
Synthesis and Anticancer Activities of Novel Guanylhydrazone and Aminoguanidine Tetrahydropyran Derivatives

Abstract
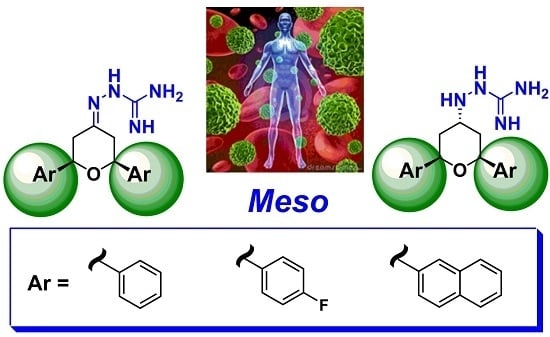
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biology
3. Materials and Methods
3.1. Chemistry
3.1.1. General Methods
3.1.2. General Procedure for Homoallylic Alcohol Synthesis
3.1.3. General Procedure for Synthesis of 4-Hydroxy-2,6-diaryl-tetrahydropyran
3.1.4. General Procedure for Oxidation of 4-Hydroxy-2,6-diaryl Tetrahydropyrans
3.1.5. General Procedure for the Preparation of Guanylhydrazone (2–4)
3.1.6. General Procedure for the Preparation of Aminoguanidine (5–7)
3.2. Pharmacology
3.2.1. Preparation of Stock Solutions
3.2.2. Cell Culture
3.2.3. Cell Viability–MTT
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Varmus, H. The New Era in Cancer Research. Science 2006, 312, 1162. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin. 2009, 59, 225–249. [Google Scholar] [PubMed]
- WHO Websites. Available online: http://www.who.int/cancer/en (accessed on 23 August 2015).
- Cancer Trends Progress Report. Available online: http://progressreport.cancer.gov (accessed on 22 June 2015).
- Verweij, J.; Jonge, M.J.A. Achievements and future of chemotherapy. Eur. J. Cancer 2000, 36, 1479–1487. [Google Scholar] [CrossRef]
- Carrillo, R.; Leon, L.G.; Martín, T.; Martín, V.S.; Padrón, J.M. Synthesis and antiproliferative activity of (2R,3R)-disubstituted tethahydropyrans. Bioorg. Med. Chem. Lett. 2006, 16, 6135–6138. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, R.; Leon, L.G.; Martín, T.; Martín, V.S.; Padrón, J.M. Synthesis and antiproliferative activity of (2R,3R)-disubstituted tetrahydropyrans. Part 2: Effect of side chain homologation. Bioorg. Med. Chem. Lett. 2007, 17, 780. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Bhardwaj, A. Mono-, Di-, and Triaryl Substituted Tetrahydropyrans as Cyclooxygenase-2 and Tumor Growth Inhibitors. Synthesis and Biological Evaluation. J. Med. Chem. 2010, 53, 3707–3717. [Google Scholar] [PubMed]
- Al-Tel, T.H. Design and synthesis of novel tetrahydro-2H-Pyrano[3,2-c]Pyridazin-3(6H)-one derivatives as potential anticancer agents. Eur. J. Med. Chem. 2010, 45, 5724–5731. [Google Scholar] [CrossRef] [PubMed]
- Al-Tel, T.H. Design, synthesis and qualitative structure–activity evaluations of novel hexahydropyrano[3,2-c][1,2]diazepin-3(4H)-one and tetrahydropyrano[3,2-b]pyrrol-2(1H)-one derivatives as anticancer agents. Eur. J. Med. Chem. 2010, 45, 4615–4621. [Google Scholar] [CrossRef] [PubMed]
- Judd, W.R.; Slattum, P.M.; Hoang, K.C; Bhoite, L.; Valppu, L.; Alberts, G.; Brown, B.; Roth, B.; Ostanin, K.; Huang, L. Discovery and SAR of Methylated Tetrahydropyranyl Derivatives as Inhibitors of Isoprenylcysteine Carboxyl Methyltransferase (ICMT). J. Med. Chem. 2011, 54, 5031–5047. [Google Scholar] [CrossRef] [PubMed]
- Vasconcellos, M.L.A.A.; Miranda, L.S.M. A reação de ciclização de Prins: Uma estratégia eficiente para síntese estereosseletiva de anéis tetraidropirânicos substituídos. Quím. Nova 2006, 29, 834–839. [Google Scholar] [CrossRef]
- Ahmeda, N.; Konduru, K.; Ahmad, S.; Owais, M. Synthesis of flavonoids based novel tetrahydropyran conjugates (Prins products) and their antiproliferative activity against human cancer cell lines. Eur. J. Med. Chem. 2014, 75, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Miranda, L.S.M.; Marinho, B.G.; Leitão, S.G.; Matheus, M.E.; Fernandes, P.D.; Vasconcellos, M.L.A.A. (±)-cis-(6-Ethyl-tetrahydropyran-2-yl)-formic acid: A novel substance with antinociceptive properties. Bioorg. Med. Chem. Lett. 2004, 14, 1573–1575. [Google Scholar] [CrossRef] [PubMed]
- Marinho, B.G.; Miranda, L.S.M.; Gomes, N.M.; Matheus, M.E.; Leitão, S.G.; Vasconcellos, M.L.A.A.; Fernandes, P.D. Antinociceptive action of (±)-cis-(6-ethyl-tetrahydropyran-2-yl)-formic acid in mice. Eur. J. Pharmacol. 2006, 550, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Marinho, B.G.; Miranda, L.S.M.; Marinho, B.G.; Vasconcellos, M.L.A.A.; Matheus, M.E.; Pereira, V.L.P.; Fernandes, P.D. Antinociceptive activity of (−)-(2S,6S)-(6-ethyl-tetrahydropyran-2-yl)-formic acid on acute pain in mice. Behav. Pharmacol 2011, 22, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Capim, S.L.; Carneiro, P.H.P.; Castro, P.C.; Barros, M.R.M.; Marinho, B.G.; Vasconcellos, M.L.A.A. Design, Prins-cyclization reaction promoting diastereoselective synthesis of 10 new tetrahydropyran derivatives and in vivo antinociceptive evaluations. Eur. J. Med. Chem. 2012, 58, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Capim, S.L.; Gonçalves, G.M.; dos Santos, G.C.M.; Marinho, B.G.; Vasconcellos, M.L.A.A. High analgesic and anti-inflammatory in vivo activities of six new hybrids NSAIAs tetrahydropyran derivatives. Bioorg. Med. Chem. 2013, 21, 6003–6010. [Google Scholar] [CrossRef] [PubMed]
- LaFrate, A.L.; Gunther, J.R.; Carlson, K.E.; Katzenellenbogen, J.A. Synthesis and biological evaluation of guanylhydrazone coactivator binding inhibitors for the estrogen receptor. Bioorg. Med. Chem. 2008, 16, 10075–10084. [Google Scholar] [CrossRef] [PubMed]
- Pignatellol, R.; Panicol, A.; Mazzonez, P.; Pinizzotto, M.R.; Garozzo, A.; Furneri, P.M. Schiff bases of N-hydroxy-N′-aminoguanidines as antiviral, antibacterial and anticancer agents. Eur. J. Med. Chem. 1994, 29, 781–785. [Google Scholar]
- Basua, A.; Sinha, B.N.; Saiko, P.; Graser, G.; Szekeres, T. N-Hydroxy-N′-aminoguanidine as anti-cancer lead molecule: QSAR, synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2011, 21, 3324–3328. [Google Scholar]
- Zhou, J.Y.; Jia, Y.; Sun, G.F.; Wu, S.H. Barbier-Type Allylation of Aldehydes and Ketones with Metallic Lead in Aqueous Media. Synth. Commun. 1997, 27, 1899–1906. [Google Scholar] [CrossRef]
- Silva, F.P.L.; Sabino, J.R.; Martins, F.T.; Vasconcellos, M.L.A.A. Diastereoselective syntheses via Prins cyclization, crystal structures determination and theoretical studies of cis-2,6-diphenyl-4-hydroxytetrahydropyran and analogues. J. Mol. Structure 2013, 1036, 478–487. [Google Scholar] [CrossRef]
- El-Azab, A.; Al-Omar, M.A.; Abdzel-Aziz, A.A.M.; Abdzel-Aziz, N.I.; El-Sayed, M.A.A.; Aleisa, A.M.; Sayed-Ahmed, M.M.; Abdel-Hamide, S.G. Design, synthesis and biological evaluation of novel quinazoline derivatives as potential antitumor agents: Molecular docking study. Eur. J. Med. Chem. 2010, 45, 4188–4198. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.D. A New Consistent Chromosomal Abnormality in Chronic Myelogenous Leukaemia identified by Quinacrine Fluorescence and Giemsa Staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Baccarani, M.; Cortes, J.; Pane, F.; Niederwieser, D.; Saglio, G.; Apperley, J.; Cervantes, F.; Deininger, M.; Gratwohl, A.; Guilhot, F.; et al. Chronic Myeloid Leukemia: An Update of Concepts and Management Recommendations of European LeukemiaNet. J. Clin. Oncol. 2009, 27, 6041–6051. [Google Scholar] [CrossRef] [PubMed]
- Slevin, M.L. The clinical pharmacology of etoposide. Cancer 1991, 67, 319–329. [Google Scholar] [CrossRef]
- Saleh, E.M. Inhibition of topoisomerase IIα sensitizes FaDu cells to ionizing radiation by diminishing DNA repair. Tumour Biol. 2015, 10, 8985–8992. [Google Scholar] [CrossRef] [PubMed]
- Torres, F.Q.J.; Díaz, J.G.; Carmona, A.J.; Estévez, F. Trifolin acetate-induced cell death in human leukemia cells is dependent on caspase-6 and activates the MAPK pathway. Apoptosis 2008, 13, 716–728. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Szücová, L.; Lukáš, S.; Dolez, K.; Zatloukal, M.; Greplová, J.; Galuszka, P.; Kryštof, V.; Voller, J.; Popa, I.; Massino, F.J.; et al. Synthesis, characterization and biological activity of ring-substituted 6-benzylamino-9-tetrahydropyran-2-yl and 9-tetrahydrofuran-2-ylpurine derivatives. Bioorg. Med. Chem. 2009, 17, 1938–1947. [Google Scholar] [CrossRef] [PubMed]
- Pedram, B.; van Oeveren, A.; Mais, D.E.; Marschke, K.B. A Tissue-Selective Nonsteroidal Progesterone Receptor Modulator: 7,9-Difluoro-5-(3-methylcyclohex-2-enyl)-2,2,4-trimethyl-1,2-dihydrochromeno[3,4-f]quinoline. J. Med. Chem. 2008, 51, 3696–3999. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Imunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 2–7 are available from the authors.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | HL-60 | K562 | HT-29 | MCF-7 | LMC | L929 | PBMC |
|---|---|---|---|---|---|---|---|
| 2 | 16.0 ± 4.6 | 13.6 ± 4.5 | 15.9 ± 3.7 | 52.0 ± 4.6 | nt | 18.1 ± 5.0 | 19.4 ± 3.2 |
| 3 | 12.5 ± 4.5 | 8.9 ± 4.0 | 11.2 ± 4.0 | 31.7 ± 4.7 | 7.5 ± 2.4 | 20.0 ± 4.2 | 14.3 ± 3.2 |
| 4 | 9.2 ± 4.6 | 7.5 ± 4.9 | 7.3 ± 4.7 | 16.7 ± 5.2 | nt | 10.8 ± 5.5 | 5.9 ± 3.3 |
| 5 | 42.0 ± 4.4 | 44.7 ± 4.8 | 46.9 ± 4.2 | >100 | nt | 65.8 ± 4.7 | 51.5 ± 3.0 |
| 6 | 85.4 ± 5.2 | 58.8 ± 4.1 | 59.5 ± 4.3 | 126.9 ± 4.3 | nt | 72.2 ± 3.9 | 72.7 ± 3.4 |
| 7 | 35.3 ± 5.2 | 15.5 ± 5.1 | 25.8 ± 4.8 | 43.3 ± 5.8 | 14.8 ± 2.4 | 39.7 ± 5.5 | 20.8 ± 3.5 |
| Etoposide | 5.8 ± 1.1 | >50 | >100 | >100 | nt | nt | >100 |
| Compound | HL-60 | K562 | HT-29 | MCF-7 | PBMCLMC | L929 | PBMC |
|---|---|---|---|---|---|---|---|
| 2 | 14.4 ± 5.2 | 31.0 ± 4.5 | 9.6 ± 4.7 | 34.8 ± 3.8 | nt | 23.9 ± 4.0 | 4.7 ± 2.9 |
| 3 | 14.9 ± 5.4 | 29.0 ± 4.9 | 6.0 ± 4.6 | 20.8 ± 4.0 | 14.6 ± 3.4 | 17.2 ± 4.7 | 6.7 ± 3.1 |
| 4 | 6.9 ± 4.9 | 19.2 ± 5.3 | 3.4 ± 4.7 | 13.7 ± 5.0 | nt | 3.9 ± 3.8 | 2.2 ± 2.9 |
| 5 | 41.4 ± 5.8 | 71.4 ± 5.0 | 19.9 ± 3.9 | 47.2 ± 3.5 | nt | 46.9 ± 4.4 | 11.4 ± 2.7 |
| 6 | 66.5 ± 5.3 | >100 | 36.9 ± 3.3 | 42.9 ± 4.2 | nt | 79.4 ± 3.8 | 12.5 ± 2.9 |
| 7 | 25.4 ± 5.3 | 43.6 ± 5.6 | 11.8 ± 4.3 | 24.14 ± 3.5 | 18.7 ± 2.7 | 16.88 ± 4.1 | 7.5 ± 3.4 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, F.P.L.; Dantas, B.B.; Faheina Martins, G.V.; De Araújo, D.A.M.; Vasconcellos, M.L.A.d.A. Synthesis and Anticancer Activities of Novel Guanylhydrazone and Aminoguanidine Tetrahydropyran Derivatives. Molecules 2016, 21, 671. https://doi.org/10.3390/molecules21060671
Silva FPL, Dantas BB, Faheina Martins GV, De Araújo DAM, Vasconcellos MLAdA. Synthesis and Anticancer Activities of Novel Guanylhydrazone and Aminoguanidine Tetrahydropyran Derivatives. Molecules. 2016; 21(6):671. https://doi.org/10.3390/molecules21060671
Chicago/Turabian StyleSilva, Fábio Pedrosa Lins, Bruna Braga Dantas, Gláucia Veríssimo Faheina Martins, Demétrius Antônio Machado De Araújo, and Mário Luiz Araújo de Almeida Vasconcellos. 2016. "Synthesis and Anticancer Activities of Novel Guanylhydrazone and Aminoguanidine Tetrahydropyran Derivatives" Molecules 21, no. 6: 671. https://doi.org/10.3390/molecules21060671
APA StyleSilva, F. P. L., Dantas, B. B., Faheina Martins, G. V., De Araújo, D. A. M., & Vasconcellos, M. L. A. d. A. (2016). Synthesis and Anticancer Activities of Novel Guanylhydrazone and Aminoguanidine Tetrahydropyran Derivatives. Molecules, 21(6), 671. https://doi.org/10.3390/molecules21060671