
Oxyresveratrol: Structural Modification and Evaluation of Biological Activities



Abstract
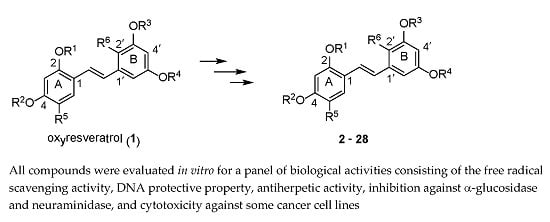
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
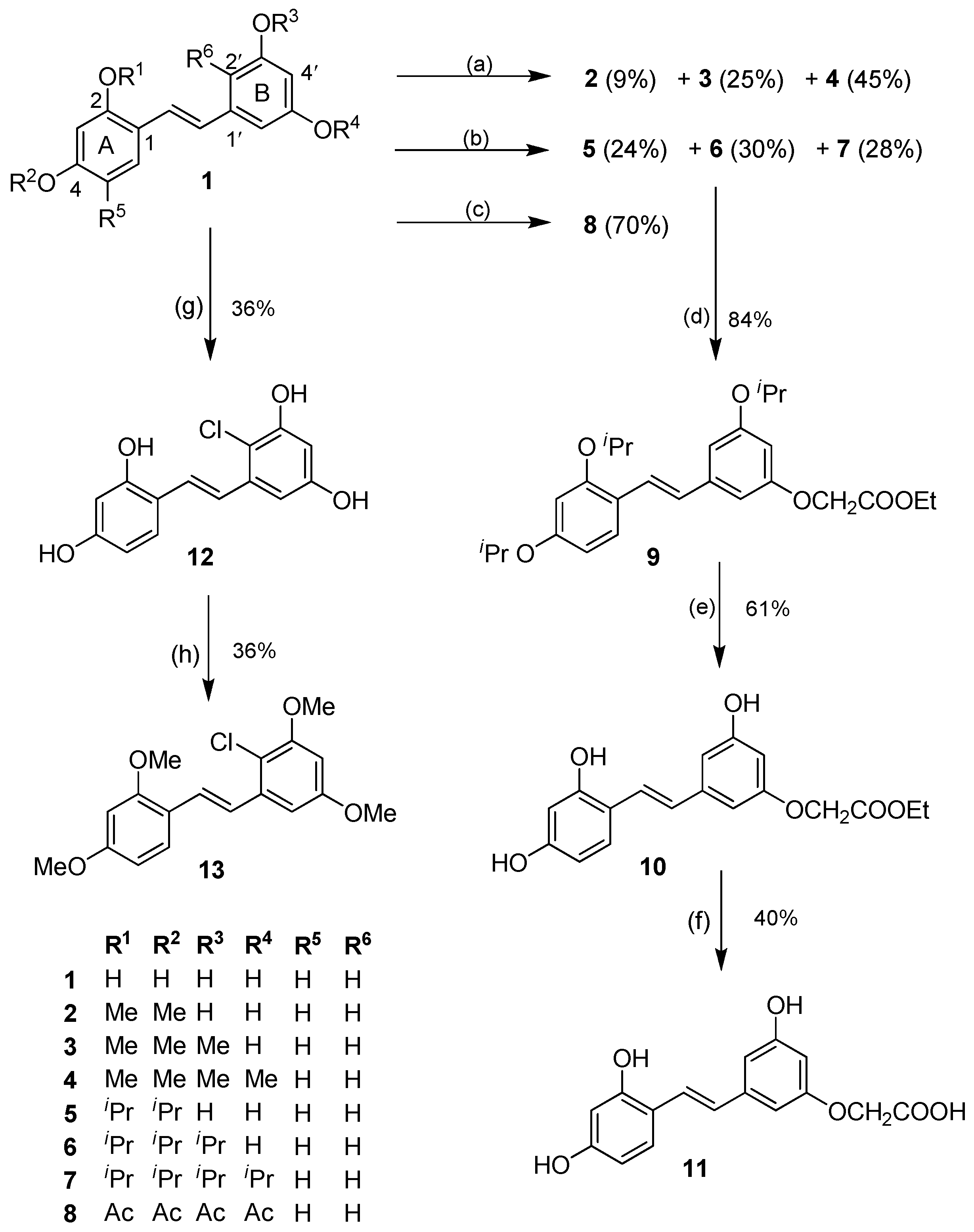
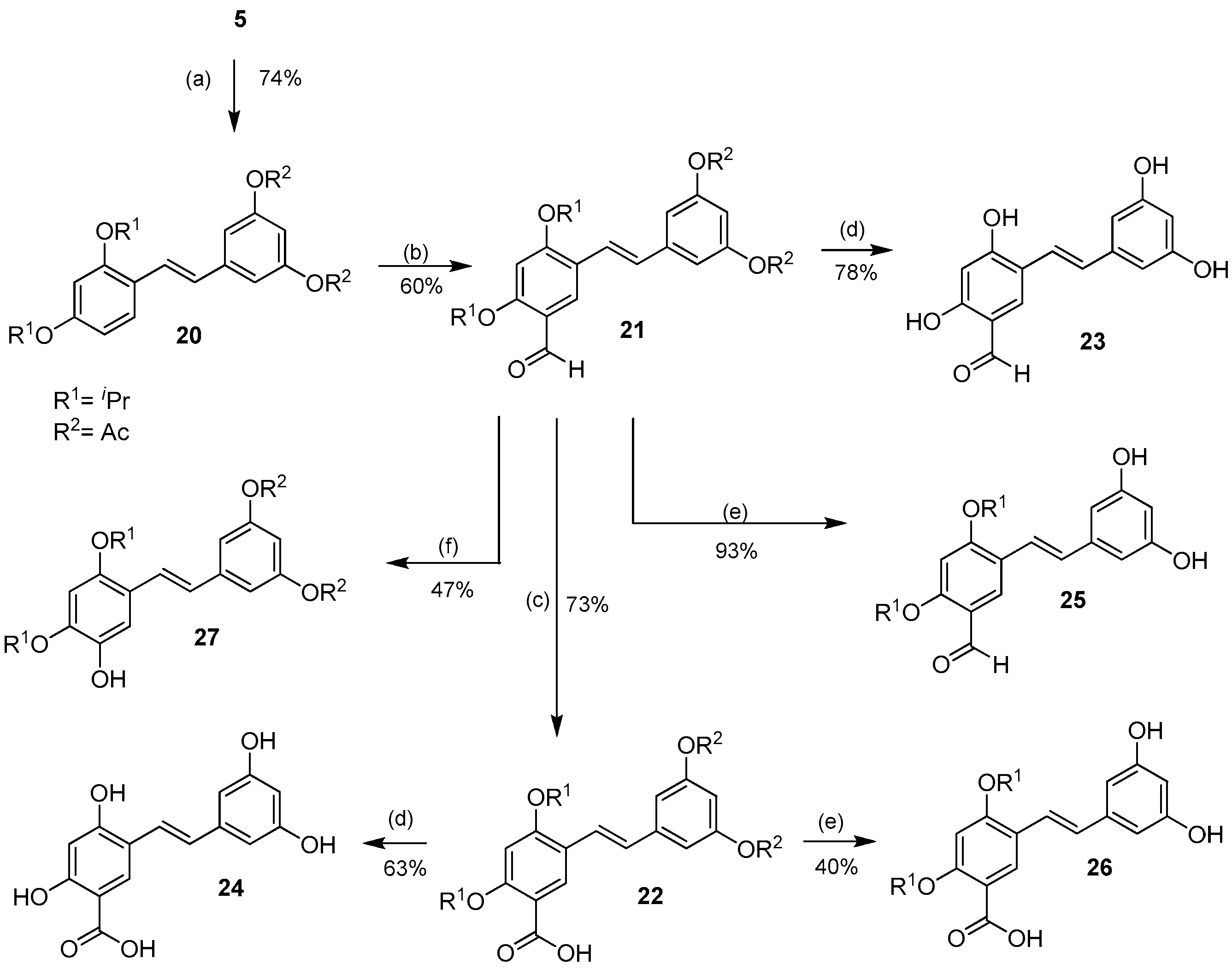
2.1.1. O-Alkylation/O-Acylation
2.1.2. Halogenation
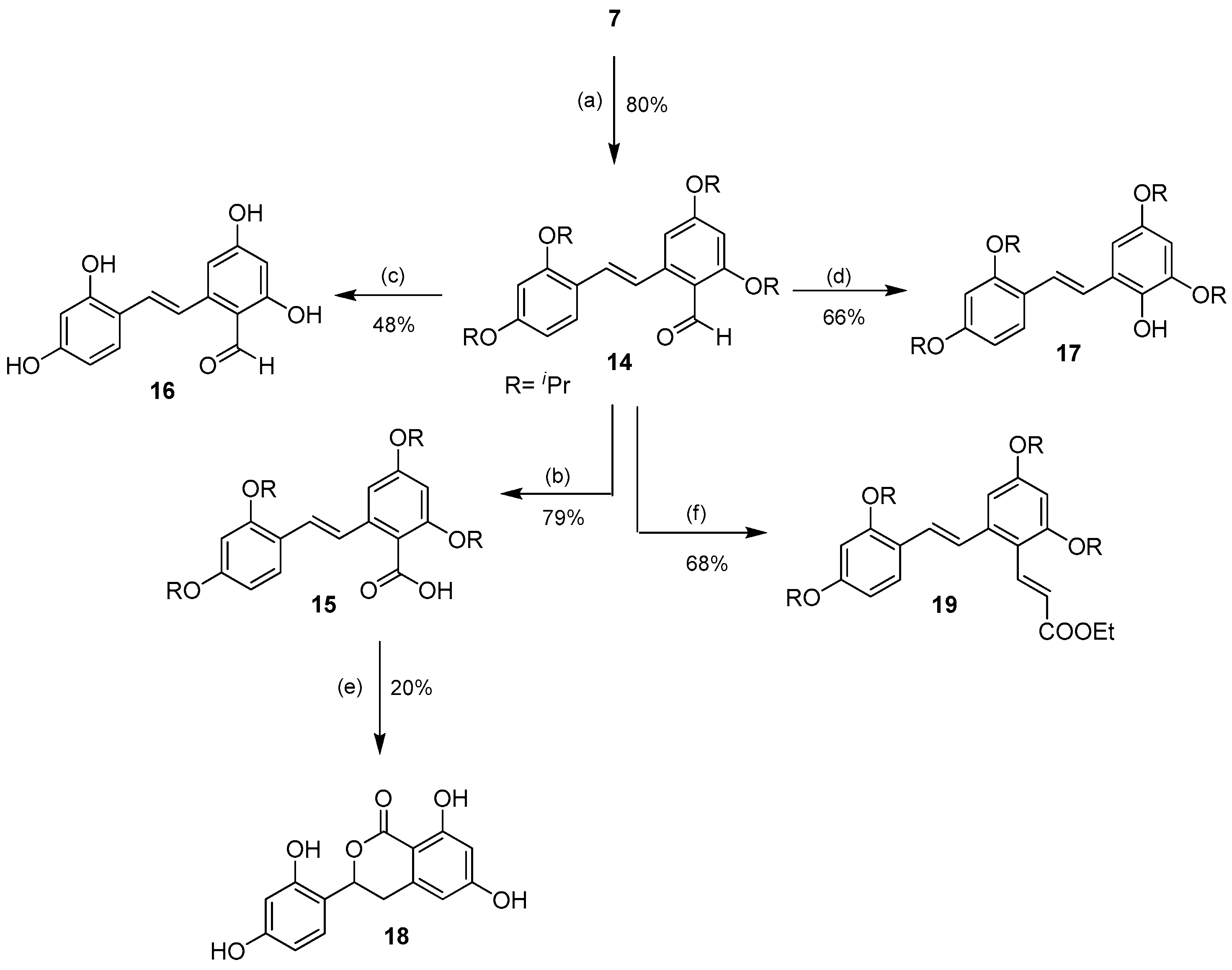
2.1.3. Aromatic Electrophilic Substitution
2.2. Biological Activity
2.2.1. Free Radical Scavenging Activity
2.2.2. DNA Protective Property
2.2.3. α-Glucosidase Inhibitory Activity
2.2.4. Neuraminidase Inhibitory Activity
2.2.5. Antiherpetic Activity
2.2.6. Cytotoxicity against Cancer Cells
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.2.1. Preparation of Compounds 2–4
3.2.2. Preparation of Compounds 5–7
3.2.3. Preparation of 2,3′,4,5′-Tetraacetoxystilbene (8)
3.2.4. Preparation of 3′-O-Carbethoxymethyl-2,4,5′-triisopropoxystilbene (9)
3.2.5. Preparation of 3′-O-Carbethoxymethyl-2,4,5′-trihydroxystilbene (10)
3.2.6. Preparation of 3′-O-Carboxymethyl-2,4,5′-trihydroxystilbene (11)
3.2.7. Preparation of 2′-Chloro-2,3′,4,5′-tetrahydroxystilbene (12)
3.2.8. Preparation of 2′-Chloro-2,3′,4,5′-tetramethoxystilbene (13)
3.2.9. Preparation of 2′-Formyl-2,3′,4,5′-tetraisopropoxystilbene (14)
3.2.10. Preparation of 2′-Carboxy-2,3′,4,5′-tetraisopropoxystilbene (15)
3.2.11. Preparation of 2′-Formyl-2,3′,4,5′-tetrahydroxystilbene (16)
3.2.12. Preparation of 2′-Hydroxy-2,3′,4,5′-tetraisopropoxystilbene (17)
3.2.13. Preparation of 3-(2,4-Dihydroxyphenyl)-6,8-dihydroxyisochroman-1-one (18)
3.2.14. Preparation of 2′-(E)-Carbethoxyethenyl-2,3′,4,5′-tetraisopropoxystilbene (19)
3.2.15. Preparation of 3′,5′-Diacetoxy-2,4-diisopropoxystilbene (20)
3.2.16. Preparation of 3′,5′-Diacetoxy-5-formyl-2,4-diisopropoxystilbene (21)
3.2.17. Preparation of 3′,5′-Diacetoxy-5-carboxy-2,4-diisopropoxystilbene (22)
3.2.18. Preparation of 5-Formyl-2,3′,4,5′-tetrahydroxystilbene (23)
3.2.19. Preparation of 5-Carboxy-2,3′,4,5′-tetrahydroxystilbene (24)
3.2.20. Preparation of 5-Formyl-3′,5′-dihydroxy-2,4-diisopropoxystilbene (25)
3.2.21. Preparation of 5-Carboxy-3′,5′-dihydroxy-2,4-diisopropoxystilbene (26)
3.2.22. Preparation of 3′,5′-Diacetoxy-5-hydroxy-2,4-diisopropoxystilbene (27)
3.3. Biological Activities
3.3.1. DPPH Radical Assay
3.3.2. Superoxide Radical Assay
3.3.3. Inhibitory Effect on Supercoiled DNA Breakage
3.3.4. Determination of Anti-Herpes Simplex Virus Activity
3.3.5. Neuraminidase (NA) Inhibition Assay
3.3.6. Determination of α-Glucosidase Inhibitory Activity
3.3.7. Determination of Cytotoxic Activity
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Riviere, C.; Pawlus, A.D.; Merillon, J.M. Natural stilbenoids: Distribution in the plant kingdom and chemotaxonomic interest in Vitaceae. Nat. Prod. Rep. 2012, 29, 1317–1333. [Google Scholar] [CrossRef] [PubMed]
- Gorham, J. The Biochemistry of the Stilbenoids; Chapman & Hall: London, UK, 1995; pp. 56–166. [Google Scholar]
- Niesen, D.B.; Hessler, C.; Seeram, N.P. Beyond resveratrol: A review of natural stilbenoids identified from 2009–2013. J. Berry Res. 2013, 3, 181–196. [Google Scholar]
- Rainsford, K.D. Influenza (“Bird Flu”), inflammation and anti-inflammatory/analgesic drugs. Inflammopharmacology 2006, 14, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.C.; Arruda, M.S.P.; Da Silva, E.A.S.; Da Silva, M.N.; Lemos, V.S.; Cortes, S.F. Inhibition of α-glucosidase and hypoglycemic effect of stilbenes from the Amazonian plant Deguelia rufescens var urucu (Ducke) A.M.G. Azevedo (Leguminosae). Planta Med. 2012, 78, 36–38. [Google Scholar] [CrossRef] [PubMed]
- Su, P.S.; Doerksen, R.J.; Chen, S.H.; Sung, W.C.; Juan, C.C.; Rawendra, R.D.S.; Chen, C.R.; Li, J.W.; Aisha; Huang, T.C.; et al. Screening and profiling stilbene-type natural products with angiotensin-converting enzyme inhibitory activity from Ampelopsis brevipedunculata var. hancei (Planch.) Rehder. J. Pharm. Biomed. 2015, 108, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.K.; Chen, S.D.; Chuang, Y.C.; Lin, H.Y.; Huang, C.R.; Chuang, J.H.; Wang, P.W.; Huang, S.T.; Tiao, M.M.; Chen, J.B.; et al. Resveratrol partially prevents rotenone-induced neurotoxicity in dopaminergic SH-SY5Y cells through induction of heme oxygenase-1 dependent autophagy. Int. J. Mol. Sci. 2014, 15, 1625–1646. [Google Scholar] [CrossRef] [PubMed]
- Maneechai, S.; Likhitwitayawuid, K.; Sritularak, B.; Palanuvej, C.; Ruangrungsi, N.; Sirisa-ard, P. Quantitative analysis of oxyresveratrol content in Artocarpus lakoocha and “Puag-Haad”. Med. Princ. Pract. 2009, 18, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Likhitwitayawuid, K.; Sornsute, A.; Sritularak, B.; Ploypradith, P. Chemical transformations of oxyresveratrol (trans-2,4,3′,5′-tetrahydroxystilbene) into a potent tyrosinase inhibitor and a strong cytotoxic agent. Bioorg. Med. Chem. Lett. 2006, 16, 5650–5653. [Google Scholar] [CrossRef] [PubMed]
- Tengamnuay, P.; Pengrungruangwong, K.; Pheansri, I.; Likhitwitayawuid, K. Artocarpus lakoocha heartwood extract as a novel cosmetic ingredient: Evaluation of the in vitro anti-tyrosinase and in vivo skin whitening activities. Int. J. Cosmetic Sci. 2006, 28, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Likhitwitayawuid, K.; Sritularak, B.; Benchanak, K.; Lipipun, V.; Mathew, J.; Schinazi, R.F. Phenolics with antiviral activity from Millettia erythrocalyx and Artocarpus lakoocha. Nat. Prod. Res. 2005, 19, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Lipipun, V.; Sasivimolphan, P.; Yoshida, Y.; Daikoku, T.; Sritularak, B.; Ritthidej, G.; Likhitwitayawuid, K.; Pramyothin, P.; Hattori, M.; Shiraki, K. Topical cream-based oxyresveratrol in the treatment of cutaneous HSV-1 infection in mice. Antivir. Res. 2011, 91, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Civitelli, L.; Marcocci, M.E.; Celestino, I.; Piacentini, R.; Garaci, E.; Grassi, C.; De Chiara, G.; Palamara, A.T. Herpes simplex virus type 1 infection in neurons leads to production and nuclear localization of APP intracellular domain (AICD): Implications for Alzheimer’s disease pathogenesis. J. Neurovirol. 2015, 21, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Ban, J.Y.; Jeon, S.Y.; Nguyen, T.T.H.; Bae, K.; Song, K.S.; Seong, Y.H. Neuroprotective effect of oxyresveratrol from Smilacis chinae rhizome on amyloid β protein (25–35)-induced neurotoxicity in cultured rat cortical neurons. Biol. Pharm. Bull. 2006, 29, 2419–2424. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.Y.; Kwon, S.H.; Seong, Y.H.; Bae, K.; Hur, J.M.; Lee, Y.Y.; Suh, D.Y.; Song, K.S. Beta-secretase (BACE1)-inhibiting stilbenoids from Smilax Rhizoma. Phytomedicine 2007, 14, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Yu, M.S.; Ho, Y.S.; Wang, M.; Chang, R.C.C. Dietary oxyresveratrol prevents parkinsonian mimetic 6-hydroxydopamine neurotoxicity. Free Radic. Biol. Med. 2008, 45, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, S.A.; Spina, M.G.; Lorenz, P.; Ebmeyer, U.; Wolf, G.; Horn, T.F.W. Oxyresveratrol (trans-2,3′,4,5′-tetrahydroxystilbene) is neuroprotective and inhibits the apoptotic cell death in transient cerebral ischemia. Brain Res. 2004, 1017, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Chatsumpun, M.; Chuanasa, T.; Sritularak, B.; Likhitwitayawuid, K. Oxyresveratrol protects against DNA damage induced by photosensitized riboflavin. Nat. Prod. Commun. 2011, 6, 41–44. [Google Scholar] [PubMed]
- Kongkamnerd, J. Development of Non-Cell Based Assays for Screening of Inhibitiors against Avian Influenza Neuraminidase. Ph.D. Thesis, Chulalongkorn University, Bangkok, Thailand, 2010. [Google Scholar]
- He, H.; Lu, Y.-H. Comparison of inhibitory activities and mechanisms of five mulberry plant bioactive components against α-glucosidase. J. Agric. Food Chem. 2013, 61, 8110–8119. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chen, G.; Lu, Z.; Zhang, J.; Guo, D.A. Identification of seven metabolites of oxyresveratrol in rat urine and bile using liquid chromatography/tandem mass spectrometry. Biomed. Chromatogr. 2010, 24, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Breuer, C.; Wolf, G.; Andrabi, S.A.; Lorenz, P.; Horn, T.F. Blood-brain barrier permeability to the neuroprotectant oxyresveratrol. Neurosci. Lett. 2006, 393, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.C. Comparison of the DNA-damaging property of photosensitised riboflavin via singlet oxygen (1O2) and superoxide radical (O2−) mechanisms. Toxicol. Lett. 1985, 26, 211–217. [Google Scholar] [CrossRef]
- Lu, C.-Y.; Wang, W.-F.; Lin, W.-Z.; Han, Z.-H.; Yao, S.-D.; Lin, N.-Y. Generation and photosensitization properties of the oxidized radical of riboflavin: A laser flash photolysis study. J. Photochem. Photobiol. B Biol. 1999, 52, 111–116. [Google Scholar] [CrossRef]
- Korycka-Dahl, M.; Richardson, T. Photogeneration of superoxide anion in serum of bovine milk and in model systems containing riboflavin and amino acids. J. Dairy Sci. 1977, 61, 400–407. [Google Scholar] [CrossRef]
- Cardoso, D.R.; Olsen, K.; Skibsted, L.H. Mechanism of deactivation of triplet-excited riboflavin by ascorbate, carotenoids, and tocopherols in homogeneous and heterogeneous aqueous food model systems. J. Agric. Food Chem. 2007, 55, 6285–6291. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Tano, K.; Takimoto, K.; Utsumi, H. Formation of 8-hydroxyguanine and 2,6-diamino-4-hydroxy-5-formamidopyrimidine in DNA by riboflavin mediated photosensitization. Biochem. Biophys. Res. Commun. 1998, 242, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Fang, J.S.; Lian, W.W.; Pang, X.C.; Liu, A.L.; Du, G.H. In vitro antiviral effects and 3D QSAR study of resveratrol derivatives as potent inhibitors of influenza H1N1 neuraminidase. Chem. Biol. Drug. Des. 2015, 85, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Sritularak, B.; De-Eknamkul, W.; Likhitwitayawuid, K. Tyrosinase inhibitors from Artocarpus lakoocha. Thai J. Pharm. Sci. 1988, 22, 149–155. [Google Scholar]
- Chuanasa, T.; Phromjai, J.; Lipipun, V.; Likhitwitayawuid, K.; Suzuki, M.; Pramyothin, P.; Hattori, M.; Shiraki, K. Anti-herpes simplex virus (HSV-1) activity of oxyresveratrol derived from Thai medicinal plant: Mechanism of action and therapeutic efficacy on cutaneous HSV-1 infection in mice. Antivir. Res. 2008, 80, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Sornsute, A. Isolation and Structure Modification of Oxyresveratrol from Artocarpus lakoocha for Tyrosinase Activity. Master’s Thesis, Chulalongkorn University, Bangkok, Thailand, 2006. [Google Scholar]
- Park, J.; Park, J.H.; Suh, H.J.; Lee, I.C.; Koh, J.; Boo, Y.C. Effects of resveratrol, oxyresveratrol, and their acetylated derivatives on cellular melanogenesis. Arch. Dermatol. Res. 2014, 306, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Likhitwitayawuid, K.; Klongsiriwet, C.; Jongbunprasert, V.; Sritularak, B.; Wongseripipatana, S. Flavones with free radical scavenging activity from Goniothalamus tenuifolius. Arch. Pharm. Res. 2006, 29, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, N.; De, B. Antioxidant activity of Piper betle L. leaf extract in vitro. Food Chem. 2004, 88, 219–224. [Google Scholar] [CrossRef]
- Lipipun, V.; Kurokawa, M.; Suttisri, R.; Taweechotipatr, P.; Pramyothin, P.; Hattori, M.; Shiraki, K. Efficacy of Thai medicinal plant extracts against herpes simplex virus type 1 infection in vitro and in vivo. Antivir. Res. 2003, 60, 175–180. [Google Scholar] [CrossRef]
- Potier, M.; Mameli, L.; Bélisle, M.; Dallaire, L.; Melançon, S.B. Fluorometric assay of neuraminidase with a sodium (4-methylumbelliferyl-α-d-N-acetylneuraminate) substrate. Anal. Biochem. 1979, 94, 287–296. [Google Scholar] [CrossRef]
- Tangdenpaisal, K.; Worayuthakarn, R.; Karnkla, S.; Ploypradith, P.; Intachote, P.; Sengsai, S.; Saimanee, B.; Ruchirawat, S.; Chittchang, M. Designing new analogs for streamlining the structure of cytotoxic lamellarin natural products. Chem. Asian J. 2015, 10, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Free Radical Scavenging Activity | DNA Protective Activity | |
|---|---|---|---|
| DPPH | Superoxide | ||
| 1 | 11.7 ± 0.4 | 303.1 ± 7.9 | 43.3 ± 6.7 |
| 2 | 77.0 ± 6.4 * | nd # | 39.4 ± 1.2 |
| 3 | nd | 120.1 ± 14.9 * | 6.3 ± 0.8 * |
| 5 | 147.7± 9.8 * | nd | 59.1 ± 9.9 |
| 10 | 9.7 ± 0.2 * | 154.9 ± 14.7 * | 19.9 ± 2.8 * |
| 11 | 19.4 ± 3.1 * | 81.9 ± 12.3 * | 28.6 ± 3.8 * |
| 12 | 14.7 ± 0.6 * | 98.4 ± 7.0 * | 32.0 ± 1.4 * |
| 16 | 16.5 ± 2.8 * | 43.4 ± 4.4 * | 32.2 ± 4.8 |
| 17 | 11.7 ± 0.3 | nd | nd |
| 18 | nd | 107.3 ± 8.7 * | 28.3 ± 4.3 * |
| 22 | nd | 17.7 ± 3.5 * | 81.4 ± 4.2 * |
| 23 | nd | 88.3 ± 9.7 * | 54.7 ± 8.3 |
| 24 | 48.3 ± 5.8 * | 38.6 ± 1.4 * | 79.4 ± 14.9 * |
| 25 | nd | nd | 111.8 ± 7.3 * |
| 26 | nd | nd | 104.7 ± 8.5 * |
| 27 | 7.0 ± 0.2 * | nd | 18.6 ± 3.7 * |
| Trolox | 8.7 ± 1.1 * | 293.5 ± 19.3 | 113.1 ± 4.6 * |
| Compounds | IC50 (µM) |
|---|---|
| 1 | 147.1 ± 16.8 |
| 2 | 40.2 ± 6.8 * |
| 3 | 32.8 ± 5.9 * |
| 6 | 107.0 ± 9.8 * |
| 10 | 182.3 ± 28.9 |
| 14 | 130.3 ± 14.9 |
| 15 | 123.5 ± 19.3 |
| 18 | 289.3 ± 40.8 * |
| 21 | 188.3 ± 25.1 |
| 22 | 207.4 ± 16.9 * |
| 23 | 344.7 ± 32.5 * |
| 26 | 226.8 ± 35.7 * |
| ACV | 1.6 ± 0.0 * |
| Compounds | IC50 (μM) | |||
|---|---|---|---|---|
| T47-D | HeLa | A549 | H69AR | |
| 1 | 152.7 ± 4.5 | 126.2 ± 0.3 | 159.8 ± 0.0 | nd # |
| 2 | 114.2 ± 2.0 * | 28.4 ± 7.0 * | 117.0 ± 11.1 * | 180.7 ± 3.4 |
| 3 | 87.1 ± 9.5 * | 13.1 ± 2.1 * | 52.4 ± 0.9 * | 36.7 ± 3.3 |
| 4 | nd | 12.1 ± 1.5 * | nd | 26.8 ± 7.6 |
| 5 | 117.4 ± 11.9 * | 11.0 ± 2.7 * | 55.5 ± 3.5 * | 39.2 ± 6.5 |
| 6 | 37.7 ± 12.9 * | 25.7 ± 2.1 * | 28.8 ± 5.1 * | 54.5 ±3.0 |
| 8 | 103.0 ± 4.3 * | nd | nd | nd |
| 10 | 88.6 ± 2.9 * | 100.9 ± 4.7 * | 137.7 ± 8.4 * | nd |
| 12 | 156.6 ± 6.0 | 33.4 ± 2.9 * | 120.7 ± 2.9 * | 111.2 ± 3.1 |
| 13 | nd | 18.7 ± 4.2 * | nd | 65.7 ± 3.2 |
| 14 | nd | 69.1 ± 3.3 * | nd | nd |
| 15 | 101.4 ± 9.1 * | 83.2 ± 9.1 * | nd | nd |
| 16 | 99.3 ± 3.1 * | 74.4 ± 4.8 * | 138.0 ± 3.1 * | nd |
| 20 | 81.3 ± 0.6 * | 5.7 ± 0.6 * | 51.7 ± 1.7 * | 38.0 ± 2.2 |
| 21 | 54.5 ± 2.0 * | 18.9 ± 0.7 * | 71.3 ± 3.2 * | nd |
| 23 | 82.9 ± 2.2 * | 77.9 ± 2.1 * | 143.2 ± 3.3 * | nd |
| 25 | 44.7 ± 2.6 * | 22.4 ± 0.4 * | 75.1 ± 0.7 * | 111.0 ± 2.3 |
| 27 | 83.2 ± 10.6 * | 79.8 ± 4.7 * | 104.4 ± 5.3 * | nd |
| Doxorubicin | 0.5 ± 0.00 * | 0.7 ± 0.2 * | 0.4 ± 0.1 * | 21.7 ± 0.9 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatsumpun, N.; Chuanasa, T.; Sritularak, B.; Lipipun, V.; Jongbunprasert, V.; Ruchirawat, S.; Ploypradith, P.; Likhitwitayawuid, K. Oxyresveratrol: Structural Modification and Evaluation of Biological Activities. Molecules 2016, 21, 489. https://doi.org/10.3390/molecules21040489
Chatsumpun N, Chuanasa T, Sritularak B, Lipipun V, Jongbunprasert V, Ruchirawat S, Ploypradith P, Likhitwitayawuid K. Oxyresveratrol: Structural Modification and Evaluation of Biological Activities. Molecules. 2016; 21(4):489. https://doi.org/10.3390/molecules21040489
Chicago/Turabian StyleChatsumpun, Nutputsorn, Taksina Chuanasa, Boonchoo Sritularak, Vimolmas Lipipun, Vichien Jongbunprasert, Somsak Ruchirawat, Poonsakdi Ploypradith, and Kittisak Likhitwitayawuid. 2016. "Oxyresveratrol: Structural Modification and Evaluation of Biological Activities" Molecules 21, no. 4: 489. https://doi.org/10.3390/molecules21040489
APA StyleChatsumpun, N., Chuanasa, T., Sritularak, B., Lipipun, V., Jongbunprasert, V., Ruchirawat, S., Ploypradith, P., & Likhitwitayawuid, K. (2016). Oxyresveratrol: Structural Modification and Evaluation of Biological Activities. Molecules, 21(4), 489. https://doi.org/10.3390/molecules21040489