
Antimicrobial Diterpenoids of Wedelia trilobata (L.) Hitchc

Abstract
:1. Introduction
2. Results and Discussion
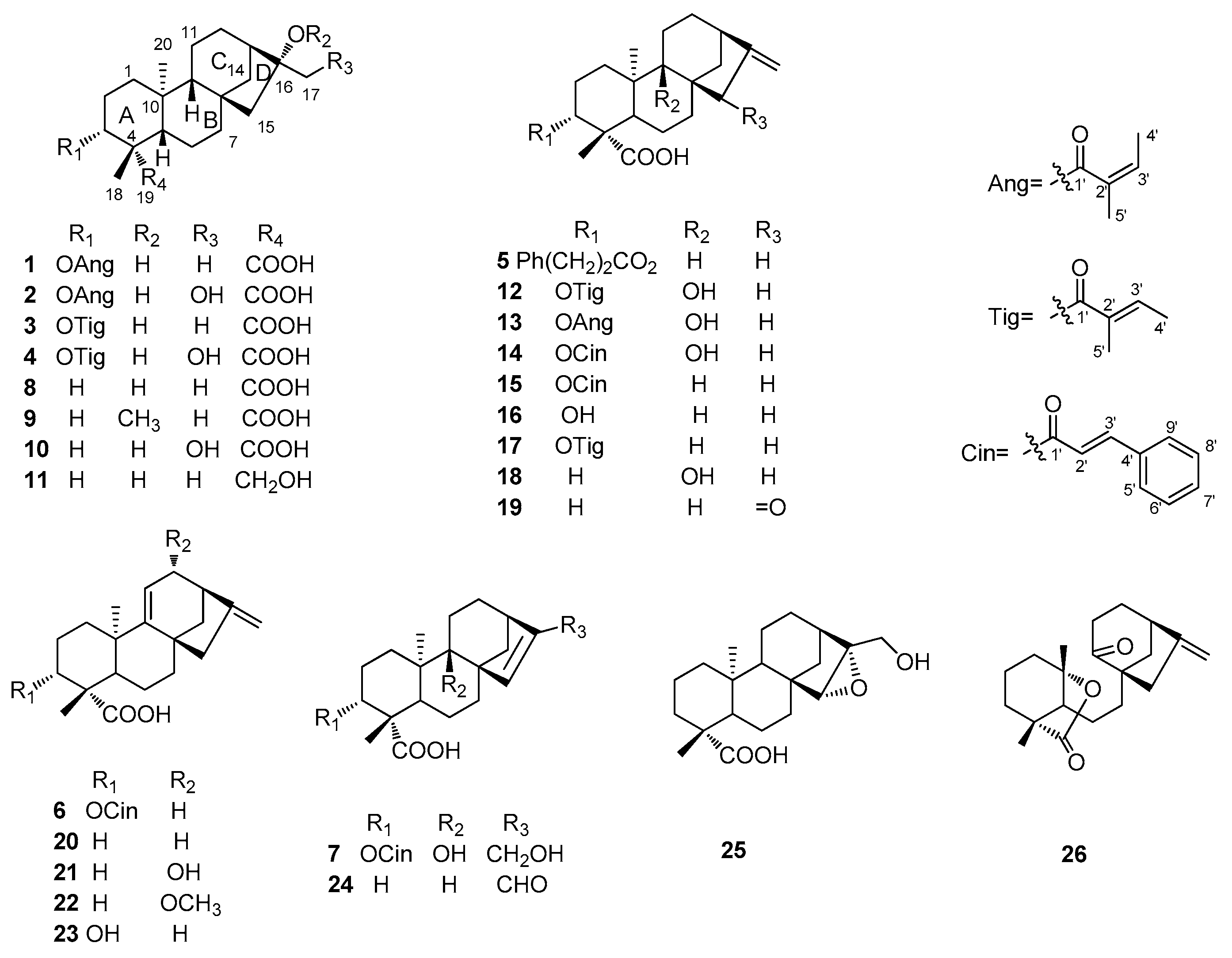
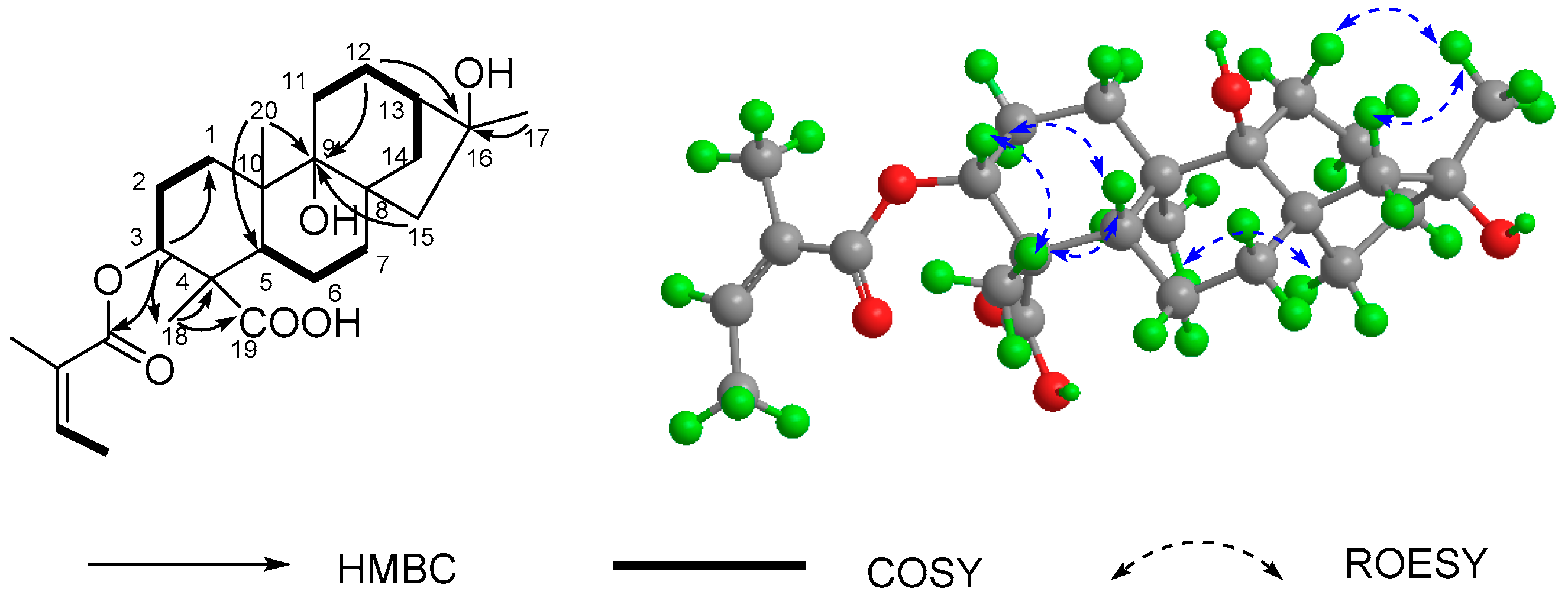
2.1. Structure Elucidation of Compounds
2.2. Evaluation of Anti-Micobial Activity
3. Experimental Section
3.1. General Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Data for 1–7
3.5. Antimicrobial Assays
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- IUCN. 100 of the World’s Worst Invasive Alien Species; Invasive Species Specialist Group: Auckland, New Zealand, 2001. [Google Scholar]
- Xie, L.J.; Zeng, R.S.; Bi, H.H.; Song, Y.Y.; Wang, R.L.; Su, Y.J.; Chen, M.; Chen, S.; Liu, Y.H. Allelochemical mediated invasion of exotic plants in China. Allelopath. J. 2010, 25, 31–50. [Google Scholar]
- Wu, J.R.; Peng, S.L.; Zhao, H.B.; Xiao, H.L. Allelopathic effects of Wedelia trilobata residues on lettuce germination and seedling growth. Allelopath. J. 2008, 22, 197–204. [Google Scholar]
- Zhang, Y.H.; Liu, M.F.; Lin, T.J.; Wei, X.Y. Allelopathic sesquiterpene lactones from Wedelia trilobata. J. Trop. Subtrop. Bot. 2004, 12, 533–537. [Google Scholar]
- Nie, C.R.; Zeng, R.S.; Luo, S.M.; Li, H.S.; Hong, M.Q.; Cheng, L.Q. Allelopathic potentials of Wedelia trilobata L. on rice. Acta Agron. Sin. 2004, 30, 942–946. [Google Scholar]
- Wu, M.L.; Zhang, D.Z. Progress of researches on the invasive plant Wedelia trilabata. Pharm. Today 2008, 6, 21–23. [Google Scholar]
- Li, Y.T.; Hao, X.J.; Li, S.F.; He, H.P.; Yan, X.H.; Chen, Y.D.; Dong, J.H.; Zhang, Z.K.; Li, S.L. Eudesmanolides from Wedelia trilobata (L.) Hitchc. as potential inducers of plant systemic acquired resistance. J. Agric. Food Chem. 2013, 61, 3884–3890. [Google Scholar] [CrossRef] [PubMed]
- Ragasa, C.Y.; Padolina, W.G.; Bowden, B.F.; Li, S.X.; Tapiolas, D.M.; Coll, J.C. New eudesmanolide sesquiterpenes from a Philippines collection of Wedelia prostata. J. Nat. Prod. 1993, 56, 386–393. [Google Scholar] [CrossRef]
- Hutchison, M.; Lewer, P.; MacMillan, J. Carbon-13 nuclear magnetic resonance spectra of eighteen derivatives of ent-kaur-16-en-19-oic acid. J. Chem. Soc. Perkin Trans. 1 1984, 10, 2363–2366. [Google Scholar] [CrossRef]
- Duan, H.; Takaishi, Y.; Momota, H.; Ohmoto, Y.; Taki, T.; Jia, Y.; Li, D. Immunosuppressive diterpenoids from Tripterygium wilfordii. J. Nat. Prod. 1999, 62, 1522–1525. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.A.; Lee, E.J.; Kim, J.S.; Kang, S.S.; Lee, J.H.; Min, B.S.; Choi, J.S. Cholinesterase and BACE1 inhibitory diterpenoids from Aralia cordata. Arch. Pharm. Res. 2009, 32, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Qiang, Y.; Du, D.L.; Chen, Y.J.; Gao, K. Ent-Kaurane diterpenes and further constituents from Wedelia trilobata. Hel. Chim. Acta 2011, 94, 817–823. [Google Scholar] [CrossRef]
- Pacheco, A.G.; Machado de Oliveira, P.; Piló-Veloso, D.; Flávio de Carvalho Alcântara, A. 13C-NMR data of diterpenes isolated from Aristolochia species. Molecules 2009, 14, 1245–1262. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.J.; Wen, C.N.; Gao, Y.; Ren, F.C.; Wang, F.; Liu, J.K. ent-Kaurane diterpenoids from the plant Wedelia trilobata. Nat. Prod. Bioprospect. 2013, 3, 107–111. [Google Scholar] [CrossRef]
- Bohlmann, F.; Ziesche, J.; King, R.M.; Robinson, H. Naturally occurring terpene derivatives. Part 300. Eudesmanolides and diterpenes from Wedelia trilobata and an ent-kaurenic acid derivative from Aspilia parvifolia. Phytochemistry 1981, 20, 751–756. [Google Scholar]
- Zhang, D.Z.; Li, X.; Zhu, T.R. Spectroscopic study of kaurane type diterpene compounds. Bopuxue Zazhi 1990, 7, 349–350. [Google Scholar]
- Murakami, T.; Iida, H.; Tanaka, N.; Saiki, Y.; Chen, C.M.; Iitaka, Y. Chemical and chemotaxonomic studies of ferns. XXXIII. Chemical studies on the constituents of Pteris longipes Don. Chem. Pharm. Bull. 1981, 29, 657–662. [Google Scholar]
- De Oliveira, A.B.; Hanson, J.R.; Takahashi, J.A. The biotransformation of ent-15-oxokaur-16-en-19-oic acid and its methyl ester by Cephalosporium aphidicola. Phytochemistry 1995, 40, 439–442. [Google Scholar] [CrossRef]
- Enriquez, R.G.; Barajas, J.; Ortiz, B.; Lough, A.J.; Reynolds, W.F.; Yu, M.; Leon, I.; Gnecco, D. Comparison of crystal and solution structures and 1H and 13C chemical shifts for grandiflorenic acid, kaurenoic acid, and monogynoic acid. Can. J. Chem. 1997, 75, 342–347. [Google Scholar] [CrossRef]
- Ahmed, M.; Jakupovic, J.; Castro, V. Kaurene derivatives from Lasianthea fruticosa, revision of stereochemistry of related compounds. Phytochemistry 1991, 30, 1712–1714. [Google Scholar] [CrossRef]
- Beattie, K.D.; Bhadbhade, M.M.; Craig, D.C.; Leach, D.N. 13C-methoxy-1,2,3,13c-tetrahydrodibenzo[a,kl]xanthan-1-one. Acta Cryst. Sect. E Struct. Rep. Online 2012, 68, 526–527. [Google Scholar] [CrossRef] [PubMed]
- Monte, F.J.; Dantas, E.M.; Braz-Filho, R. New diterpenoids from Croton argyrophylloides. Phytochemistry 1988, 27, 3209–3212. [Google Scholar] [CrossRef]
- Xu, S.Y.; Bian, R.L.; Chen, X. Pharmacological Experiment Methodology, 3rd ed.; People’s Medical Publishing House: Beijing, China, 2002; pp. 1647–1719. [Google Scholar]
- Sample Availability: Samples of the compounds 4, 7, 16, 17, 24, and 26 are available from the authors.




{kind=link}
{kind=link}
{kind=link}
| NO. | 1 a | 2 b | 3 a | 4 b | 5 a | 6 c | 7 d |
|---|---|---|---|---|---|---|---|
| 1a | 0.97 (1H, *) | 1.05 (1H, s) | 0.97 (1H, *) | 1.04 (1H, m) | 1.03 (1H, d, 9.6) | 1.55 (1H, m) | 1.56 (1H, m) |
| 1b | 1.88 (1H, br d, 13.5) | 1.96 (1H, m) | 1.87 (1H, br d, 13.7) | 1.95 (1H, m) | 1.94 (1H, br d) | 2.01 (1H, *) | 2.18 (1H, td, 13.7, 4.3) |
| 2a | 1.72 (1H, m) | 1.70 (1H, m) | 1.69 (1H, m) | 1.65 (1H, m) | 1.68 (1H, m) | 1.87 (1H, m) | 1.71 (1H, m) |
| 2b | 2.30 (1H, m) | 2.46 (1H, m) | 2.27 (1H, m) | 2.42 (1H, m) | 2.33 (1H, m) | 2.55 (1H, m) | 2.57 (1H, m) |
| 3 | 4.50 (1H, dd, 12.2, 4.7) | 4.56 (1H, dd, 12.1, 4.6) | 4.50 (1H, dd, 12.2, 4.7) | 4.50 (1H, dd, 12.1, 4.6) | 4.52 (1H, dd, 12.2, 4.6) | 4.77 (1H, dd, 12.0, 4.8) | 4.61 (1H, dd, 12.5, 4.5) |
| 5 | 1.04 (1H, br d, 11.9) | 1.12 (1H, d, 6.4) | 1.04 (1H, br d, 11.9) | 1.11 (1H, m) | 1.08 (1H, m) | 1.85 (1H, m) | 1.93 (1H, dd, 12.5, 2.2) |
| 6a | 1.61 (1H, *) | 1.67 (1H, m) | 1.61 (1H, *) | 1.68 (1H, m) | 1.63 (1H, m) | 2.01 (1H, *) | 1.66 (1H, m) |
| 6b | 1.80 (1H, *) | 1.87 (1H, m) | 1.79 (1H, *) | 1.86 (1H, m) | 1.84 (1H, m) | 2.01 (1H, *) | 1.85 (1H, dd, 13.9, 2.4) |
| 7a | 1.37 (1H, *) | 1.49 (1H, m) | 1.37 (1H, *) | 1.47 (1H, dd, 10.3, 3.3) | 1.45 (1H, m) | 1.56 (1H, m) | 1.29 (1H, t, 3.2) |
| 7b | 1.59 (1H, *) | 1.65 (1H, m) | 1.59 (1H, *) | 1.64 (1H, m) | 1.53 (1H, *) | 2.10 (1H, m) | 2.11 (1H, m) |
| 9 | 0.90 (1H, br s) | 1.04 (1H, br d) | 0.91 (1H, br s) | 1.03 (1H, br s) | 1.04 (1H, br s) | ||
| 11a | 1.49 (1H, *) | 1.63 (2H, *) | 1.49 (1H, *) | 1.62 (2H, m) | 1.53 (1H, *) | 5.28 (1H, s) | 1.47 (1H, m) |
| 11b | 1.49 (1H, *) | 1.49 (1H, *) | 1.65 (1H, d, 4.9) | 1.62 (1H, m) | |||
| 12a | 1.44 (1H, m) | 1.51 (1H, m) | 1.43 (1H, m) | 1.51 (1H, d, 3.8) | 1.47 (1H, m) | 2.03 (1H, m) | 1.42 (1H, dd, 12.9, 5.3) |
| 12b | 1.51 (1H, *) | 1.63 (1H, *) | 1.50 (1H, *) | 1.62 (1H, m) | 1.56 (1H, m) | 2.44 (1H, m) | 2.01 (1H, m) |
| 13 | 1.77 (1H, *) | 2.03 (1H, br s) | 1.77 (1H, *) | 2.02 (1H, br s) | 2.62 (1H, s) | 2.80 (1H, s) | 2.53 (1H, m) |
| 14a | 1.55 (1H, m) | 1.63 (1H, *) | 1.55 (1H, m) | 1.63 (1H, m) | 1.12 (1H, m) | 1.52 (1H, m) | 1.48 (1H, dd, 5.0, 2.0) |
| 14b | 1.81 (1H, *) | 1.89 (1H, m) | 1.80 (1H, *) | 1.89 (1H, d, 11.3) | 1.91 (1H, d, 11.0) | 1.65 (1H, m) | 2.24 (1H, d, 10.5) |
| 15a | 1.50 (1H, *) | 1.41 (1H, d, 14.4) | 1.50 (1H, *) | 1.40 (1H, d, 14.2) | 2.05 (1H, br s) | 2.24 (1H, d, 15.6) | 5.58 (1H, br s) |
| 15b | 1.50 (1H, *) | 1.53 (1H, d, 14.4) | 1.50 (1H, *) | 1.54 (1H, d, 14.2) | 2.05 (1H, br s) | 2.64 (1H, d, 15.6) | |
| 17a | 1.30 (3H, s) | 3.60 (1H, d, 11.4) | 1.30 (3H, s) | 3.60 (1H, d, 11.3) | 4.74 (1H, s) | 4.84 (1H, s) | 4.10 (1H, d, 14.3) |
| 17b | 3.70 (1H, d, 11.4) | 3.70 (1H, d, 11.3) | 4.80 (1H, s) | 4.95 (1H, s) | 4.14 (1H, d, 14.3) | ||
| 18 | 1.22 (3H, s) | 1.23 (3H, s) | 1.20 (3H, s) | 1.20 (3H, s) | 1.15 (3H, s) | 1.35 (3H, s) | 1.26 (3H, s) |
| 20 | 0.97 (3H, s) | 1.07 (3H, s) | 0.97 (3H, s) | 1.07 (3H, s) | 1.01 (3H, s) | 1.19 (3H, s) | 1.18 (3H, s) |
| 3-ester | 6.02 (1H, q, 7.0) | 6.11 (1H, dq, 7.0, 1.4) | 6.80 (1H, q, 7.1) | 6.89 (1H, dq, 7.0, 1.2) | 2.68 (2H, dd, 15.0, 7.2) | 6.46 (1H, d, 16.2) | 6.53 (1H, d, 15.8) |
| 1.92 (3H, d, 7.0) | 1.96 (3H, dd, 7.2, 1.5) | 1.72 (3H, d, 7.1) | 1.79 (3H, d, 7.2) | 2.95 (2H, m) | 7.70 (1H, d, 16.2) | 7.68 (1H, d, 15.8) | |
| 1.82 (3H, s) | 1.86 (3H, s) | 1.76 (3H, s) | 1.81 (3H, s) | 7.26 (2H, m) | 7.53 (2H, m) | 7.67 (2H, m) | |
| 7.19 (2H, *) | 7.40 (2H, *) | 7.43 (2H, *) | |||||
| 7.19 (1H, *) | 7.40 (1H, *) | 7.43 (1H, *) |
| No. | 1 a | 2 b | 3 a | 4 b | 5 a | 6 c | 7 d |
|---|---|---|---|---|---|---|---|
| 1 | 38.9 t | 40.0 t | 38.9 t | 40.0 t | 38.7 t | 39.5 t | 30.7 t |
| 2 | 24.3 t | 25.3 t | 24.2 t | 25.1 t | 25.3 t | 25.0 t | 24.7 t |
| 3 | 78.9 d | 80.7 t | 79.0 d | 80.8 t | 79.0 d | 79.5 d | 79.9 d |
| 4 | 48.0 s | 48.8 s | 48.1 s | 49.0 s | 47.8 s | 49.6 s | 48.3 s |
| 5 | 56.4 d | 57.3 d | 56.4 d | 57.3 d | 56.3 d | 46.0 d | 49.6 d |
| 6 | 21.9 t | 23.0 t | 22.0 t | 23.0 t | 21.4 t | 18.9 t | 21.5 t |
| 7 | 41.9 t | 43.0 t | 41.9 t | 43.0 t | 40.9 t | 38.1 t | 34.8 t |
| 8 | 45.2 s | 45.5 s | 45.2 s | 45.5 s | 43.8 s | 42.3 s | 54.2 s |
| 9 | 56.1 d | 57.3 d | 56.0 d | 57.3 d | 55.1 d | 155.6 s | 75.6 s |
| 10 | 39.5 s | 40.5 s | 39.5 s | 40.5 s | 39.3 s | 38.4 s | 44.4 s |
| 11 | 18.5 t | 19.7 t | 18.5 t | 19.7 t | 18.5 t | 115.2 t | 26.6 t |
| 12 | 26.9 t | 27.2 t | 26.9 t | 27.2 t | 33.0 t | 38.0 t | 31.5 t |
| 13 | 49.0 d | 46.2 d | 48.9 d | 46.2 d | 43.7 d | 41.2 d | 41.1 d |
| 14 | 37.6 t | 38.0 t | 37.5 t | 38.0 t | 39.4 t | 44.9 t | 45.1 t |
| 15 | 57.5 t | 53.5 t | 57.5 t | 53.5 t | 48.7 t | 51.0 d | 134.2 d |
| 16 | 79.6 s | 82.8 s | 79.7 s | 82.8 s | 155.0 s | 158.2 s | 149.2 s |
| 17 | 24.1 q | 66.8 t | 24.1 q | 66.8 t | 103.0 t | 106.0 t | 60.9 t |
| 18 | 24.7 q | 24.5 q | 24.7 q | 24.5 q | 23.6 q | 24.3 q | 24.4 q |
| 19 | 178.1 s | 177.9 s | 178.3 s | 178.1 s | 180.0 s | 178.1 s | 176.0 s |
| 20 | 15.7 q | 16.1 q | 15.7 q | 16.1 q | 15.3 q | 23.3 q | 17.4 q |
| 3-ester | 167.9 s | 169.4 s | 167.9 s | 169.4 s | 173.0 s | 166.8 s | 166.9 s |
| 128.1 s | 129.3 s | 128.9 s | 129.5 s | 36.0 t | 118.4 d | 119.5 d | |
| 138.6 d | 139.0 d | 137.8 d | 138.8 d | 30.9 t | 145.3 d | 145.2 d | |
| 16.0 q | 16.0 q | 14.7 q | 14.4 q | 140.0 s | 134.5 s | 135.4 s | |
| 20.9 q | 20.9 q | 12.3 q | 12.1 q | 128.0 d | 128.4 d | 129.0 d | |
| 128.0 d | 129.1 d | 131.1 d | |||||
| 126.0 d | 130.5 d | 129.8 d |
| Compounds | Antimicrobial Activities (MIC in μg/mL) | ||||||
|---|---|---|---|---|---|---|---|
| M. albicans | 1R b | 2R | 3R | 4R | 5R | 535R | |
| 2 | 125 | >250 | >250 | >250 | >250 | >250 | >250 |
| 4 | 125 | 125 | >250 | >250 | >250 | >250 | >250 |
| 7 | 125 | >250 | >250 | >250 | >250 | >250 | >250 |
| 10 | 125 | >250 | >250 | >250 | >250 | >250 | >250 |
| 12 | 125 | >250 | >250 | >250 | >250 | >250 | >250 |
| 13 | 125 | >250 | >250 | >250 | >250 | >250 | >250 |
| Fluconazole a | 125 | >250 | >250 | >250 | >250 | >250 | >250 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.-F.; Ding, J.-Y.; Li, Y.-T.; Hao, X.-J.; Li, S.-L. Antimicrobial Diterpenoids of Wedelia trilobata (L.) Hitchc. Molecules 2016, 21, 457. https://doi.org/10.3390/molecules21040457
Li S-F, Ding J-Y, Li Y-T, Hao X-J, Li S-L. Antimicrobial Diterpenoids of Wedelia trilobata (L.) Hitchc. Molecules. 2016; 21(4):457. https://doi.org/10.3390/molecules21040457
Chicago/Turabian StyleLi, Shi-Fei, Jia-Yin Ding, Ya-Ting Li, Xiao-Jiang Hao, and Shun-Lin Li. 2016. "Antimicrobial Diterpenoids of Wedelia trilobata (L.) Hitchc" Molecules 21, no. 4: 457. https://doi.org/10.3390/molecules21040457
APA StyleLi, S. -F., Ding, J. -Y., Li, Y. -T., Hao, X. -J., & Li, S. -L. (2016). Antimicrobial Diterpenoids of Wedelia trilobata (L.) Hitchc. Molecules, 21(4), 457. https://doi.org/10.3390/molecules21040457