
Conformational and Molecular Structures of α,β-Unsaturated Acrylonitrile Derivatives: Photophysical Properties and Their Frontier Orbitals

Abstract
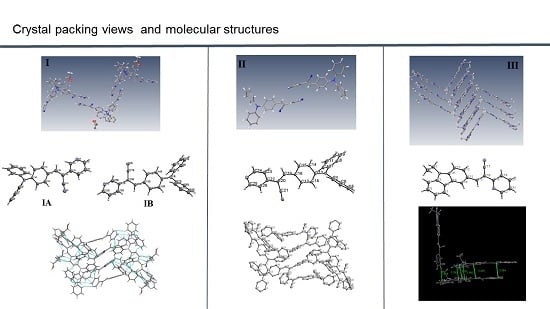
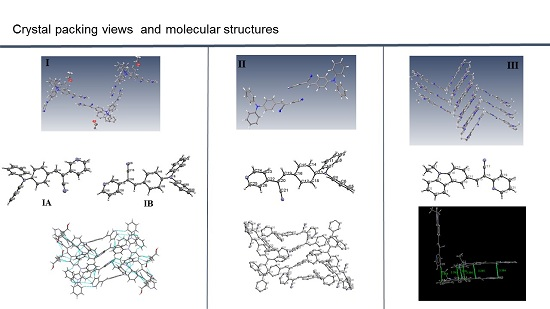
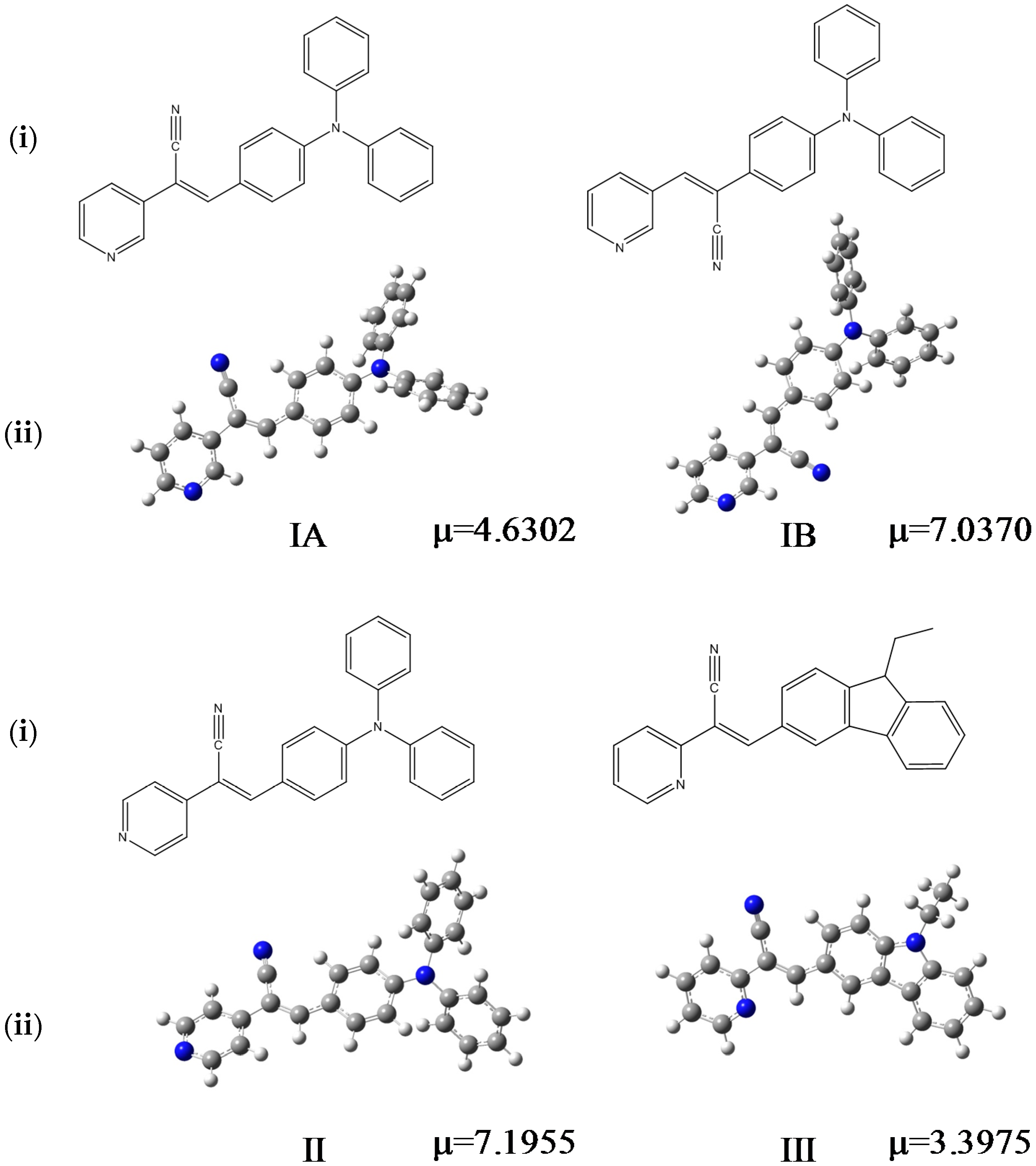
:
1. Introduction
2. Results and Discussion
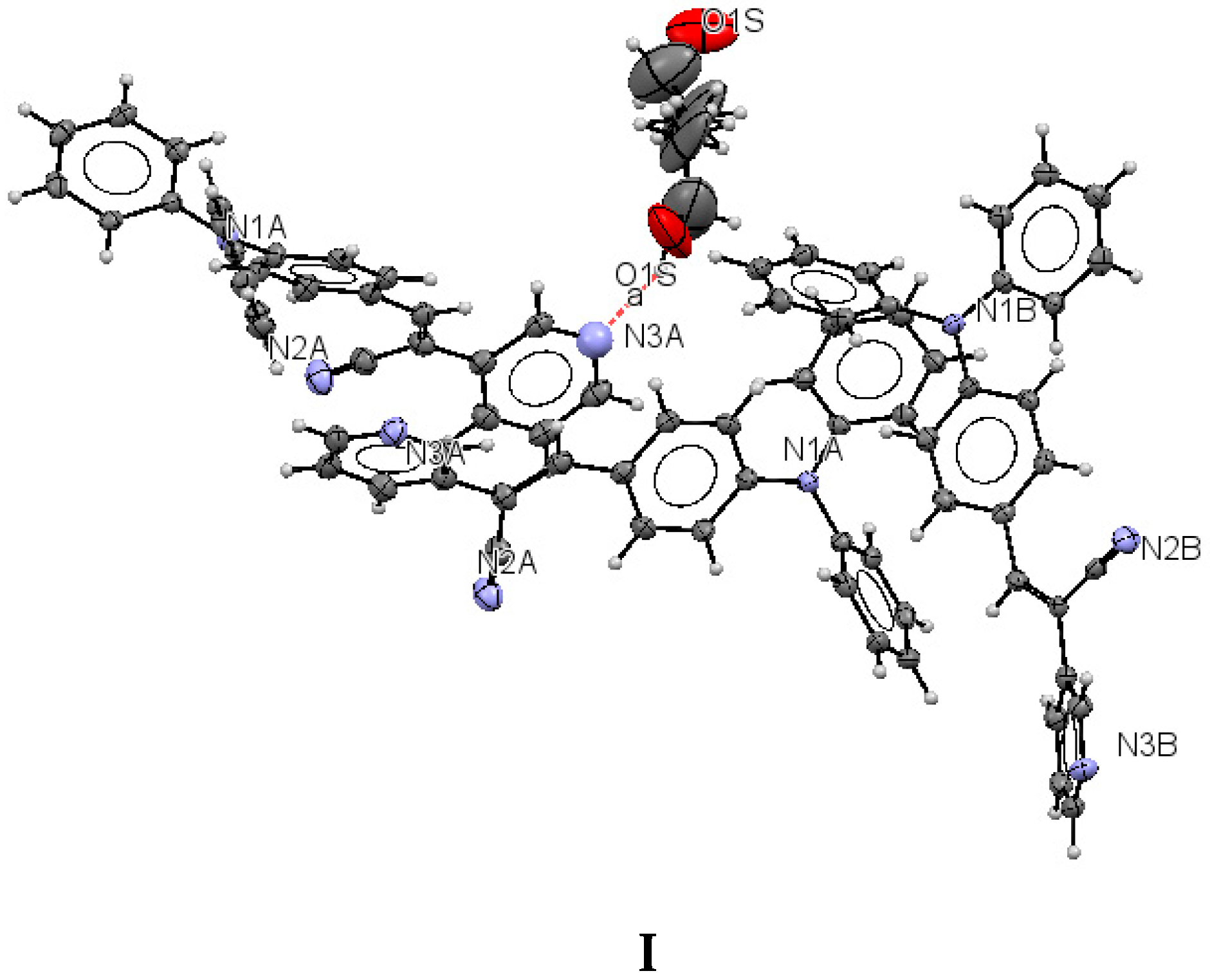
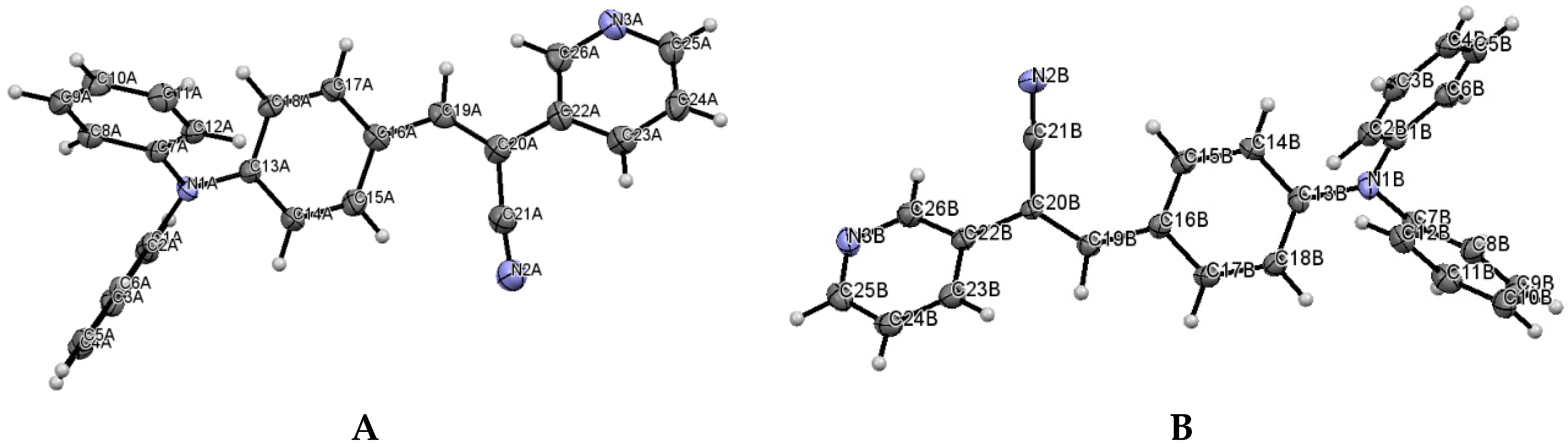
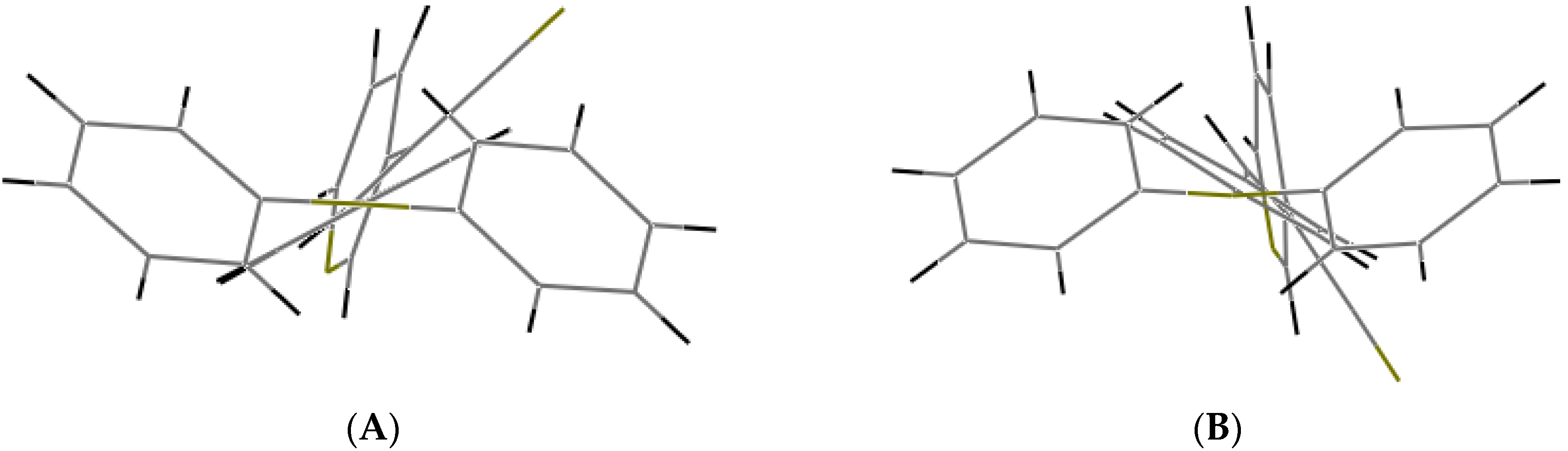
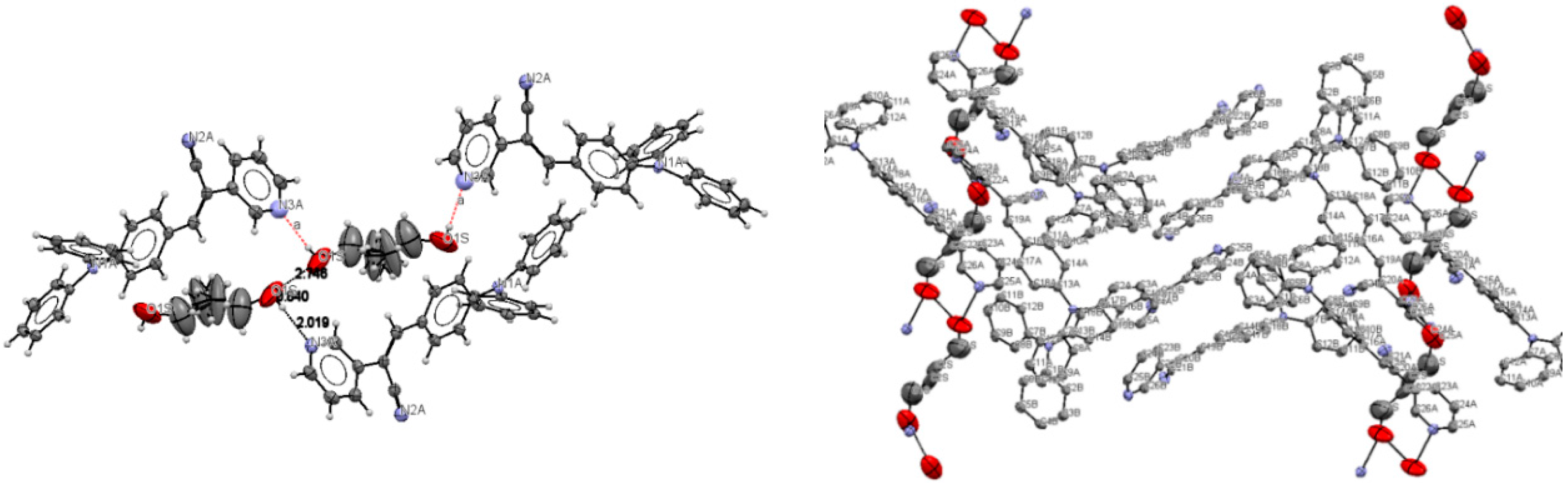
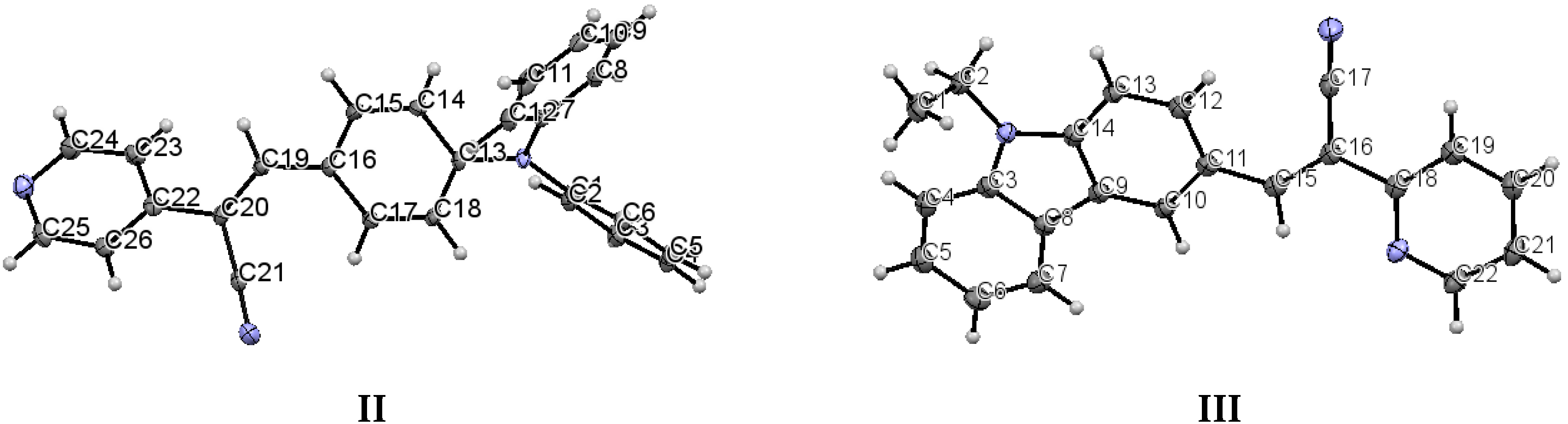
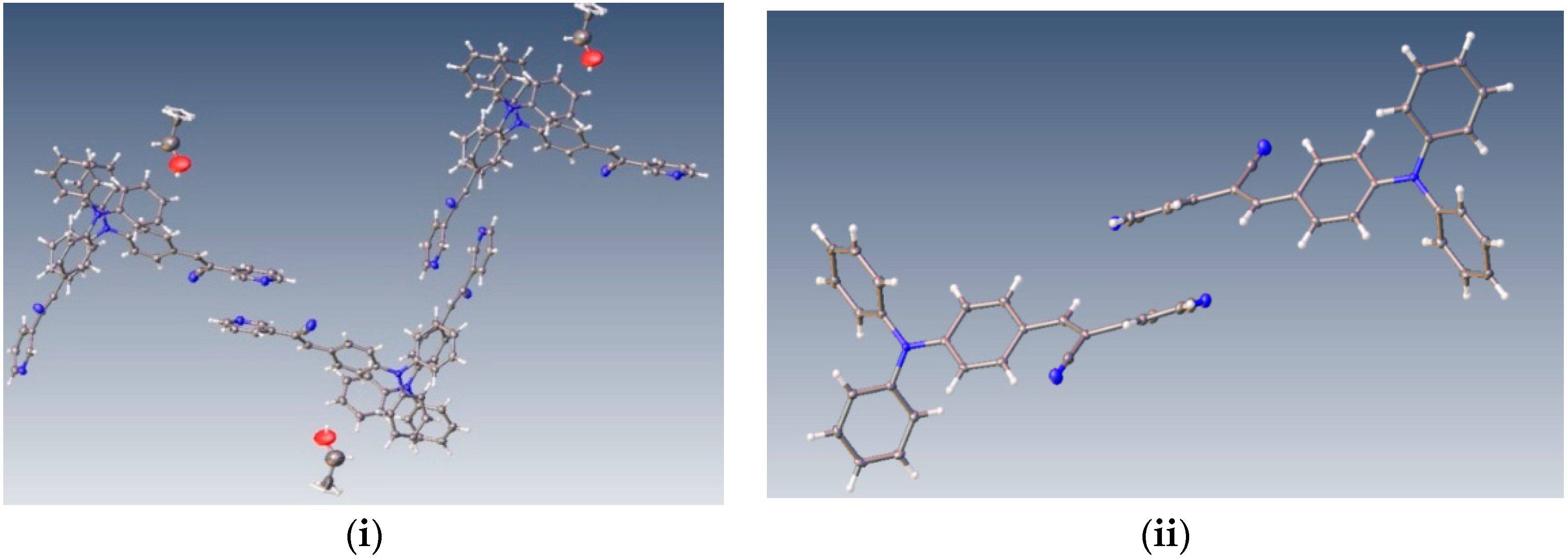
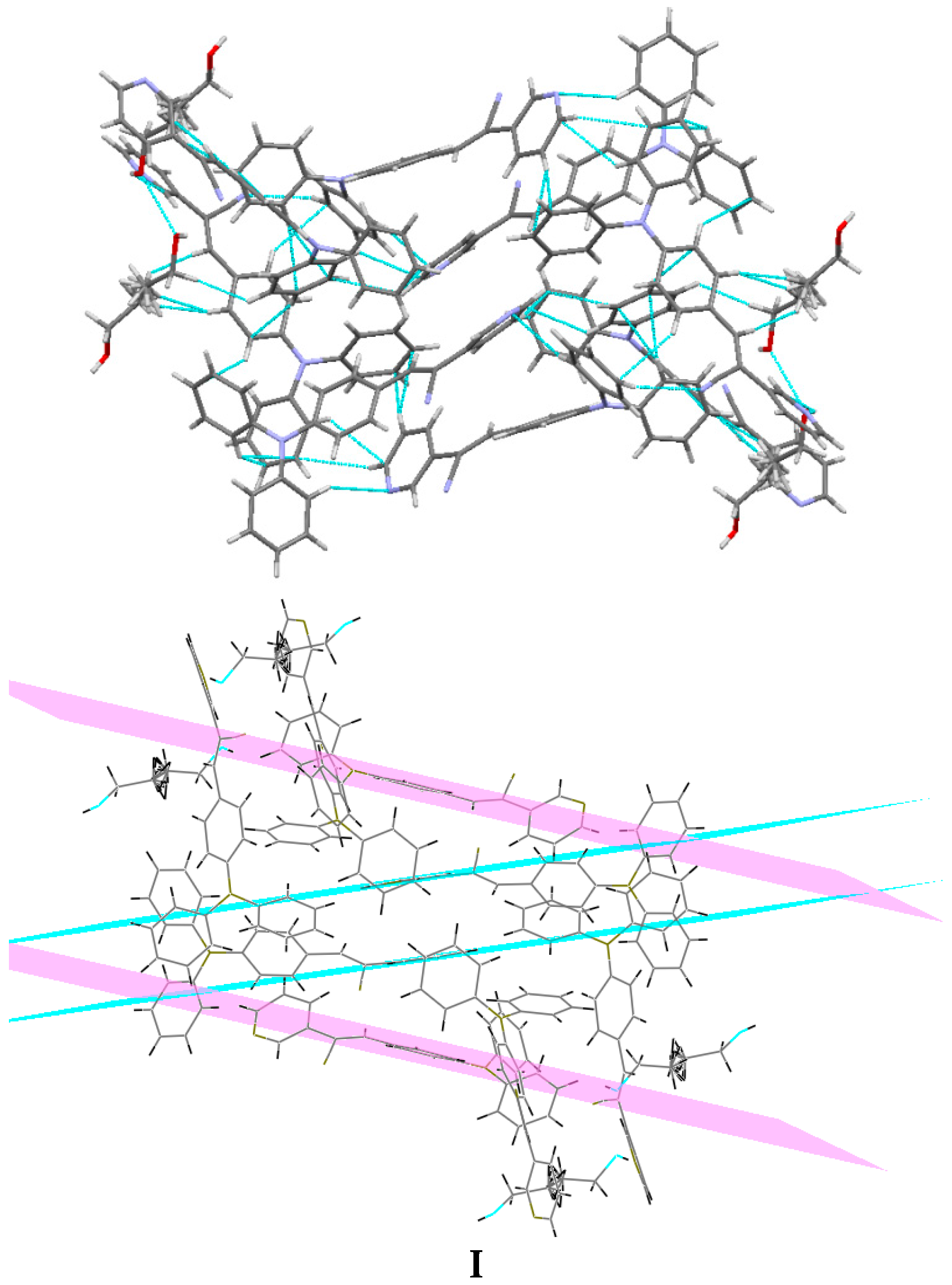
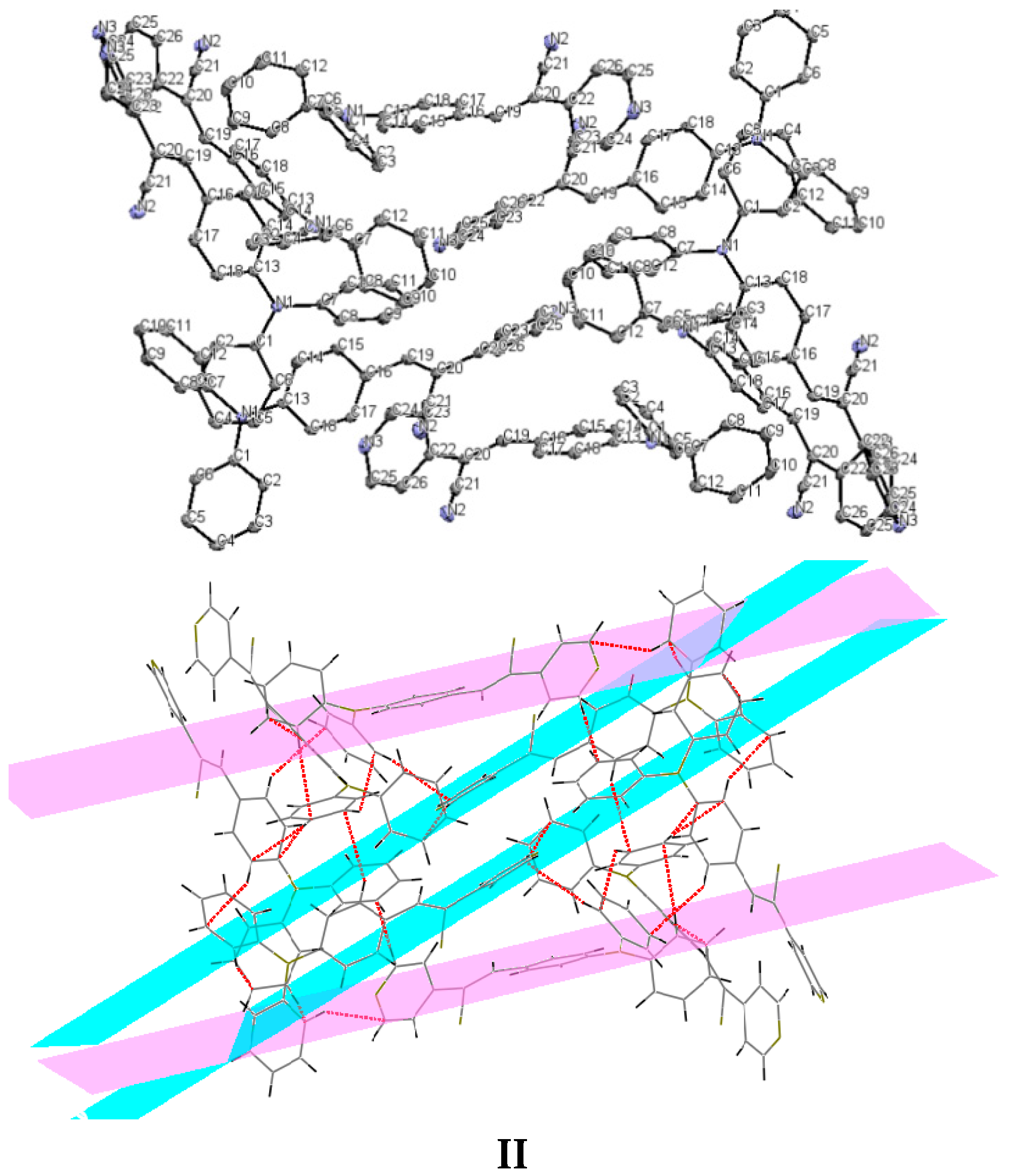
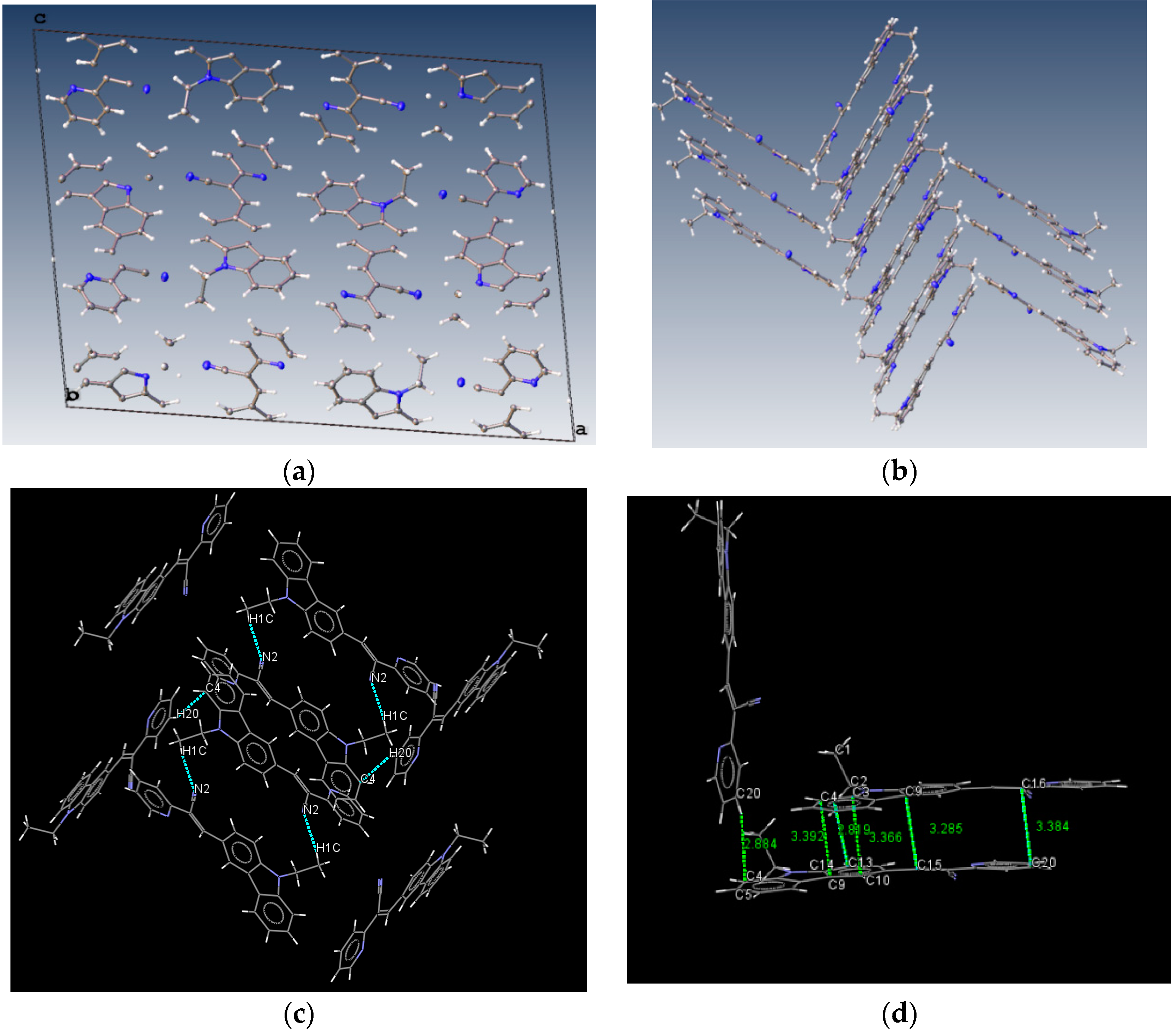
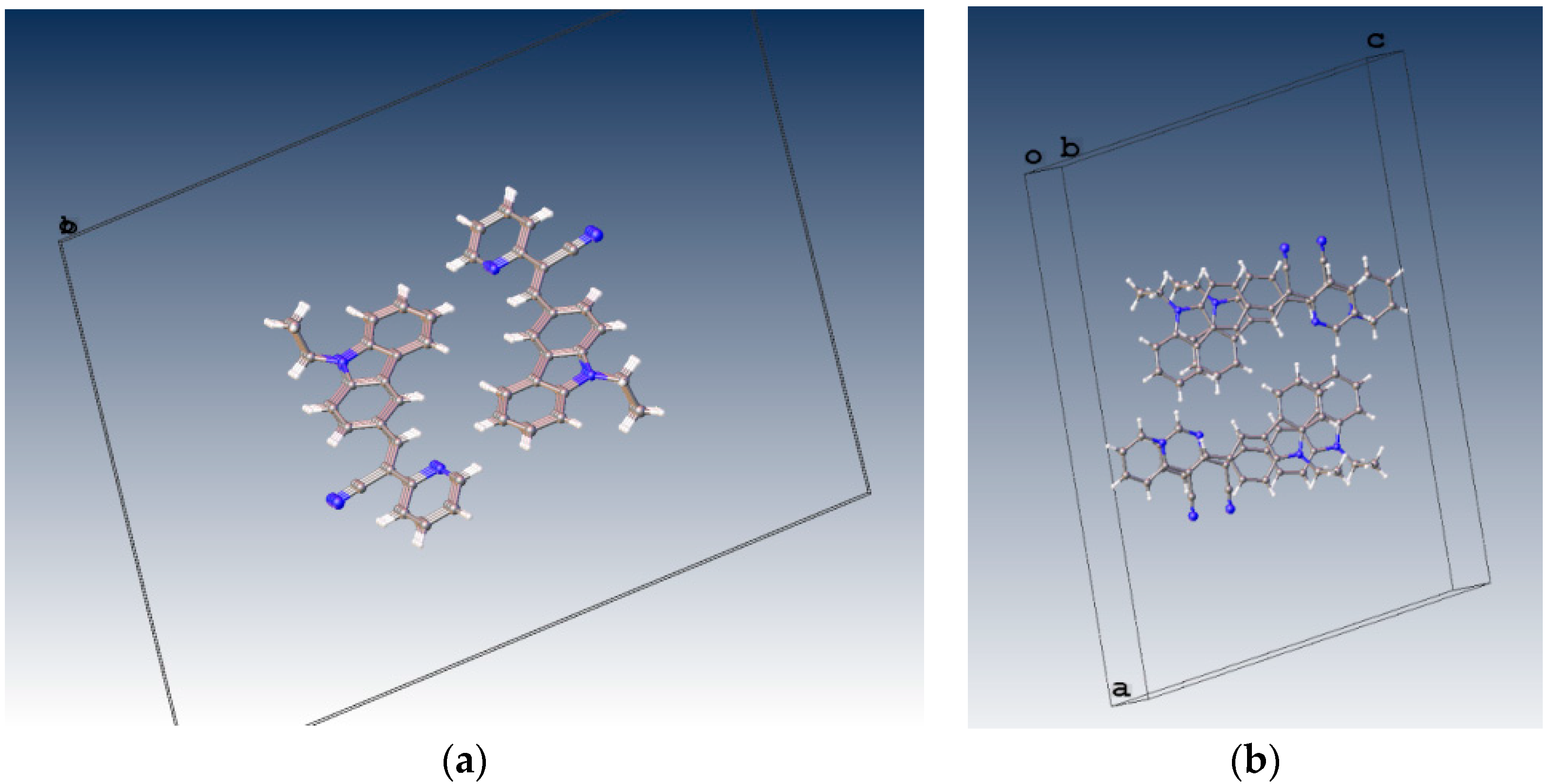
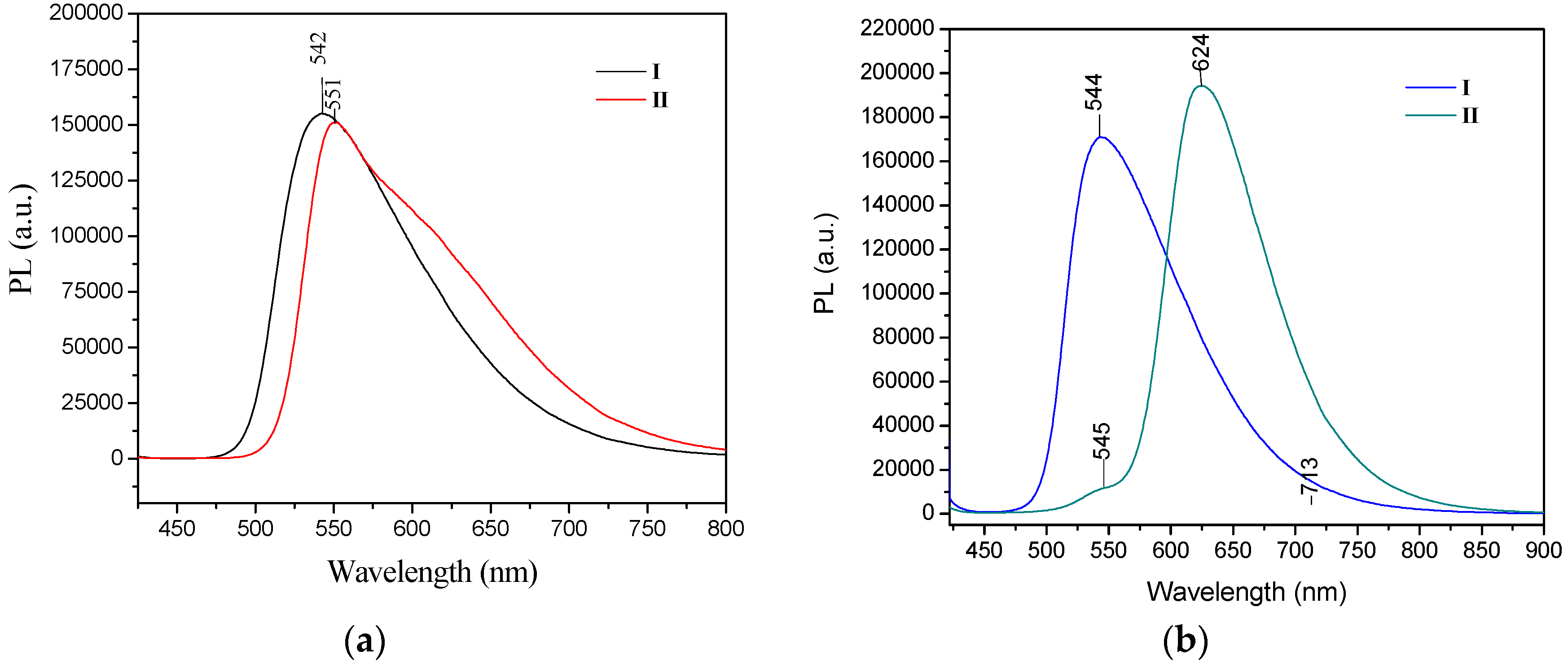
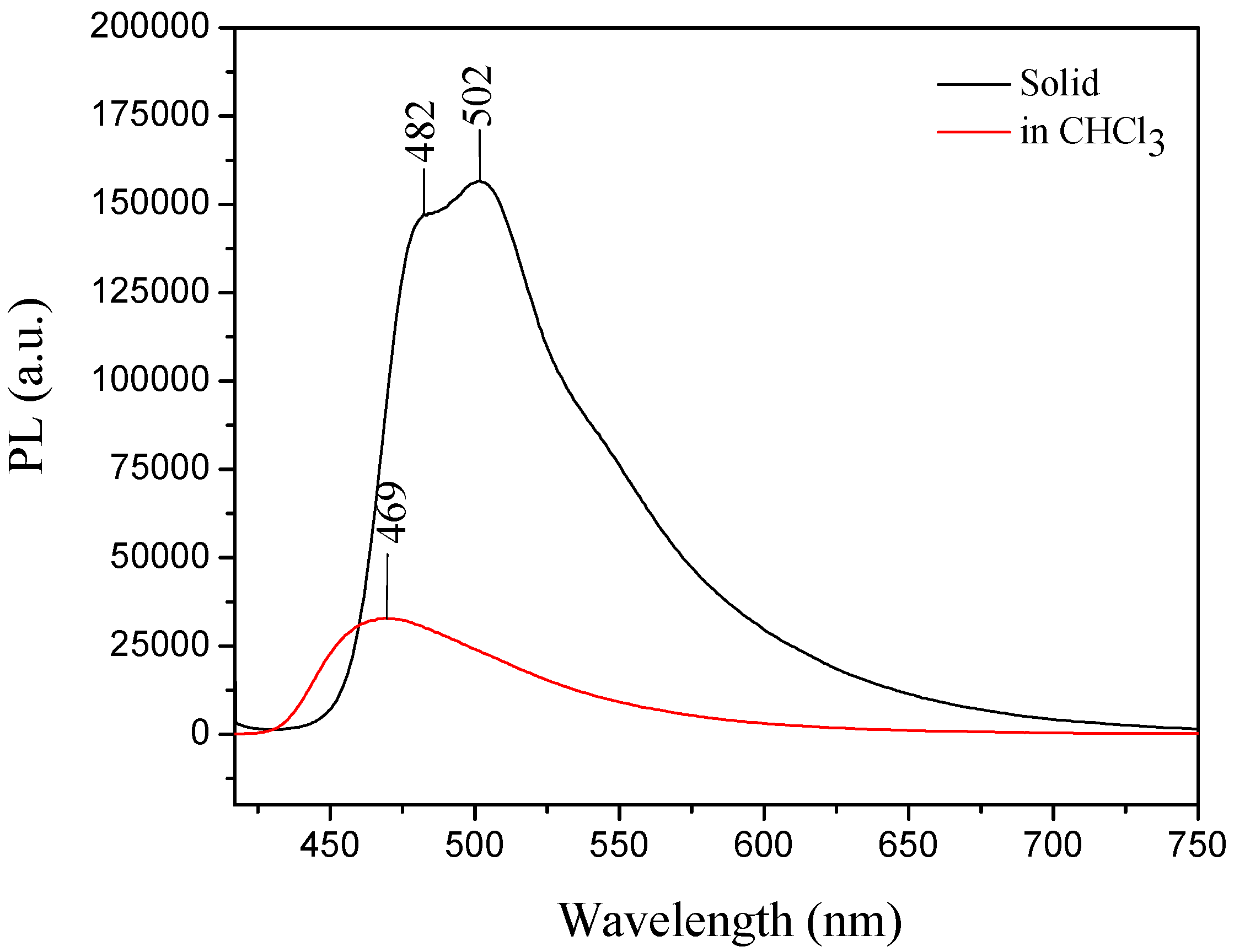
2.1. X-ray Crystallography and Photolysis Properties of I–III
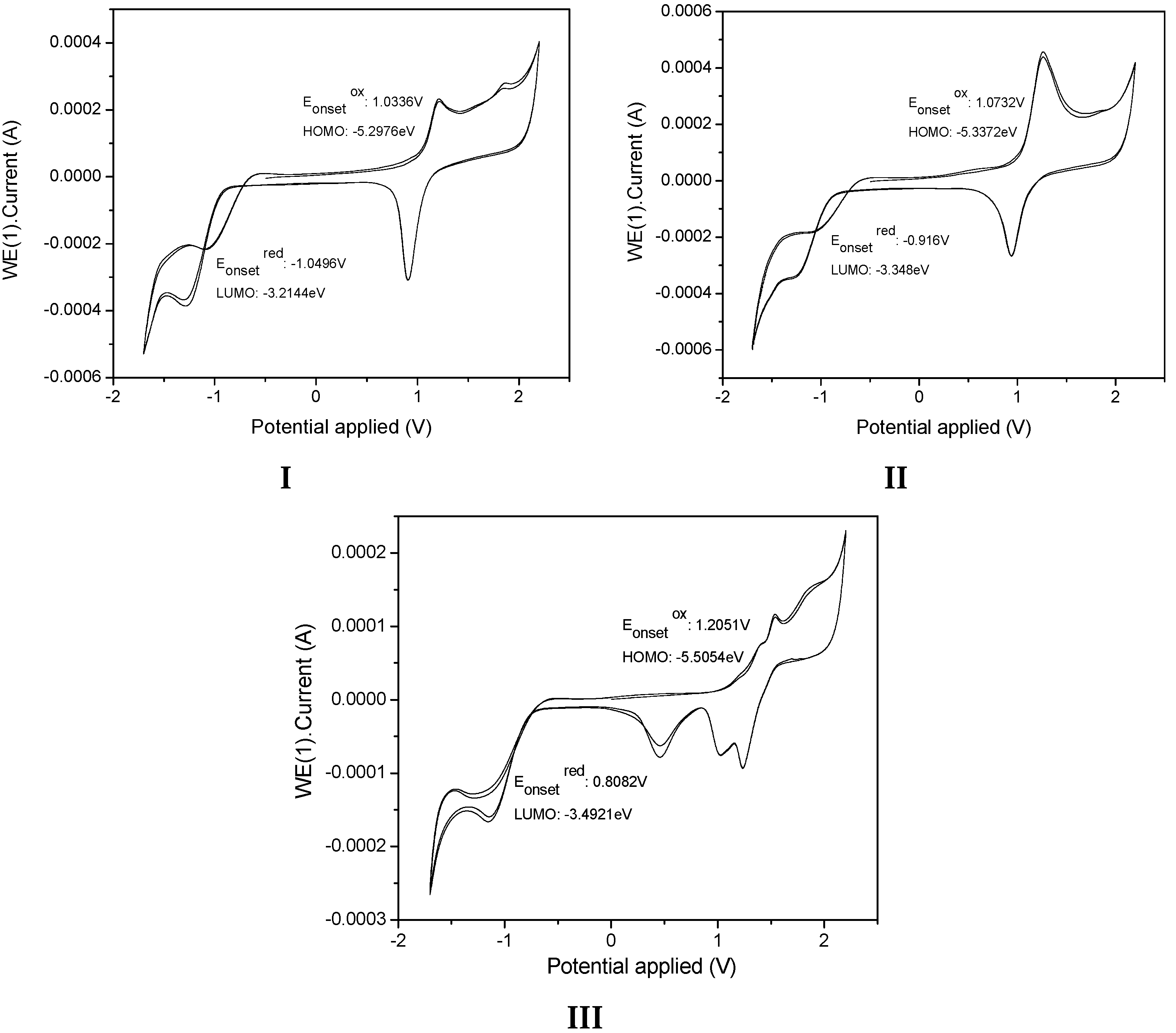
2.2. Electrochemical Properties.
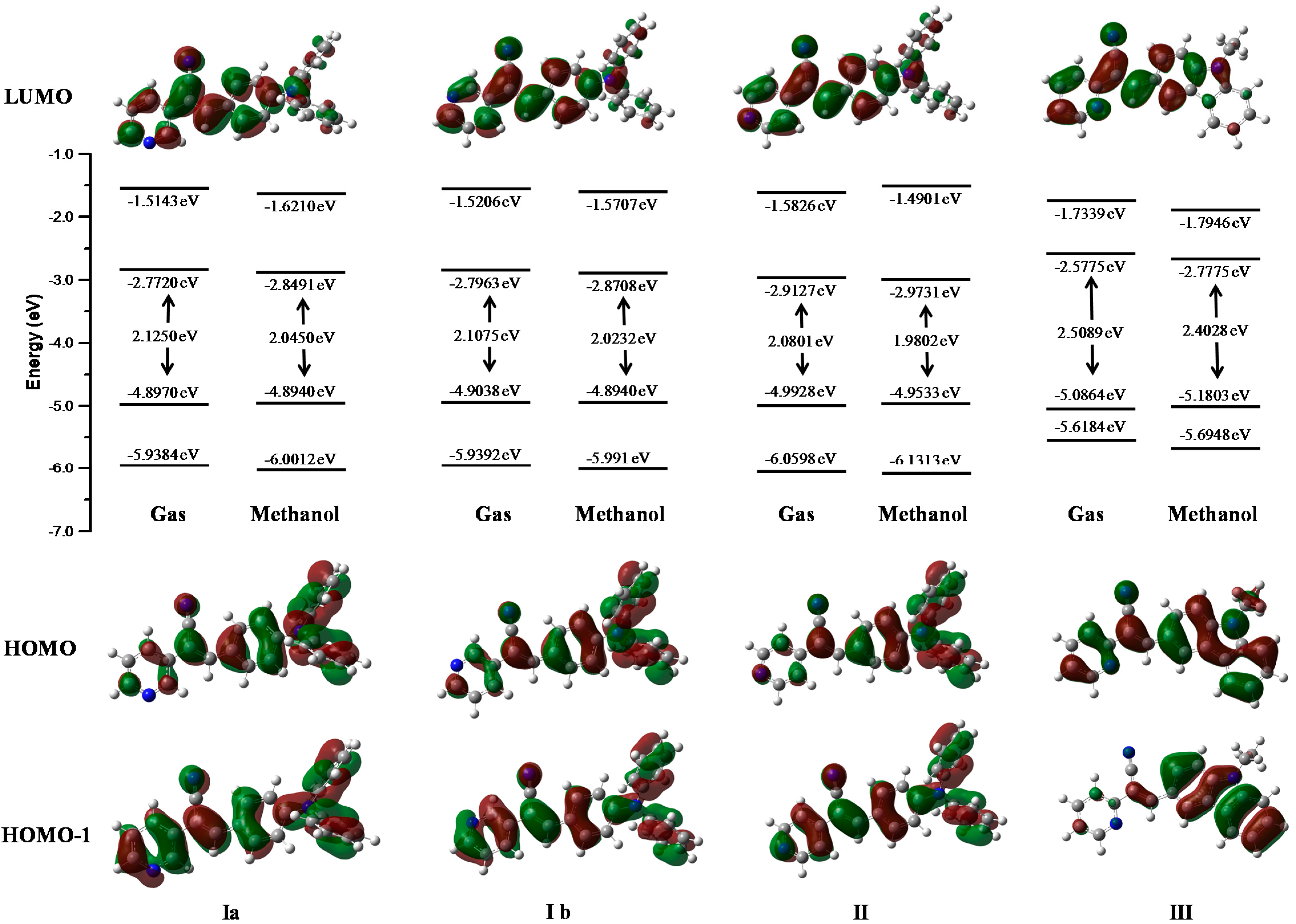
2.3. DFT Calculations
3. Experimental Section
3.1. Single Crystal X-ray Diffraction (SCXRD)
3.2. Absorbance and Emission (UV-VIS and PL)
3.3. Cyclic Voltammetry (CV)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bader, M.A.; Marowsky, G.; Bahtiar, A.; Koynov, K.; Bubeck, C.; Tillmann, H.; Hörhold, H.H.; Pereira, S. Poly(p-phenylenevinylene) derivatives: New promising materials for nonlinear all-optical waveguide switching. J. Opt. Soc. Am. B 2002, 19, 2250–2262. [Google Scholar] [CrossRef]
- Braga, D.; Grepioni, F.; Maini, L. The growing world of crystal forms. Chem. Commun. 2010, 46, 6232–6242. [Google Scholar] [CrossRef] [PubMed]
- Schwoerer, M.; Wolf, H.C. Organic Molecular Solids; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2007. [Google Scholar]
- Cornil, J.; Beljonne, D.; Calbert, J.-P.; Brédas, J.-L. Interchain interactions in organic π-conjugated materials: Impact on electronic structure, optical response, and charge transport. Adv. Mater. 2001, 13, 1053–1067. [Google Scholar] [CrossRef]
- Datta, A.; Pati, S.K. Dipolar interactions and hydrogen bonding in supramolecular aggregates: Understanding cooperative phenomena for 1st hyperpolarizability. Chem. Soc. Rev. 2006, 35, 1305–1323. [Google Scholar] [CrossRef] [PubMed]
- Chow, T.J.; Lin, R.; Ko, C.-W.; Tao, Y.-T. Photo and electroluminescence of 2-anilino-5-phenylpenta-2,4-dienenitrile derivatives. J. Mater. Chem. 2002, 12, 42–46. [Google Scholar]
- Sun, H.; Zhao, Z.; Spano, F.C.; Beljonne, D.; Cornil, J.; Shuai, Z.; Brédas, J.-L. Absorption and emission in quaterthienyl thin films. Adv. Mater. 2003, 15, 818–822. [Google Scholar] [CrossRef]
- Dreuw, A.; Plötner, J.; Lorenz, L.; Wachtveitl, J.; Djanhan, J.E.; Brüning, J.; Metz, T.; Bolte, M.; Schmidt, M.U. Molecular mechanism of the solid-state fluorescence behavior of the organic pigment yellow 101 and its derivatives. Angew. Chem. Int. Ed 2005, 44, 7783–7786. [Google Scholar] [CrossRef] [PubMed]
- Langhals, H.; Potrawa, T.; Nöth, H.; Linti, G. The influence of packing effects on the solid-state fluorescence of diketopyrrolopyrroles. Angew. Chem. Int. Ed. 1989, 28, 478–480. [Google Scholar] [CrossRef]
- Datta, A.; Terenziani, F.; Painelli, A. Cooperative interactions in supramolecular aggregates: Linear and nonlinear responses in calix[4]arenes. Chem. Phys. Chem. 2006, 7, 2168–2174. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Swager, T.M. Control of conformational and interpolymer effects in conjugated polymers. Nature 2001, 411, 1030–1034. [Google Scholar] [CrossRef] [PubMed]
- Jenekhe, S.A.; Osaheni, J.A. Excimers and exciplexes of conjugated polymers. Science 1994, 265, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Yang, B.; Li, F.; Cheng, G.; Liu, L.; Yang, G.; Xu, H.; Ye, L.; Hanif, M.; Liu, S.; et al. Cross dipole stacking in the crystal of distyrylbenzene derivative: The approach toward high solid-state luminescence efficiency. J. Am. Chem. Soc. 2005, 127, 14152–14153. [Google Scholar] [CrossRef] [PubMed]
- Mizobe, Y.; Ito, H.; Hisaki, I.; Miyata, M.; Hasegawa, Y.; Tohnai, N. A novel strategy for fluorescence enhancement in the solid-state: Affording rigidity to fluorophores packing. Chem. Commun. 2006, 20, 2126–2128. [Google Scholar]
- Desiraju, G. Crystal Engineering: A holistic view. Angew. Chem. Int. Ed. 2007, 46, 8342–8356. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Hu, W.; Liu, Y.; Zhu, D. Micro- and nanocrystals of organic semiconductors. Acc. Chem. Res. 2010, 43, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Gsanger, M.; Oh, J.H.; Konemann, M.; Hoffken, H.W.; Krause, A.-M.; Bao, Z.; Wurthner, F. A crystal-engineered hydrogen-bonded octachloroperylene diimide with a twisted core: An n-channel organic semiconductor. Angew. Chem. Int. Ed. 2010, 49, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jiang, X.; Luo, J.; Chi, C.; Chen, H.; Wu, J. A Cruciform 6,6′-dipentacenyl: Synthesis, solid-state packing and applications in thin-film transistors. Chem. Eur. J. 2010, 16, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.D.; Cao, J.; Kampf, J.W. Solid-state packing of conjugated oligomers: From π-stacks to the herringbone structure. J. Am. Chem. Soc. 2004, 126, 4318–4328. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Dong, H.; Li, H.; Zhao, H.; Meng, Q.; Hu, W. Dibenzothiophene Derivatives: From Herringbone to Lamellar Packing Motif. Cryst. Growth Des. 2010, 10, 4155–4160. [Google Scholar] [CrossRef]
- Mattheus, C.C.; de Wijs, G.A.; de Groot, R.A.; Palstra, T.T.M. Modeling the polymorphism of pentacene. J. Am. Chem. Soc. 2003, 125, 6323–6330. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Zeis, R.; Borkent, E.J.; Besnard, C.; Lovinger, A.J.; Siegris, T.; Kloc, C.; Bao, Z.N. Synthesis, Crystal Structure, and Transistor Performance of Tetracene Derivatives. J. Am. Chem. Soc. 2004, 126, 15322–15323. [Google Scholar] [CrossRef] [PubMed]
- Sundar, V.C.; Zaumescil, J.; Podzorov, V.; Menard, E.; Willeett, R.L.; Someya, T.; Gershenson, M.E.; Rogers, J.A. Elastomeric transistor stamps: Reversible probing of charge transport in organic crystals. Science 2004, 303, 1644–1646. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, F.; Bacchi, S.; Filippini, G.; Pilati, T.; Gavezzotti, A. Synthesis, X-ray diffraction and computational study of the crystal packing of polycyclic hydrocarbons featuring aromatic and perfluoroaromatic rings condensed in the same molecule: 1,2,3,4-Tetrafluoronaphthalene, -anthracene and -phenanthrene. Chem. Eur. J. 2007, 13, 7177–7184. [Google Scholar] [PubMed]
- Anthony, J.E.; Brooks, J.S.; Eaton, D.J.; Parkin, S.R. Functionalized pentacene: Improved electronic properties from control of solid-state order. J. Am. Chem. Soc. 2001, 123, 9482–9483. [Google Scholar] [CrossRef] [PubMed]
- Coropceanu, V.; Cornil, J.; da Silva, D.A.; Yoann, F.; Silbey, O.R.; Brédas, J.L. Charge transport in organic semiconductors. Chem. Rev. 2007, 107, 926–952. [Google Scholar] [CrossRef] [PubMed]
- Valeev, E.F.; Coropceanu, V.; da Silva, D.A.; Salman, S.; Bredas, J.L. Effect of Electronic Polarization on Charge-Transport Parameters in Molecular Organic Semiconductors. J. Am. Chem. Soc. 2006, 128, 9882–9886. [Google Scholar] [CrossRef] [PubMed]
- Percino, M.J.; Chapela, V.M.; Cerón, M.; Soriano-Moro, G.; Castro, M.E.; Meléndez, F.J. Fluorescence improvement of pyridylacrylonitrile by dimethylaminophenyl-substitutions: The effect of packing modes of conjugated compounds. J. Mol. Struct. 2013, 1034, 238–248. [Google Scholar] [CrossRef]
- Percino, M.J.; Cerón, M.; Soriano-Moro, G.; Castro, M.E.; Chapela, V.M.; Bonilla, J.; Reyes-Reyes, M.; López-Sandoval, R. The effect of the supramolecular network of (Z)-3-(4-(diphenylamino)phenyl)-2-(pyridin-2-yl)-acrylonitrile on the fluorescence behavior of a single crystal: Experimental and theoretical studies. CrystEngComm 2014, 16, 8591–8604. [Google Scholar] [CrossRef]
- Percino, M.J.; Chapela, V.M.; Cerón, M.; Soriano-Moro, G.; Castro, M.E.; Melendez, F.J. Molecular packing and solid-state fluorescence of conjugated compounds of carbazole-acrylonitrile derivatives. Curr. Phys. Chem. 2014, 4, 137–150. [Google Scholar] [CrossRef]
- Percino, M.J.; Cerón, M.; Coca, P.; Soriano, G.; Castro, M.E.; Chapela, V.M.; Bonilla, J.; Reyes-Reyes, M.; López-Sandoval, R.; Siegler, M.A. Important role of molecular packing and intermolecular interactions in two polymorphs of (Z)-2-phenyl-3-(4-(pyridin-2-yl)phenyl)acrylonitrile. Preparation, structures, and optical propertie. J. Mol. Struct. 2014, 1078, 74–82. [Google Scholar] [CrossRef]
- Hong, Y.; Lam, J.W.Y.; Tang, B.Z. Aggregation-induced emission. Chem. Soc. Rev. 2011, 40, 536–5388. [Google Scholar] [CrossRef] [PubMed]
- An, B.-K.; Kwon, S.-K.; Jung, S.-D.; Park, S.Y. Enhanced emission and its switching in fluorescent organic nanoparticles. J. Am. Chem. Soc. 2002, 124, 14401–14415. [Google Scholar] [CrossRef]
- Li, Z.; Dong, Y.Q.; Lam, J.W.Y.; Sun, J.; Qin, A.; Haeussler, M.; Dong, Y.P.; Sung, H.H.Y.; Williams, I.D.; Kwok, H.S.; et al. Functionalized siloles: Versatile synthesis, aggregation-induced emission, and sensory and device applications. Adv. Funct. Mater. 2009, 19, 905–917. [Google Scholar] [CrossRef]
- Zhang, X.; Chi, Z.; Li, H.; Xu, B.; Li, X.; Liu, S.; Zhang, Y.; Xu, J. Synthesis and properties of novel aggregation-induced emission compounds with combined tetraphenylethylene and dicarbazolyl triphenylethylene moieties. J. Mater. Chem. 2011, 21, 1788–1796. [Google Scholar] [CrossRef]
- Zhang, G.-F.; Aldred, M.P.; Gong, W.-L.; Li, C.; Zhu, M.-Q. Utilising tetraphenylethene as a dual activator for intramolecular charge transfer and aggregation induced emission. Chem. Commun. 2012, 48, 7711–7713. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.W.; Yoon, S.-J.; An, B.-K.; Park, S.Y. High-contrast on/off fluorescence switching via reversible E–Z isomerization of diphenylstilbene containing the α-cyanostilbenic moiety. J. Phys. Chem. C 2013, 117, 11285–11291. [Google Scholar] [CrossRef]
- Palakollu, V.; Kanvah, S. α-Cyanostilbene based fluorophores: Aggregation-induced enhanced emission, solvatochromism and the pH effect. New J. Chem. 2014, 38, 5736–5746. [Google Scholar] [CrossRef]
- Bredas, J.L.; Calbert, J.P.; da Silva, D.A.; Cornil, J. Organic semiconductors: A theoretical characterization of the basic parameters governing charge transport. Proc. Natl. Acad. Sci. USA 2002, 99, 5804–5809. [Google Scholar] [CrossRef] [PubMed]
- Kazmaier, P.M.; Hoffmann, R. A theoretical study of crystallochromy. Quantum interference effects in the spectra of perylene pigments. J. Am. Chem. Soc. 1994, 116, 9684–9691. [Google Scholar] [CrossRef]
- Wurthner, F. Perylene bisimide dyes as versatile building blocks for functional supramolecular architectures. Chem. Commun. 2004, 1564–1579. [Google Scholar] [CrossRef] [PubMed]
- Bishop, R.; Scudder, M.L. Multiple Molecules in the Asymmetric Unit (Z′ > 1) and the Formation of False Conglomerate Crystal Structures. Cryst. Growth Des. 2009, 9, 2890–2894. [Google Scholar] [CrossRef]
- Plötner, J.; Drew, A. Solid state fluorescence of Pigment Yellow 101 and derivatives: A conserved property of the individual molecules. Phys. Chem. Chem. Phys. 2006, 8, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Steed, K.M.; Steed, J.W. Packing problems: High Z' crystal structures and their relationship to cocrystals, inclusion compounds, and polymorphism. Chem. Rev. 2015, 115, 2895–2933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, T. The hydrogen bond in the solid state. Angew. Chem. Int. Ed. 2002, 41, 49–76. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourkos, L.J.; Gilda, R.J.; Howard, J.A.K.; Puschman, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Molčanov, K.; Sabljić, I.; Kojić-Prodić, B. Face-to-face π-stacking in the multicomponent crystals of chloranilic acid, alkali hydrogenchloranilates, and water. CrystEngComm 2011, 13, 4211–4217. [Google Scholar] [CrossRef]
- Li, X.C.; Sirringhaus, H.; Garnier, F.; Holmes, A.B.; Moratti, S.C.; Feeder, M.N.; Clegg, F.W.; Teat, S.J.; Friend, R.H. A highly π-stacked organic semiconductor for thin film transistors based on fused thiophenes. J. Am. Chem. Soc. 1998, 120, 2206–2207. [Google Scholar] [CrossRef]
- Zhang, X.; Cote, A.P.; Matzger, A.J. Synthesis and structure of fused α-oligothiophenes with up to seven rings. J. Am. Chem. Soc. 2005, 127, 10502–10503. [Google Scholar] [CrossRef] [PubMed]
- Mercury 3.5.1. Available online: http://www.ccdc.cam.ac.uk/mercury/ (accessed on 13 December 2014).
- Nagarajan, K.; Rajagopal, S.K.; Hariharan, M. C–H⋯H–C and C–H⋯π contacts aid transformation of dimeric to monomeric anthracene in the solid state. CrystEngComm 2014, 16, 8946–8949. [Google Scholar] [CrossRef]
- Percino, M.J.; Chapela, V.M.; Cerón, M.; Castro, M.E.; Soriano-Moro, G.; Pérez-Gutiérrez, E.; Meléndez-Bustamante, F. Synthesis and characterization of conjugated pyridine-(N-diphenylamino) acrylonitrile derivatives: Photophysical properties. J. Mater. Res. 2012, 1, 181–192. [Google Scholar]
- Upamali, K.A.N.; Estrada, L.A.; De, P.K.; Cai, X.C.; Krause, J.A.; Neckers, D.C. Carbazole-based cyano-stilbene highly fluorescent microcrystals. Langmuir 2011, 27, 1573–1580. [Google Scholar] [CrossRef] [PubMed]
- Pomrnerehne, J.; Vestweber, H.; Guss, W.; Mahrt, R.F.; Bassler, H.; Porsch, M.; Daub, J. Efficient two layer leds on a polymer blend basis. Adv. Mater. 1995, 7, 551–554. [Google Scholar] [CrossRef]
- Weissbuch, I.; Torbeev, V.Y.; Leiserowitz, L.; Lahav, M. Solvent Effect on Crystal Polymorphism: Why Addition of Methanol or Ethanol to Aqueous Solutions Induces the Precipitation of the Least Stable Beta Form of Glycine. Angew. Chem. Int. Ed. 2005, 44, 3226–3229. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Lee, I.S.; Myerson, A. Factor affecting the polymorphic outcome of gycine crystals constrained on patterned substrates. Chem. Eng. Technol. 2006, 29, 281–285. [Google Scholar] [CrossRef]
- Blagden, N.; Davey, R.J.; Lieberman, H.F.; Williams, L.; Payne, R.; Roberts, R.; Rowe, R.; Docherty, R. Crystal Chemistry and Solvent Effects in Polymorphic Systems Sulfathiazole. J. Chem. Soc. Faraday Trans. 1998, 94, 1035–1044. [Google Scholar] [CrossRef]
- Kitamura, M.; Horimoto, K. Role of Kinetic Process in the Solvent Effect on Crystallization of BPT Propyl Ester Polymorph. J. Cryst. Growth 2013, 373, 151–155. [Google Scholar] [CrossRef]
- Percino, M.J.; Chapela, V.M.; Perez-Gutierrez, E.; Ceron, M.; Soriano, G. Synthesis, optical, and spectroscopic characterisation of substituted 3-phenyl-2-arylacrylonitriles. Chem. Pap. 2011, 65, 42–51. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of compounds I, II and III are available from the authors.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical Formula | I | II | III |
|---|---|---|---|
| C26H19N3, 0.395 (C2H6O) | C26H19N3 | C22H17N3 | |
| Color, habit | yellow, irregular shape | yellow, plate | yellow, thin needle |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic |
| Formula weight | 391.68 | 373.44 | 323.38 |
| Space group | P2/c | Pbcn | C2/c |
| T (K) | 110(2) | 110(2) | 110(2) |
| A (Å) | 24.9448(10) | 24.4603(9) | 30.4066(6) |
| b (Å) | 10.5094(5) | 10.1791(3) | 4.81650(10) |
| c (Å) | 15.4015(6) | 15.4796(6) | 22.6451(5) |
| α (°) | 90 | 90 | 90 |
| β (°) | 94.300(4) | 90 | 98.965(2) |
| γ (°) | 90 | 90 | 90 |
| V (Å3) | 4026.2(3) | 3854.2(2) | 3275.94(12) |
| Z, Z′ | 8, 2 | 8, 1 | 8, 1 |
| Dc (g·cm−3) | 1.292 | 1.287 | 1.311 |
| F (000) | 1650.4 | 1568 | 1360 |
| μ (mm−1) | 0.078 | 0.077 | 0.612 |
| λ (Å) | 0.71073 | 0.71073 | 1.54178 |
| Crystal size (mm3) | 0.44 × 0.20 × 0.08 | 0.25 × 0.21 × 0.06 | 0.73 × 0.06 × 0.063 |
| 2θmax (°) | 48.6 | 50 | 134.5 |
| Ntotal, Nunique | 23,831, 7364 | 17,555, 3395 | 9198, 2944 |
| Nobs (I > 2.0 σ(I)) | 5257 | 2671 | 2497 |
| R1 (I > 2.0 σ(I)) | 6.10 | 4.23 | 3.66 |
| wR2 (I > 2.0 σ(I)) | 13.77 | 8.94 | 8.72 |
| goodness-of-fit | 1.055 | 1.028 | 1.045 |
| Largest diff peak and hole (e·Å−3) | 0.61 and −0.57 | 0.19 and −0.21 | 0.22 and −0.21 |
| D | A | Symmetry for A | d(D···A) | D(A···H) | D(D-H) | D-H···A |
|---|---|---|---|---|---|---|
| O1S | O1S | x, 1 − y,−1/2 + z | 2.746 | - | - | 101.21 |
| O1S | N3A | x, y, −1 + z | 2.770 | N(3)······H(1S) 2.019 | O(1S)-H(1S) 0.8400 | 148.57 |
| Bond Length | I | II | III | |
|---|---|---|---|---|
| A | B | |||
| N(1)-C(1) | 1.429(5) | 1.429(5) | 1.424(2) | |
| N(1)-C(2) | 1.522(2) | |||
| N(1)-C(3) | 1.3910(17) | |||
| N(1)-C(7) | 1.431(5) | 1.409(5) | 1.4279(19) | |
| N(1)-C(13) | 1.397(5) | 1.407(5) | 1.4042(19) | |
| N(1)-C(14) | 1.3800(18) | |||
| C(19)-C(20) | 1.352(6) | 1.339(6) | 1.348(2) | |
| C(19)-C(16) | 1.459(6) | 1.456(6) | 1.454(2) | |
| C(15)-C(11) | 1.449(2) | |||
| C(15)-C(16) | 1.365(6) | 1.386(6) | 1.398(2) | 1.351(2) |
| C(16)-C(17) | 1.397(6) | 1.390(6) | 1.404(2) | 1.4381(19) |
| C(16)-C(18) | 1.483(2) | |||
| C(20)-C(22) | 1.483(6) | 1.476(6) | 1.487(2) | |
| C(23)-C(22) | 1.393(6) | 1.394(6) | 1.392(2) | |
| C(17)-N(2) | 1.1493(19) | |||
| C(18)-N(3) | 1.3460(18) | |||
| C(22)-N(3) | 1.3318(19) | |||
| C(26)-N(3) | 1.330(6) | 1.333(6) | ||
| C(20)-C(21) | 1.429(6) | 1.447(5) | 1.437(2) | |
| C(21)-N(2) | 1.147(6) | 1.135(5) | 1.152(2) | |
| C(25)-N(3) | 1.331(6) | 1.333(6) | 1.336(2) | |
| C(24)-N(3B) | 1.341(2) | |||
| Torsion angle | ||||
| C(14)-C(13)-N(1)-C(7) | 150.0(4) | −154.6(4) | 30.0(2) | |
| C(14)-C(13)-N(1)-C(1) | −31.3(6) | 35.6(6) | −148.58(15) | |
| C(8)-C(7)-N(1)-C(1) | −41.5(5) | 39.7(5) | 46.5(2) | |
| C(12)-C(7)-N(1)-C(13) | −42.7(6) | 48.4(6) | 48.6(2) | |
| C(18)-C(13)-N(1)-C(1) | 138.5(4) | −144.8(4) | 31.9(2) | |
| C(18)-C(13)-N(1)-C(7) | −30.1(6) | 24.9(6) | −149.59(15) | |
| C(2)-C(1)-N(1)-C(13) | −45.0(5) | 36.8(6) | 37.5(2) | |
| C(21)-C(20)-C(22)-C(23) | −30.0(6) | −152.4(4) | −151.57(15) | |
| C(16)-C(19)-C(20)-C(22) | −179.0(4) | −178.6(4) | −178.98(15) | |
| C(15)-C(16)-C(19)-C(20) | −12.7(8) | 22.2(7) | −161.32(17) | |
| C(16)-C(19)-C(20)-C(21) | −2.8(8) | 4.4(7) | 6.9(3) | |
| C(19)-C(20)-C(22)-C(23) | 30.3(6) | 33.9(2) | ||
| C(19)-C(20)-C(22)-C(26) | −34.7(7) | −146.6(4) | ||
| C(21)-C(20)-C(22)-C(26) | 30.7(6) | 28.9(2) | ||
| C(17)-C(16)-C(19)-C(20) | 20.5(3) | |||
| C(17)-C(16)-C(18)-C(19) | 1.68(19) | |||
| C(11)-C(15)-C(16)-C(17) | −3.1(2) | |||
| C(12)-C(11)-C(15)-C(16) | −13.8(2) | |||
| C(15)-C(16)-C(18)-N(3) | −1.53(19) | |||
| C(10)-C(11)-C(15)-C(16) | 167.60(14) | |||
| C(13)-C(14)-N(1)-C(3) | 178.78(14) | |||
| C(4)-C(3)-N(1)-C(2) | 1.7(2) | |||
| Cyclic voltammetry | Theoretical calculations | Optical data | |||||||
|---|---|---|---|---|---|---|---|---|---|
| (V) | (V) | HOMO (eV) | LUMO (eV) | ΔE (eV) | HOMO (eV) | LUMO (eV) | ΔE (eV) | ΔEopt (eV) | |
| I | 1.0336 | −1.0496 | −5.2976 | −3.2144 | 2.0832 | −4.897000 | −2.772046 | 2.124954 | 2.68 |
| II | 1.072 | −0.916 | −5.3372 | −3.348 | 1.9892 | −4.992784 | −2.912730 | 2.080055 | 2.72 |
| III | 1.2051 | −0.8082 | −5.5054 | −3.4921 | 2.0133 | −5.086392 | −2.577483 | 2.508910 | 2.63 |
| HOMO energy (eV) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Gas | C6H12 | CHCl3 | THF | CH2Cl2 | C2H5OH | CH3OH | DMSO |
| A | −4.8970 | −4.8837 | −4.8856 | −4.8880 | −4.8891 | −4.8932 | −4.8940 | −4.8945 |
| B | −4.9038 | −4.8886 | −4.8883 | −4.8899 | −4.8907 | −4.8935 | −4.8940 | −4.8945 |
| II | −4.9928 | −4.9664 | −4.9560 | −4.9544 | −4.9541 | −4.9533 | −4.9533 | −4.9533 |
| III | −5.0864 | −5.1117 | −5.1446 | −5.1579 | −5.1623 | −5.1781 | −5.1803 | −5.1827 |
| LUMO energy (eV) | ||||||||
| Compound | Gas | C6H12 | CHCl3 | THF | CH2Cl2 | C2H5OH | CH3OH | DMSO |
| A | −2.7720 | −2.7965 | −2.8237 | −2.8338 | −2.8368 | −2.8474 | −2.8490 | −2.8507 |
| B | −2.7963 | −2.8197 | −2.8461 | −2.8556 | −2.8588 | −2.8692 | −2.8708 | −2.8722 |
| II | −2.9127 | −2.9326 | −2.9541 | −2.9617 | −2.9639 | −2.9720 | −2.9731 | −2.9742 |
| III | −2.5775 | −2.6455 | −2.7130 | −2.7380 | −2.7459 | −2.7734 | −2.7775 | −2.7813 |
| Gap energy (eV) | ||||||||
| Compound | Gas | C6H12 | CHCl3 | THF | CH2Cl2 | C2H5OH | CH3OH | DMSO |
| A | 2.1249 | 2.0871 | 2.0618 | 2.0542 | 2.0523 | 2.0458 | 2.0449 | 2.0439 |
| B | 2.1075 | 2.0689 | 2.0422 | 2.0343 | 2.0319 | 2.0243 | 2.0232 | 2.0224 |
| II | 2.0800 | 2.0338 | 2.0019 | 1.9927 | 1.9902 | 1.9813 | 1.9802 | 1.9791 |
| III | 2.5089 | 2.4662 | 2.4316 | 2.4199 | 2.4164 | 2.4047 | 2.4028 | 2.4014 |
| μ (Debye) | ||||||||
| Compound | Gas | C6H12 | CHCl3 | THF | CH2Cl2 | C2H5OH | CH3OH | DMSO |
| A | 4.6302 | 5.3812 | 5.9549 | 6.1395 | 6.1962 | 6.3849 | 6.4112 | 6.4370 |
| B | 7.0370 | 8.0966 | 8.9946 | 9.3050 | 9.4029 | 9.7378 | 9.7858 | 9.8333 |
| II | 7.1955 | 8.3345 | 9.2504 | 9.5546 | 9.6490 | 9.9654 | 10.0098 | 10.0535 |
| III | 3.3975 | 3.9375 | 4.3492 | 4.4809 | 4.5212 | 4.6546 | 4.6731 | 4.6913 |
| I | II | III |
|---|---|---|
| Ethylacetate:hexane (1:2) | Ethylacetate:hexane (1:2) | Ethylacetate:hexane (1:2) |
| Ethanol–cyclohexane (1:3) | Acetone:water (1:1) | Ethanol-cyclohexane 1:3 |
| Ethanol–DMSO 1:5 | Ethanol:DMF 1:5 | Ethanol-DMSO 1:5 |
| Ethanol:cyclohexane (3:2)√ | DMSO√ | Cyclohexane√ |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Percino, M.J.; Cerón, M.; Rodríguez, O.; Soriano-Moro, G.; Castro, M.E.; Chapela, V.M.; Siegler, M.A.; Pérez-Gutiérrez, E. Conformational and Molecular Structures of α,β-Unsaturated Acrylonitrile Derivatives: Photophysical Properties and Their Frontier Orbitals. Molecules 2016, 21, 389. https://doi.org/10.3390/molecules21040389
Percino MJ, Cerón M, Rodríguez O, Soriano-Moro G, Castro ME, Chapela VM, Siegler MA, Pérez-Gutiérrez E. Conformational and Molecular Structures of α,β-Unsaturated Acrylonitrile Derivatives: Photophysical Properties and Their Frontier Orbitals. Molecules. 2016; 21(4):389. https://doi.org/10.3390/molecules21040389
Chicago/Turabian StylePercino, María Judith, Margarita Cerón, Oscar Rodríguez, Guillermo Soriano-Moro, María Eugenia Castro, Víctor M. Chapela, Maxime A. Siegler, and Enrique Pérez-Gutiérrez. 2016. "Conformational and Molecular Structures of α,β-Unsaturated Acrylonitrile Derivatives: Photophysical Properties and Their Frontier Orbitals" Molecules 21, no. 4: 389. https://doi.org/10.3390/molecules21040389
APA StylePercino, M. J., Cerón, M., Rodríguez, O., Soriano-Moro, G., Castro, M. E., Chapela, V. M., Siegler, M. A., & Pérez-Gutiérrez, E. (2016). Conformational and Molecular Structures of α,β-Unsaturated Acrylonitrile Derivatives: Photophysical Properties and Their Frontier Orbitals. Molecules, 21(4), 389. https://doi.org/10.3390/molecules21040389