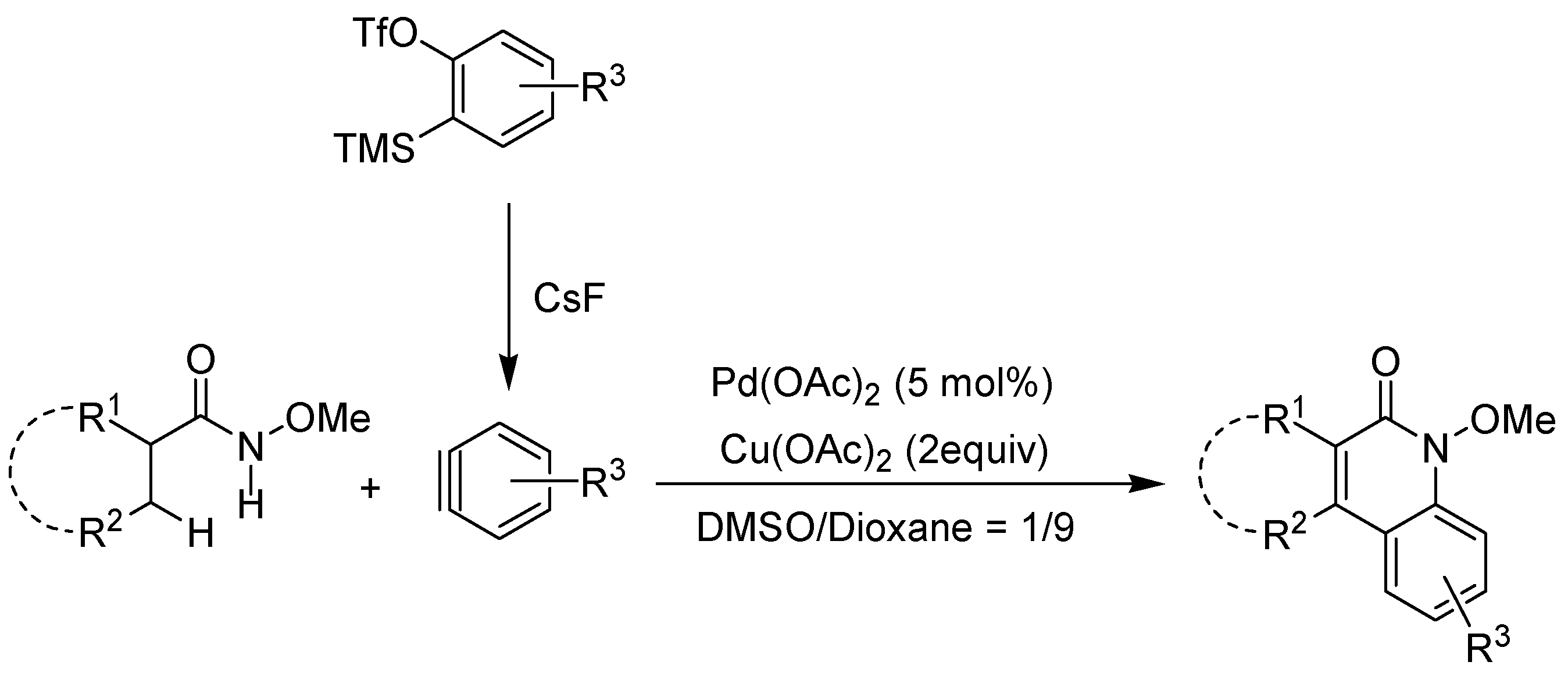
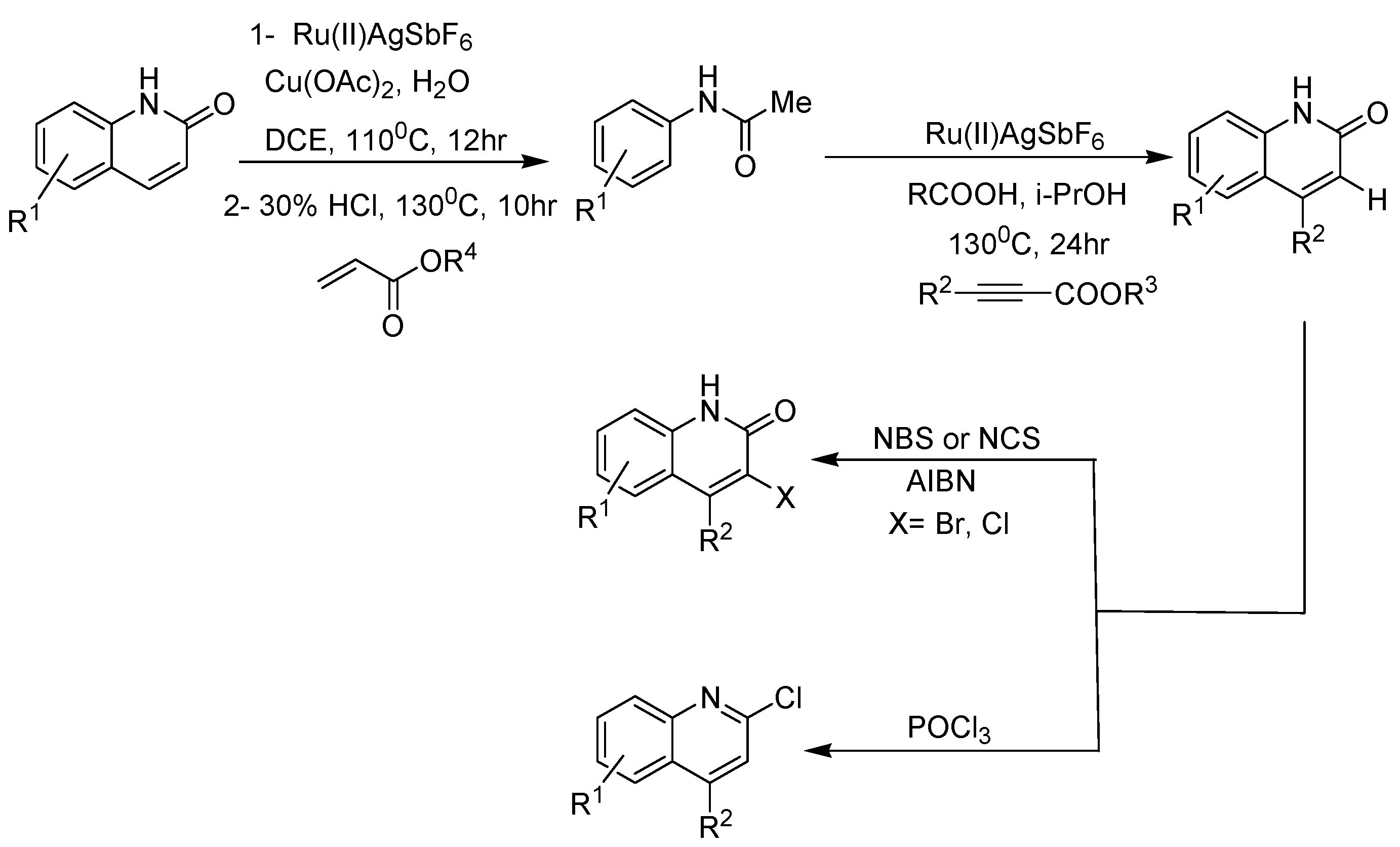
2. Recent Developments in the Synthesis of Quinolones
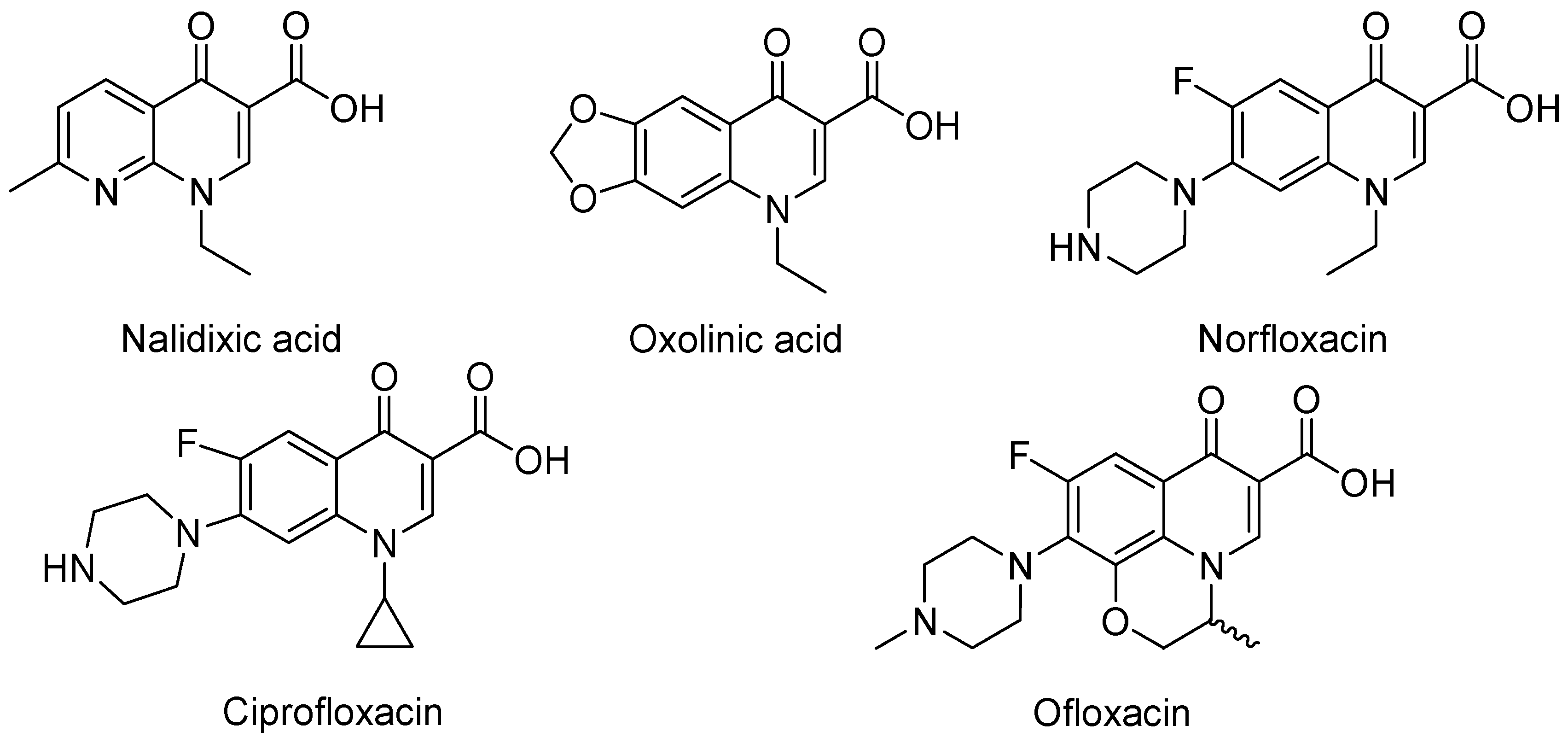
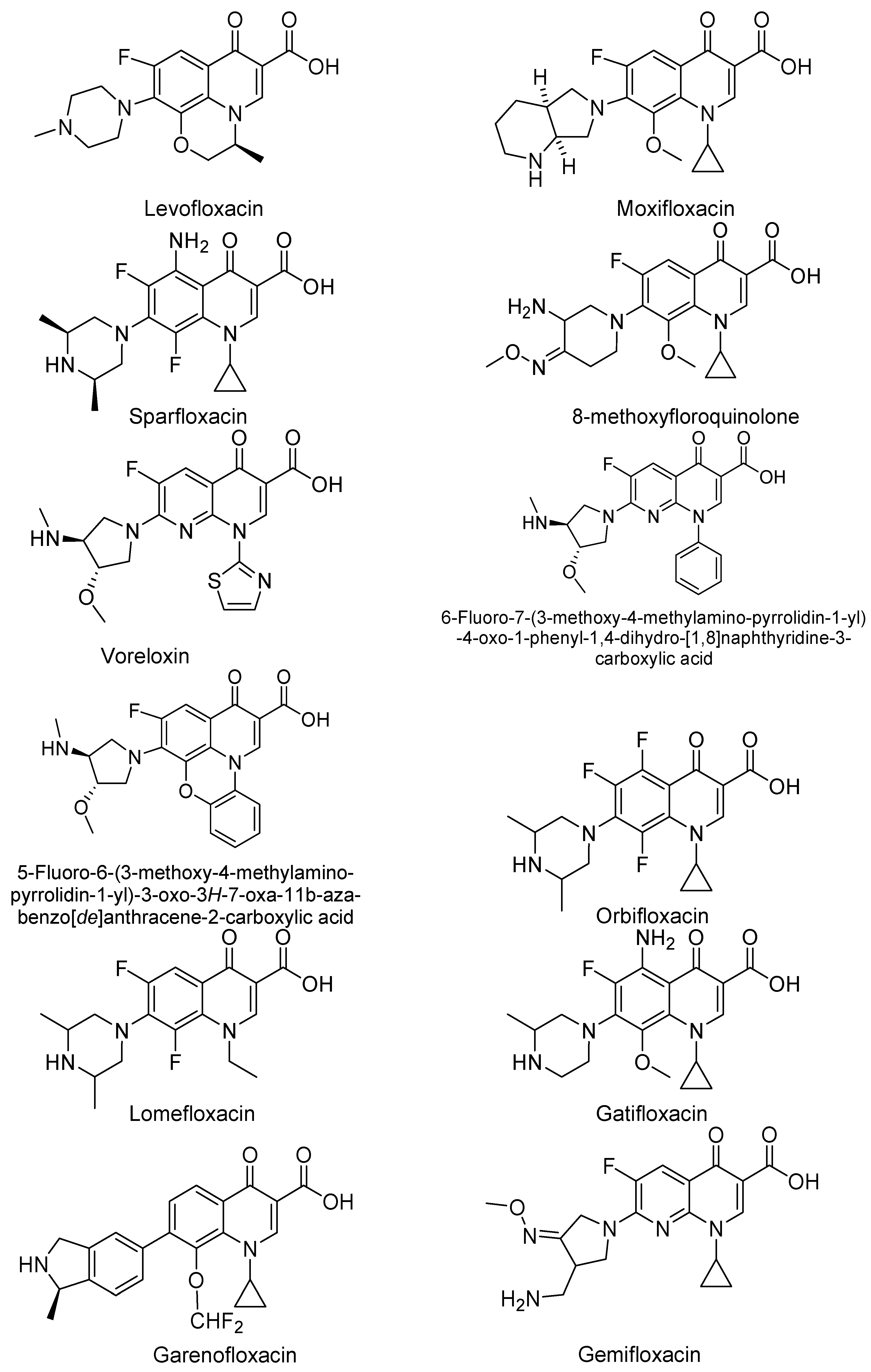
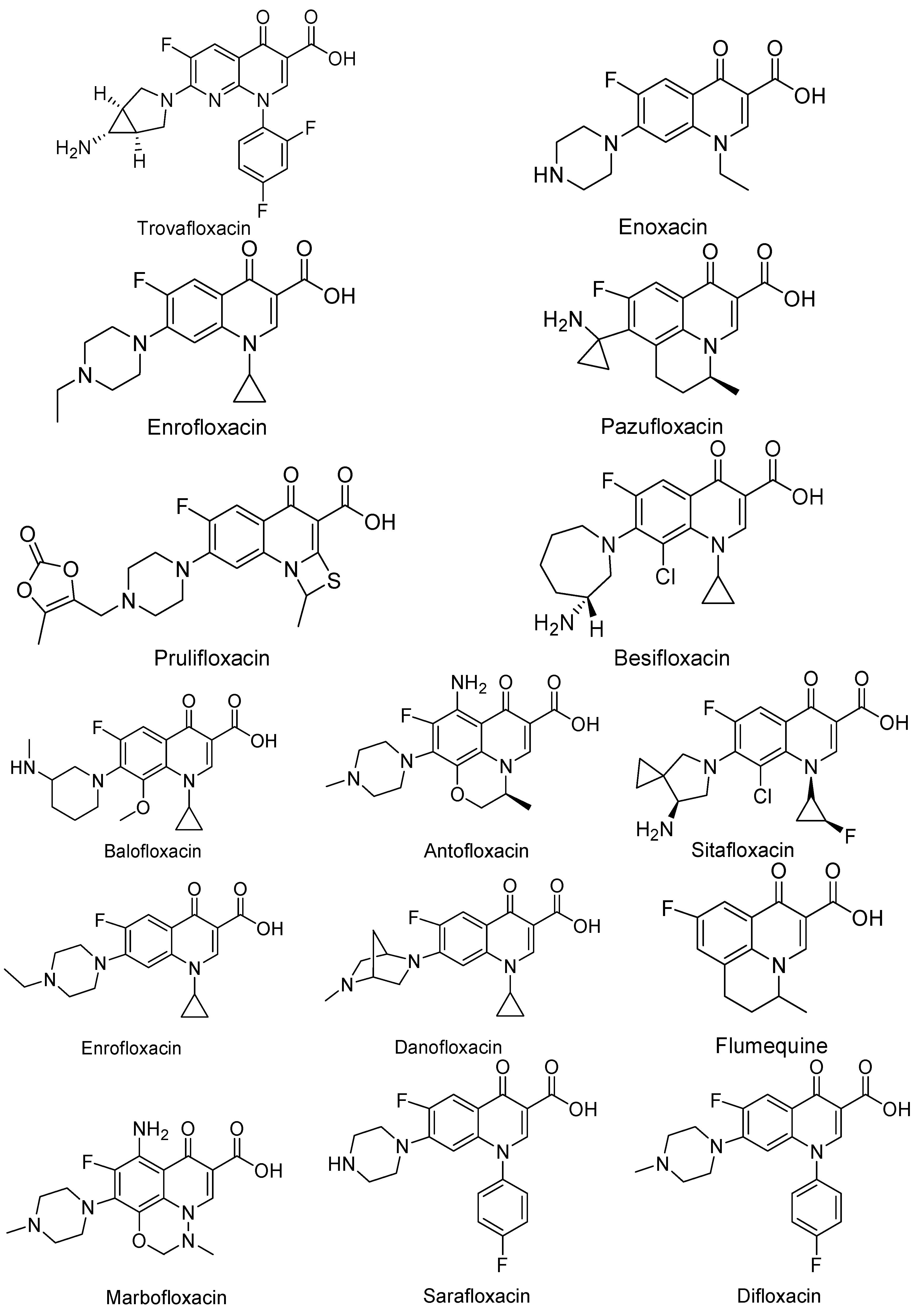
3. Analogues of Quinolones
4. Clinical Use
5. Targets of Quinolones
6. Mode of Action of Quinolones
7. Interaction of Quinolones with Topoisomerases
8. Bacterial Resistance to Quinolones
9. Target-Mediated Quinolones Resistance
10. Plasmid-Mediated Quinolone Resistance
11. Chromosome-Mediated Quinolone Resistance
12. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Armstrong, G.L.; Conn, L.A.; Pinner, R.W. Trends in infectious disease mortality in the united states during the 20th century. JAMA 1999, 281, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Cross, R.M.; Flanigan, D.L.; Monastyrskyi, A.; LaCrue, A.N.; Saenz, F.E.; Maignan, J.R.; Mutka, T.S.; White, K.L.; Shackleford, D.M.; Bathurst, I. Orally bioavailable 6-chloro-7-methoxy-4(1H)-quinolones efficacious against multiple stages of plasmodium. J. Med. Chem. 2014, 57, 8860–8879. [Google Scholar] [CrossRef] [PubMed]
- Baharoglu, Z.; Garriss, G.; Mazel, D. Multiple pathways of genome plasticity leading to development of antibiotic resistance. Antibiotics 2013, 2, 288–315. [Google Scholar] [CrossRef]
- Walsh, C. Where will new antibiotics come from? Nat. Rev. Microbiol. 2003, 1, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M. Has the era of untreatable infections arrived? J. Antimicrob. Chemother. 2009, 64, i29–i36. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Lin, J.-P.; Song, L.-R.; Long, Y.-Q. Direct synthesis of 2-aryl-4-quinolones via transition-metal-free intramolecular oxidative C (sp3)-H/C(sp3)-H coupling. Org. Lett. 2015, 17, 1268–1271. [Google Scholar] [CrossRef] [PubMed]
- Oliphant, C.M.; Green, G.M. Quinolones: A comprehensive review. Am. Fam. Physician 2002, 65, 455–464. [Google Scholar] [PubMed]
- Lesher, G.Y.; Froelich, E.J.; Gruett, M.D.; Bailey, J.H.; Brundage, R.P. 1,8-naphthyridine derivatives. A new class of chemotherapeutic agents. J. Med. Chem. 1962, 5, 1063–1065. [Google Scholar] [CrossRef]
- Wu, J.; Xiang, S.; Zeng, J.; Leow, M.; Liu, X.-W. Practical route to 2-quinolinones via a pd-catalyzed C-H bond activation/C-C bond formation/cyclization cascade reaction. Org. Lett. 2014, 17, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Emmerson, A.M.; Jones, A.M. The quinolones: Decades of development and use. J. Antimicrob. Chemother. 2003, 51, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Andriole, V.T. The quinolones: Past, present, and future. Clin. Infect. Dis. 2005, 41, S113–S119. [Google Scholar] [CrossRef] [PubMed]
- Bisacchi, G.S. Origins of the quinolone class of antibacterials: An expanded “discovery story”. J. Med. Chem. 2015, 58, 4874–4882. [Google Scholar] [CrossRef] [PubMed]
- Mitscher, L.A. Bacterial topoisomerase inhibitors: Quinolone and pyridone antibacterial agents. Chem. Rev. 2005, 105, 559–592. [Google Scholar] [CrossRef] [PubMed]
- Domagala, J.M. Structure-activity and structure-side-effect relationships for the quinolone antibacterials. J. Antimicrob. Chemother. 1994, 33, 685–706. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Liu, M.L.; Guo, H.Y.; Wang, Y.C.; Wang, J.X. Synthesis and in vitro antibacterial activity of 7-(3-amino-6,7-dihydro-2-methyl-2H-pyrazolo[4,3-c]pyridin-5(4H)-yl) fluoroquinolone derivatives. Molecules 2011, 16, 2626–2635. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; Kerns, R.J.; Osheroff, N. Mechanism of quinolone action and resistance. Biochemistry 2014, 53, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.C.; Jain, A.; Jain, S. Fluoroquinolone antibacterials: A review on chemistry, microbiology and therapeutic prospects. Acta. Pol. Pharm. 2009, 66, 587–604. [Google Scholar] [PubMed]
- Liu, M.L.; Guo, H.Y. Evolution of the quinolones. World Notes Antibiot. 2006, 27, 69–75. [Google Scholar]
- Blasco, C.; PicoÌ, Y. Development of an improved method for trace analysis of quinolones in eggs of laying hens and wildlife species using molecularly imprinted polymers. J. Agric. Food Chem. 2012, 60, 11005–11014. [Google Scholar] [CrossRef] [PubMed]
- Cross, R.M.; Maignan, J.R.; Mutka, T.S.; Luong, L.; Sargent, J.; Kyle, D.E.; Manetsch, R. Optimization of 1,2,3,4-tetrahydroacridin-9(10H)-ones as antimalarials utilizing structure-activity and structure-property relationships. J. Med. Chem 2011, 54, 4399–4426. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, J.; Zeng, F.; Alwis, N.; Alper, H. Synthesis of 2 (1H)-quinolinones via Pd-catalyzed oxidative cyclocarbonylation of 2-vinylanilines. Org. Lett. 2013, 15, 1998–2001. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Hong, S. Rh (III) and Ru (II)-catalyzed site-selective C-H alkynylation of quinolones. Org. Lett. 2015, 17, 1938–1941. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Moriya, Y.; Akiyama, T. Chiral phosphoric acid catalyzed asymmetric synthesis of 2-substituted 2, 3-dihydro-4-quinolones by a protecting-group-free approach. Org. Lett. 2015, 17, 3202–3205. [Google Scholar] [CrossRef] [PubMed]
- Kanagaraj, K.; Pitchumani, K. Per-6-amino-β-cyclodextrin as a chiral base catalyst promoting one-pot asymmetric synthesis of 2-aryl-2,3-dihydro-4-quinolones. J. Org. Chem. 2013, 78, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Åkerbladh, L.; Nordeman, P.; Wejdemar, M.; Odell, L.R.; Larhed, M. Synthesis of 4-quinolones via a carbonylative Sonogashira cross-coupling using molybdenum hexacarbonyl as a CO source. J. Org. Chem. 2015, 80, 1464–1471. [Google Scholar] [CrossRef] [PubMed]
- Monastyrskyi, A.; Namelikonda, N.K.; Manetsch, R. Metal-free arylation of ethyl acetoacetate with hypervalent diaryliodonium salts: An immediate access to diverse 3-aryl-4 (1H)-quinolones. J. Org. Chem. 2015, 80, 2513–2520. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Peng, X.; Qin, X.; Zhao, X.; Ma, C.; Tung, C.-H.; Xu, Z. Synthesis of quinolinones with palladium-catalyzed oxidative annulation between acrylamides and arynes. J. Org. Chem. 2015, 80, 2835–2841. [Google Scholar] [CrossRef] [PubMed]
- Manikandan, R.; Jeganmohan, M. Ruthenium-catalyzed cyclization of anilides with substituted propiolates or acrylates: An efficient route to 2-quinolinones. Org. Lett. 2014, 16, 3568–3571. [Google Scholar] [CrossRef] [PubMed]
- Leung, S.C.; Gibbons, P.; Amewu, R.; Nixon, G.L.; Pidathala, C.; Hong, W.D.; Pacorel, B.; Berry, N.G.; Sharma, R.; Stocks, P.A.; et al. Identification, design and biological evaluation of heterocyclic quinolones targeting Plasmodium falciparum type II NADH: Quinone oxidoreductase (PfNDH2). J. Med. Chem. 2012, 55, 1844–1857. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.B.; Pathak, K.K. Pyridoquinolones containing azetidinones: Synthesis and their biological evaluation. Med. Chem. Res. 2011, 21, 2044–2055. [Google Scholar] [CrossRef]
- Patel, N.B.; Chauhan, H.I. Novel pyridoquinolones of sulfonamides, thioureas and amines and their antimicrobial activity. Indian J. Heterocycl. Chem. 2005, 15, 39–42. [Google Scholar]
- Patel, N.B.; Modi, S.H. Synthesis and antimicrobial activity of novel 6-hydroxy-4-oxo-pyrido[2,3-H]quinoline-3[(substituted aryl ureido/piperazinyl)carbonyl]. Indian J. Pharm. Educational Res. 2010, 44, 8–21. [Google Scholar]
- Patel, N.B.; Patel, J.C. Synthesis and antimicrobial study of fluoroquinolone based 4-thiozolidinones. Med. Chem. Res. 2010, 19, 757–770. [Google Scholar] [CrossRef]
- Patel, N.B.; Patel, J.C. Synthesis and antimicrobial activities of 2-azetidinyl-4-quinazolinone derivatives of diclofenac analogue. Med. Chem. Res. 2011, 20, 511–521. [Google Scholar] [CrossRef]
- Patel, N.B.; Patel, J.C.; Modi, S.H. Synthesis and antimicrobial activity of carbonyl pyridoquinolones containing urea and piperazine residue. J. Saudi Chem. Soc. 2010, 15, 167–176. [Google Scholar] [CrossRef]
- Patel, N.B.; Patel, S.D.; Chauhan, H.I. Synthesis and in vitro microbial activities of amides of pyridoquinolone. Med. Chem. Res. 2011, 20, 1054–1067. [Google Scholar] [CrossRef]
- Komarnicka, U.K.; Starosta, R.; Guz-Regner, K.; Bugla-Płoskońska, G.; Kyzioł, A.; Jeżowska-Bojczuk, M. Phosphine derivatives of sparfloxacin—Synthesis, structures and in vitro activity. J. Mol. Struct. 2015, 1096, 55–63. [Google Scholar] [CrossRef]
- Plech, T.; Wujec, M.; Kosikowska, U.; Malm, A.; Rajtar, B.; Polz-Dacewicz, M. Synthesis and in vitro activity of 1,2,4-triazole-ciprofloxacin hybrids against drug-susceptible and drug-resistant bacteria. Eur. J. Med. Chem. 2013, 60, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, E.I.; Bair, J.S.; Nakamura, B.A.; Lee, H.Y.; Kuttab, H.I.; Southgate, E.H.; Lezmi, S.; Lau, G.W.; Hergenrother, P.J. Deoxynybomycins inhibit mutant DNA gyrase and rescue mice infected with fluoroquinolone-resistant bacteria. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Kern, G. Inhibition of Neisseria gonorrhoeae type II topoisomerases by the novel spiropyrimidinetrione azd0914. J. Biol. Chem. 2015, 290, 20984–20994. [Google Scholar] [CrossRef] [PubMed]
- Soni, K. Fluoroquinolones: Chemistry & action—A review. Indo Global J. Pharm. Sci. 2012, 2, 43–53. [Google Scholar]
- Mohammadhosseini, N.; Alipanahi, Z.; Alipour, E.; Emami, S.; Faramarzi, M.; Samadi, N.; Khoshnevis, N.; Shafiee, A.; Foroumadi, A. Synthesis and antibacterial activity of novel levofloxacin derivatives containing a substituted thienylethyl moiety. DARU J. Pharm. Sci. 2012, 20. [Google Scholar] [CrossRef] [PubMed]
- Vijan, L.E.; Giosanu, D. Binding of norfloxacin, enoxacin and enrofloxacin to calf thymus DNA. Rev. Roum. Chim. 2012, 57, 823–827. [Google Scholar]
- Mdluli, K.; Ma, Z. Mycobacterium tuberculosis DNA gyrase as a target for drug discovery. Infect. Disord. Drug Targets 2007, 7, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Hawtin, R.E.; Stockett, D.E.; Byl, J.A.; McDowell, R.S.; Nguyen, T.; Arkin, M.R.; Conroy, A.; Yang, W.; Osheroff, N.; Fox, J.A. Voreloxin is an anticancer quinolone derivative that intercalates DNA and poisons topoisomerase II. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Cazedey, E.C.L.; Salgado, H.R. Development and validation of a microbiological agar assay for determination of orbifloxacin in pharmaceutical preparations. Pharmaceutics 2011, 3, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Panunzio, M.; Biondi, S. Beta-lactam antibiotics renaissance. Antibiotics 2014, 3, 193–215. [Google Scholar] [CrossRef]
- Zhao, S.; Li, X.; Ra, Y.; Li, C.; Jiang, H.; Li, J.; Qu, Z.; Zhang, S.; He, F.; Wan, Y. Developing and optimizing an immunoaffinity cleanup technique for determination of quinolones from chicken muscle. J. Agric. Food Chem. 2009, 57, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Owens, R.C.; Ambrose, P.G. Clinical use of the fluoroquinolones. Med. Clin. N. Am. 2000, 84, 1447–1469. [Google Scholar] [CrossRef]
- Long, T.E.; Keding, L.C.; Lewis, D.D.; Anstead, M.I.; Ryan Withers, T.; Yu, H.D. Anionic fluoroquinolones as antibacterials against biofilm-producing Pseudomonas aeruginosa. Bioorg. Med. Chem. Lett. 2016, 26, 1305–1309. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.; Jackson, M. The use of systemic and topical fluoroquinolones. Pediatrics 2011, 128, e1034–e1045. [Google Scholar] [CrossRef] [PubMed]
- Takiff, H.; Guerrero, E. Current prospects for the fluoroquinolones as first-line tuberculosis therapy. Antimicrob. Agents Chemother. 2011, 55, 5421–5429. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhang, J.; Zhang, X.; Wang, S.; Zhang, Y.; Li, C. Comparison of gyrA gene mutations between laboratory-selected ofloxacin-resistant Mycobacterium tuberculosis strains and clinical isolates. Int. J. Antimicrob. Agents 2008, 31, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Avalos, E.; Catanzaro, D.; Catanzaro, A.; Ganiats, T.; Brodine, S.; Alcaraz, J.; Rodwell, T. Frequency and geographic distribution of gyrA and gyrB mutations associated with fluoroquinolone resistance in clinical Mycobacterium tuberculosis isolates: A systematic review. PLoS ONE 2015, 10, e0120470. [Google Scholar]
- Nosova, E.Y.; Bukatina, A.A.; Isaeva, Y.D.; Makarova, M.V.; Galkina, K.Y.; Moroz, A.M. Analysis of mutations in the gyrA and gyrB genes and their association with the resistance of Mycobacterium tuberculosis to levofloxacin, moxifloxacin and gatifloxacin. J. Med. Microbiol. 2013, 62, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Liu, M.; Wang, Y.; Pang, Y.; Zhao, Z. Mechanisms of fluoroquinolone monoresistance in mycobacterium tuberculosis. FEMS Microbiol. Lett. 2014, 353, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lu, J.; Wang, Y.; Pang, Y.; Zhao, Y. Prevalence and molecular characterization of fluoroquinolone-resistant Mycobacterium tuberculosis isolates in china. Antimicrob. Agents Chemother. 2014, 58, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. Advances in the treatment of tuberculosis. Clin. Pharmacol. Ther. 2007, 82, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Zhang, Y.; Shen, Y.; Siu, G.K.H.; Wu, W.; Qian, X.; Deng, G.; Xu, Y.; Lau, R.; Fan, X.; et al. Molecular characterization of fluoroquinolone-resistant Mycobacterium tuberculosis clinical isolates from Shanghai, China. Diagn. Microbiol. Infect. Dis. 2012, 73, 260–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, R.; Zhang, J.; Li, C.; Kazumi, Y.; Sugawara, I. Emergence of ofloxacin resistance in Mycobacterium tuberculosis clinical isolates from china as determined by gyrA mutation analysis using denaturing high-pressure liquid chromatography and DNA sequencing. J. Clin. Microbiol. 2006, 44, 4566–4568. [Google Scholar] [CrossRef] [PubMed]
- Umubyeyi, A.N. Limited fluoroquinolone resistance among Mycobacterium tuberculosis isolates from rwanda: Results of a national survey. J. Antimicrob. Chemother. 2007, 59, 1031–1033. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Yin, L.; Ali, A.; Cooke, A.J.; Bennett, J.; Ratcliffe, P.; Lo, M.M.-C.; Metzger, E.; Hoyt, S.; Hartmann, R.W. Novel pyridyl substituted 4,5-dihydro-[1,2,4]triazolo[4,3-a]quinolines as potent and selective aldosterone synthase inhibitors with improved in vitro metabolic stability. J. Med. Chem. 2015, 58, 2530–2537. [Google Scholar] [CrossRef] [PubMed]
- Pescatori, L.; Métifiot, M.; Chung, S.; Masoaka, T.; Cuzzucoli Crucitti, G.; Messore, A.; Pupo, G.; Madia, V.N.; Saccoliti, F.; Scipione, L.; et al. N-substituted quinolinonyl diketo acid derivatives as HIV integrase strand transfer inhibitors and their activity against RNase H function of reverse transcriptase. J. Med. Chem. 2015, 58, 4610–4623. [Google Scholar] [CrossRef] [PubMed]
- Di Santo, R.C.R.; Roux, A.; Artico, M.; Lavecchia, A.; Marinelli, L.; Novellino, E.; Palmisano, L.; Andreotti, M.; Amici, R.; Galluzzo, C.M.; et al. Novel bifunctional quinolonyl diketo acid derivatives as HIV-1 integrase inhibitors: Design, synthesis, biological activities, and mechanism of action. J. Med. Chem. 2006, 49, 1939–1945. [Google Scholar] [CrossRef] [PubMed]
- Di Santo, R. Diketo acids derivatives as dual inhibitors of human immunodeficiency virus type 1 integrase and the reverse transcriptase RNase H domain. Curr. Med. Chem. 2011, 18, 3335–3342. [Google Scholar] [CrossRef] [PubMed]
- Costi, R.M.M.; Chung, S.; Cuzzucoli Crucitti, G.; Maddali, K.; Pescatori, L.; Messore, A.; Madia, V.N.; Pupo, G.; Scipione, L.; Tortorella, S.; et al. Basic quinolinonyl diketo acid derivatives as inhibitors of hiv integrase and their activity against RNase H function of reverse transcriptase. J. Med. Chem. 2014, 57, 3223–3234. [Google Scholar] [CrossRef] [PubMed]
- Di Santo, R.C.R.; Roux, A.; Miele, G.; Cuzzucoli Crucitti, G.; Iacovo, A.; Rosi, F.; Lavecchia, A.; Marinelli, L.; Di Giovanni, C.; Novellino, E.; et al. Novel quinolinonyl diketo acid derivatives as HIV-1 integrase inhibitors: Design, synthesis, and biological activities. J. Med. Chem. 2008, 51, 4744–4750. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Yang, Y.; Zhou, S.; Liu, Y.; Yuan, Y.; Li, S.; Li, C. Ciprofloxacin containing Mannich base and its copper complex induce antitumor activity via different mechanism of action. Int. J. Oncol. 2014, 45, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Boothe, D.; Boeckh, A.; Simpson, B.; Dubose, K. Comparison of pharmacodynamics and pharmacokinetic indices of efficacy for 5 fluoroquinolones toward pathogens of dogs and cats. J. Vet. Inter. Med. 2006, 20, 1297–1306. [Google Scholar] [CrossRef]
- Mueller, R.S.; Stephan, B. Pradofloxacin in the treatment of canine deep pyoderma: A multicentred, blinded, randomized parallel trial. Vet. Dermatol. 2007, 18, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed]
- Dale, A.G.; Hinds, J.; Mann, J.; Taylor, P.W.; Neidle, S. Symmetric bis-benzimidazoles are potent anti-staphylococcal agents with dual inhibitory mechanisms against DNA gyrase. Biochemistry 2012, 51, 5860–5871. [Google Scholar] [CrossRef] [PubMed]
- Deweese, J.E.; Osheroff, N. The DNA cleavage reaction of topoisomerase II: Wolf in sheep’s clothing. Nucleic Acids Res. 2009, 37, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.D.; Shultzaberger, R.K.; Berger, J.M. The c-terminal domain of DNA gyrase A adopts a DNA-bending beta-pinwheel fold. Proc. Natl. Acad. Sci. USA 2004, 101, 7293–7298. [Google Scholar] [CrossRef] [PubMed]
- Levine, C.; Hiasa, H.; Marians, K.J. DNA gyrase and topoisomerase IV: Biochemical activities, physiological roles during chromosome replication, and drug sensitivities. Biochim. Biophys. Acta 1998, 1400, 29–43. [Google Scholar] [CrossRef]
- Schoeffler, A.J.; Berger, J.M. DNA topoisomerases: Harnessing and constraining energy to govern chromosome topology. Q. Rev. Biophys. 2008, 41, 41–101. [Google Scholar] [CrossRef] [PubMed]
- Anderson, V.E.; Osheroff, N. Type II topoisomerases as targets for quinolone antibacterials turning Dr. Jekyll into Mr. Hyde. Curr. Pharm. Des. 2001, 7, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Schoeffler, A.J.; Berger, J.M. Recent advances in understanding structure—function relationships in the type II topoisomerase mechanism. Biochem. Soc. Trans. 2005, 33, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Schoeffler, A.J.; May, A.P.; Berger, J.M. A domain insertion in Escherichia coli gyrB adopts a novel fold that plays a critical role in gyrase function. Nucleic Acids Res. 2010, 38, 7830–7844. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.H.; Burgin, A.B.; Deweese, J.E.; Osheroff, N.; Berger, J.M. A novel and unified two-metal mechanism for DNA cleavage by type II and Ia topoisomerases. Nature 2010, 465, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Pitts, S.L.; Liou, G.F.; Mitchenall, L.A.; Burgin, A.B.; Maxwell, A.; Neuman, K.C.; Osheroff, N. Use of divalent metal ions in the DNA cleavage reaction of topoisomerase IV. Nucleic Acids Res. 2011, 39, 4808–4817. [Google Scholar] [CrossRef] [PubMed]
- Deweese, J.E.; Osheroff, M.A.; Osheroff, N. DNA topology and topoisomerases. Biochem. Mol. Biol. Educ. 2009, 37, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Laponogov, I.; Pan, X.-S.; Veselkov, D.A.; McAuley, K.E.; Fisher, L.M.; Sanderson, M.R. Structural basis of gate-DNA breakage and resealing by type II topoisomerases. PLoS ONE 2010, 5, e11338. [Google Scholar] [CrossRef]
- Laponogov, I.; Sohi, M.K.; Veselkov, D.; Pan, X.; Sawhney, R.; Thompson, A.; McAuley, K.; Fisher, L.; Sanderson, M. Structural insight into the quinolone–DNA cleavage complex of type IIa topoisomerases. Nat. Struct. Mol. Biol. 2009, 16, 667–669. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.M.; Weigand, S.; Maar-Mathias, S.; Mondragon, A. Solution structures of DNA-bound gyrase. Nucleic Acids Res. 2011, 39, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; Schwanz, H.A.; Li, G.; Williamson, B.H.; McPherson, S.A.; Turnbough, C.L., Jr.; Kerns, R.J.; Osheroff, N. Activity of quinolone cp-115,955 against bacterial and human type II topoisomerases is mediated by different interactions. Biochemistry 2015, 54, 1278–1286. [Google Scholar] [CrossRef] [PubMed]
- Kreuzer, K.N.; Cozzarelli, N.R. Escherichia coli mutants thermosensitive for deoxyribonucleic acid gyrase subunit A: Effects on deoxyribonucleic acid replication, transcription, and bacteriophage growth. J. Bacteriol. 1979, 140, 424–435. [Google Scholar] [PubMed]
- Hooper, D.C. Mechanisms of Quinolone Resistance. In Quinolone Antimicrobial Agents, 3rd ed.; ASM Press: Washington, DC, USA, 2003; pp. 41–67. [Google Scholar]
- Hooper, D.C.; Jacoby, G.A. Mechanisms of drug resistance: Quinolone resistance. Ann. N. Y. Acad. Sci. 2015, 1354, 12–31. [Google Scholar] [CrossRef] [PubMed]
- Fournier, B.; Zhao, X.; Lu, T.; Drlica, K.; Hooper, D.C. Selective targeting of topoisomerase iv and DNA gyrase in Staphylococcus aureus: Different patterns of quinolone-induced inhibition of DNA synthesis. Antimicrob. Agents Chemother. 2000, 44, 2160–2165. [Google Scholar] [CrossRef] [PubMed]
- Price, L.B.; Vogler, A.; Pearson, T.; Busch, J.D.; Schupp, J.M.; Keim, P. In vitro selection and characterization of Bacillus anthracis mutants with high-level resistance to ciprofloxacin. Antimicrob. Agents Chemother. 2003, 47, 2362–2365. [Google Scholar] [CrossRef] [PubMed]
- Morgan-Linnell, S.K.; Boyd, L.B.; Steffen, D.; Zechiedrich, L. Mechanisms accounting for fluoroquinolone resistance in Escherichia coli clinical isolates. Antimicrob. Agents Chemother. 2009, 53, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Sissi, C.; Perdona, E.; Domenici, E.; Feriani, A.; Howells, A.J.; Maxwell, A.; Palumbo, M. Ciprofloxacin affects conformational equilibria of DNA gyrase A in the presence of magnesium ions. J. Mol. Biol. 2001, 311, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Sissi, C.; Palumbo, M. Effects of magnesium and related divalent metal ions in topoisomerase structure and function. Nucleic Acids Res. 2009, 37, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Barnard, F.M.; Maxwell, A. Interaction between DNA gyrase and quinolones: Effects of alanine mutations at gyrA subunit residues Ser83 and Asp87. Antimicrob. Agents Chemother. 2001, 45, 1994–2000. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.F.; Srikannathasan, V.; Huang, J.; Cui, H.; Fosberry, A.P.; Gu, M.; Hann, M.M.; Hibbs, M.; Homes, P.; Ingraham, K.; et al. Structural basis of DNA gyrase inhibition by antibacterial QPT-1, anticancer drug etoposide and moxifloxacin. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Bax, B.C.P.; Eggleston, D.; Fosberry, A.; Gentry, D.; Gorrec, F.; Giordano, I.; Hann, M.; Hennessy, A.; Hibbs, M.; Huang, J.; et al. Type IIa topoisomerase inhibition by a new class of antibacterial agents. Nature 2010, 466, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Srikannathasan, V.; Wohlkonig, A.; Shillings, A.; Singh, O.; Chan, P.F.; Huang, J.; Gwynn, M.N.; Fosberry, A.P.; Homes, P.; Hibbs, M.; et al. Crystallization and initial crystallographic analysis of covalent DNA-cleavage complexes of Staphyloccocus aureus DNA gyrase with QPT-1, moxifloxacin and etoposide. Acta Crystallogr. F Struct. Biol. Commun. 2015, 71, 1242–1246. [Google Scholar] [CrossRef] [PubMed]
- Wohlkonig, A. Structural basis of quinolone inhibition of type IIa topoisomerases and target-mediated resistance. Nat. Struct. Mol. Biol. 2010, 17, 1152–1153. [Google Scholar] [CrossRef] [PubMed]
- Collin, F.; Karkare, S.; Maxwell, A. Exploiting bacterial DNA gyrase as a drug target: Current state and perspectives. Appl. Microbiol. Biotechnol. 2011, 92, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; Breland, E.J.; Vlckova, V.; Strub, M.-P.; Neuman, K.C.; Kerns, R.J.; Osheroff, N. Role of the water metal ion bridge in mediating interactions between quinolones and Escherichia coli topoisomerase IV. Biochemistry 2014, 53, 5558–5567. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; McPherson, S.A.; Turnbough, C.L., Jr.; Kerns, R.J.; Osheroff, N. Topoisomerase IV-quinolone interactions are mediated through a water-metal ion bridge: Mechanistic basis of quinolone resistance. Nucleic Acids Res. 2013, 41, 4628–4639. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; McPherson, S.A.; Wang, P.; Kerns, R.J.; Graves, D.E.; Turnbough, C.L., Jr.; Osheroff, N. Drug interactions with Bacillus anthracis topoisomerase IV: Biochemical basis for quinolone action and resistance. Biochemistry 2012, 51, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; Schwanz, H.A.; Li, G.; McPherson, S.A.; Turnbough, C.L., Jr.; Kerns, R.J.; Osheroff, N. Overcoming target-mediated quinolone resistance in topoisomerase IV by introducing metal-ion-independent drug-enzyme interactions. ACS Chem. Biol. 2013, 8, 2660–2668. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, E.L.; Osheroff, N. Etoposide, topoisomerase ii and cancer. Curr. Med. Chem. Anticancer Agents 2005, 5, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Hsiung, Y.; Elsea, S.H.; Osheroff, N.; Nitiss, J.L. A mutation in yeast top2 homologous to a quinolone-resistant mutation in bacteria. Mutation of the amino acid homologous to Ser83 of Escherichia coli GyrA alters sensitivity to eukaryotic topoisomerase inhibitors. J. Biol. Chem. 1995, 270, 20359–20364. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jung, S.-R.; Heo, K.; Byl, J.A.W.; Deweese, J.E.; Osheroff, N.; Hohng, S. DNA cleavage and opening reactions of human topoisomerase iiα are regulated via Mg2+-mediated dynamic bending of gate-DNA. Proc. Natl. Acad. Sci. USA 2012, 109, 2925–2930. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.H.; Osheroff, N.; Berger, J.M. Structure of a topoisomerase ii-DNA-nucleotide complex reveals a new control mechanism for ATPase activity. Nat. Struct. Mol. Biol. 2012, 19, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, G.A. Mechanisms of resistance to quinolones. Clin. Infect. Dis. 2005, 41, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Redgrave, L.S.; Sutton, S.B.; Webber, M.A.; Piddock, L.J. Fluoroquinolone resistance: Mechanisms, impact on bacteria, and role in evolutionary success. Trends Micobiol. 2014, 22, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.-S.; Gould, K.A.; Fisher, L.M. Probing the differential interactions of quinazolinedione pd 0305970 and quinolones with gyrase and topoisomerase IV. Antimicrob. Agents Chemother. 2009, 53, 3822–3831. [Google Scholar] [CrossRef] [PubMed]
- Yague, G.; Morris, J.E.; Pan, X.-S.; Gould, K.A.; Fisher, L.M. Cleavable-complex formation by wild-type and quinolone-resistant streptococcus pneumoniae type II topoisomerases mediated by gemifloxacin and other fluoroquinolones. Antimicrob. Agents Chemother. 2002, 46, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, E.S.; Hiasa, H. Determination of the primary target of a quinolone drug and the effect of quinolone resistance-conferring mutations by measuring quinolone sensitivity based on its mode of action. Antimicrob. Agents Chemother. 2007, 51, 3410–3412. [Google Scholar] [CrossRef] [PubMed]
- Drlica, K.; Hiasa, H.; Kerns, R.; Malik, M.; Mustaev, A.; Zhao, X. Quinolones: Action and resistance updated. Curr. Top. Med. Chem. 2009, 9, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Tran, J.H.; Jacoby, G.A.; Zhang, Y.; Wang, F.; Hooper, D.C. Plasmid-mediated quinolone resistance in clinical isolates of Escherichia coli from shanghai, china. Antimicrob. Agents Chemother. 2003, 47, 2242–2248. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Martinez, J.M.; Cano, M.E.; Velasco, C.; Martinez-Martinez, L.; Pascual, A. Plasmid-mediated quinolone resistance: An update. J. Infect. Chemother. 2011, 17, 149–182. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Deguchi, T.; Yasuda, M.; Kawamura, T.; Kanematsu, E.; Nishino, Y.; Ishihara, S.; Kawada, Y. Alteration in the gyra subunit of DNA gyrase and the parc subunit of DNA topoisomerase IV in quinolone-resistant clinical isolates of Staphylococcus epidermidis. Antimicrob. Agents Chemother. 1998, 42, 3293–3295. [Google Scholar] [PubMed]
- Robicsek, A.; Jacoby, G.A.; Hooper, D.C. The worldwide emergence of plasmid-mediated quinolone resistance. Lancet Infect. Dis. 2006, 6, 629–640. [Google Scholar] [CrossRef]
- Strahilevitz, J.; Jacoby, G.A.; Hooper, D.C.; Robicsek, A. Plasmid-mediated quinolone resistance: A multifaceted threat. Clin. Microbiol. Rev. 2009, 22, 664–689. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martinez, L.; Pascual, A.; Jacoby, G.A. Quinolone resistance from a transferable plasmid. Lancet 1998, 351, 797–799. [Google Scholar] [CrossRef]
- Tran, J.H.; Jacoby, G.A. Mechanism of plasmid-mediated quinolone resistance. Proc. Natl. Acad. Sci. USA 2002, 99, 5638–5642. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Bromley, E.H.C.; Oelschlaeger, P.; Woolfson, D.N.; Spencer, J. Structural insights into quinolone antibiotic resistance mediated by pentapeptide repeat proteins: Conserved surface loops direct the activity of a qnr protein from a gram-negative bacterium. Nucleic Acids Res. 2011, 39, 3917–3927. [Google Scholar] [CrossRef] [PubMed]
- Guillard, T.; Cambau, E.; Chau, F.; Massias, L.; de Champs, C.; Fantin, B. Ciprofloxacin treatment failure in a murine model of pyelonephritis due to an AAC (6′)-Ib-cr-producing Escherichia coli strain susceptible to ciprofloxacin in vitro. Antimicrob. Agents Chemother. 2013, 57, 5830–5835. [Google Scholar] [CrossRef] [PubMed]
- Poole, K. Efflux pumps as antimicrobial resistance mechanisms. Ann. Med. 2007, 39, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.D.; White, D.G.; Levy, S.B. Multiple antibiotic resistance (mar) locus protects Escherichia coli from rapid cell killing by fluoroquinolones. Antimicrob. Agents Chemother. 1996, 40, 1266–1269. [Google Scholar] [PubMed]













© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naeem, A.; Badshah, S.L.; Muska, M.; Ahmad, N.; Khan, K. The Current Case of Quinolones: Synthetic Approaches and Antibacterial Activity. Molecules 2016, 21, 268. https://doi.org/10.3390/molecules21040268
Naeem A, Badshah SL, Muska M, Ahmad N, Khan K. The Current Case of Quinolones: Synthetic Approaches and Antibacterial Activity. Molecules. 2016; 21(4):268. https://doi.org/10.3390/molecules21040268
Chicago/Turabian StyleNaeem, Abdul, Syed Lal Badshah, Mairman Muska, Nasir Ahmad, and Khalid Khan. 2016. "The Current Case of Quinolones: Synthetic Approaches and Antibacterial Activity" Molecules 21, no. 4: 268. https://doi.org/10.3390/molecules21040268
APA StyleNaeem, A., Badshah, S. L., Muska, M., Ahmad, N., & Khan, K. (2016). The Current Case of Quinolones: Synthetic Approaches and Antibacterial Activity. Molecules, 21(4), 268. https://doi.org/10.3390/molecules21040268