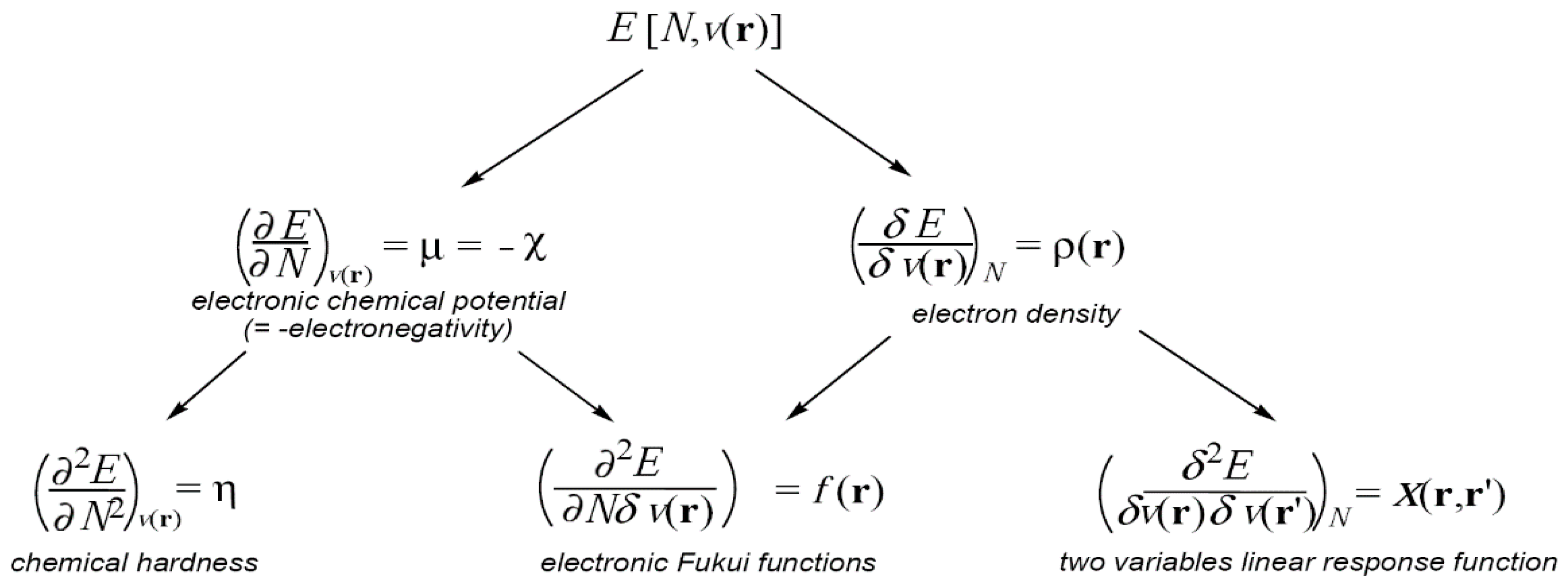
Conceptual DFT essentially relies on the fact that the ground state energy of an N-electron system as given by the Hohenberg-Kohn theorem (Equation (1)) can be considered as depending upon the number of electrons N and the external potential v(r), which are themselves determined solely by the density, in other words E[ρ(r)] = E[N;v(r)]. In this context, the responses of the system to changes of the number of its electrons, of the external potential or of both, provide information about its reactivity.
The E[N;
v(
r)] derivatives with respect to N and
v(
r) constitute a first series of reactivity indicators, the electronic chemical potential μ, which is the opposite of the electronegativity χ, the chemical hardness η, the Fukui function
f(
r) and the two variables linear response function χ(
r,
r′), as shown in the diagram given in
Figure 1. The left side properties in the diagram are global properties,
i.e., their values are the same wherever the position they are calculated, whereas the right side ones are local functions of one or two coordinate variables,
i.e., their values depend on the position where they are evaluated.
Herein, the electronic chemical potential μ, the electronegativity χ, the chemical hardness η, the electrophilicity ω and nucleophilicity N indices, and the Fukui f(r) and Parr P(r) functions, as well as their applications in the study of organic chemical reactivity, will be discussed. Non-local response functions such as X(r,r′) are out of the scope of this review.
2.1. Electronic Chemical Potential μ and Mulliken Electronegativity χ
In 1983, Parr defined the electronic chemical potential μ as the energy changes of the system with respect to the electron number N at a fixed external potential
v(
r),
i.e., the potential created by the nuclei (see Equation (3)) [
19,
20]. The electronic chemical potential μ is associated with the feasibility of a system to exchange electron density with the environment at the ground state.
Applying the finite difference approximation, the following simple expression is obtained:
where I and A are the ionisation potential and the electron affinity of an atom or molecule, respectively. Although a large number of experimental I values for organic molecules can be obtained, a very small number of experimental A values can be found. Using Koopmans theorem [
21] and Kohn–Sham formalism [
8] within the DFT, these energies can be approached by the frontier HOMO and LUMO energies as I by −
and A by −
. Consequently, the electronic chemical potential μ can be expressed as:
The electronic chemical potential μ of some reagents involved in Diels-Alder reactions is given in
Table 1. In this short series, while reagents such as tetracyanoethylene
1a, μ = −7.04 eV, located at the top of
Table 1, will act as strong electron-acceptor molecules, reagents located at the bottom such as dimethylvinyl amine
1j, μ = −1.85 eV, will act as strong electron-donor molecules. As can be seen, butadiene
1f, μ = −3.46 eV, and cyclopentadiene
1h, μ = −3.01 eV, present a similar electronic chemical potential μ as ethylene
1g, μ = −3.37 eV. Consequently, it is expected that the Diels-Alder reactions of dienes
1f or
1h with ethylene
1g will present a low polar character [
22]. The increase of the electron-withdrawing character of the substituent present in ethylene
1g, –CHO < –CN < –NO
2, decreases the electronic chemical potential μ of the corresponding ethylene derivative,
1e <
1d <
1c, thus increasing the polar character of the reactions towards dienes
1f or
1h.
The identification of the electronic chemical potential μ with the negative of Mulliken electronegativity, −χ, which is a measure of the resistance to electron density loss, offers a way to calculate electronegativity values for atoms and molecules. In this sense, it was an important step forward, as there was not a systematic way to evaluate electronegativities for atoms and molecules with the existing scales established by Pauling [
23,
24].
Electronegativity Equalisation Principle
According to the electronegativity equalisation principle, primarily formulated by Sanderson [
25,
26,
27,
28], “when two or more atoms initially different in electronegativity combine chemically, their electronegativities become equalised in the molecule”. The correctness of Sanderson’s principle immediately comes from the fact that the electronic chemical potential μ is a property of an equilibrium state. Consequently, when two atoms A and B, with
<
, approach one another, there is a flux of electron density from B, the less electronegative atom, towards A, the more electronegative one, to equilibrate the electronic chemical potential
in the new interacting system.
Accordingly, the electronic chemical potential μ allows the establishment of the flux direction of the GEDT [
15] along a polar reaction. Likewise, in a polar reaction involving two molecules, A and B, with
<
, the electron density flux will take place from molecule B, which has the higher μ, towards molecule A, which has the lower μ. Therefore, along a polar process, while A will act as the electron-acceptor,
i.e., the electrophile, B will act as the electron-donor,
i.e., the nucleophile. The larger the electronic chemical potential difference, Δμ, the larger the GEDT.
2.4. The Electrophilicity ω Index
In 1999, Parr defined the electrophilicity ω index [
36], which gives a measure of the energy stabilisation of a molecule when it acquires an additional amount of electron density, ΔN, from the environment. The electrophilicity ω index is given by the simple expression:
The electrophilicity ω index encompasses the tendency of an electrophile to acquire an extra amount of electron density, given by μ, and the resistance of a molecule to exchange electron density with the environment, given by η. Thus, a good electrophile is a species characterised by a high |μ| value and a low η value.
Besides, the maximum number of electrons that an electrophile can acquire is given by the expression [
36]:
The electrophilicity ω index has become a powerful tool for the study of the reactivity of organic molecules participating in polar reactions [
37]. A comprehensive study carried out in 2002 on the electrophilicity of a series of reagents involved in Diels-Alder reactions allowed establishing a single electrophilicity ω scale (see
Table 2) [
38]. In 2003, the three-atom-components (TACs) participating in [3+2] cycloaddition (32CA) reactions were studied using the electrophilicity ω index, allowing a rationalisation of their reactivity in polar processes [
39]. The electrophilicity ω scale allowed the classification of organic molecules as strong electrophiles with ω > 1.5 eV, moderate electrophiles with 0.8 < ω < 1.5 eV and marginal electrophiles with ω < 0.8 eV [
38].
Strong electrophiles such as 1,1-dicyanoethylene
2d, ω = 2.82 eV, and nitroethylene
2e, ω = 2.61 eV, presenting an electrophilicity ω index higher than 2.0 eV, participate easily in polar Diels-Alder reactions [
40]. Ethylene
2n, ω = 0.73 eV, is a marginal electrophile that is not able to participate in polar processes. Substitution of a hydrogen atom of ethylene
2n by an electron-withdrawing carbonyl group increases the electrophilicity ω index of acrolein
2f to 1.84 eV. Although acrolein is classified as a strong electrophile, it needs an additional electrophilic activation to participate in polar processes under mild conditions. Thus, even the coordination of a weak Lewis acid such as BH
3 to the carbonyl oxygen atom of acrolein
2f notably increases the electrophilicity ω index of the acrolein:BH
3 complex
2c to 3.20 eV. This electrophilic activation of acrolein in the complex accounts for the role of Lewis acid catalysts in organic reactions favouring the reactions to take place
via a more polar process [
40]. Cationic species such as
N-dimethylmethyleneammonium cation
2a, ω = 8.25 eV, are the most electrophilic organic species, participating in ionic organic reactions [
41].
Species such as ethylene
2n, ω = 0.73 eV, methyl vinyl ether
2q, ω = 0.42 eV or dimethylvinylamine
2r, ω = 0.27 eV, with an electrophilicity ω index below 0.80 eV, are classified as marginal electrophiles. Interestingly, diene species such as
N,
N′-dimethyl-1,3-buta-1,3-dien-1-amine
2o, or ethylenes such as
N,
N′-dimethyl-vinylamine
2r, participate in polar reactions as good nucleophiles. Consequently, for the short series of simple marginal electrophiles given in
Table 2, a good correlation between the inverse of the electrophilicity ω index and the nucleophilicity was established; for these marginal electrophilic species, the less electrophilic they are, the more nucleophilic they are.
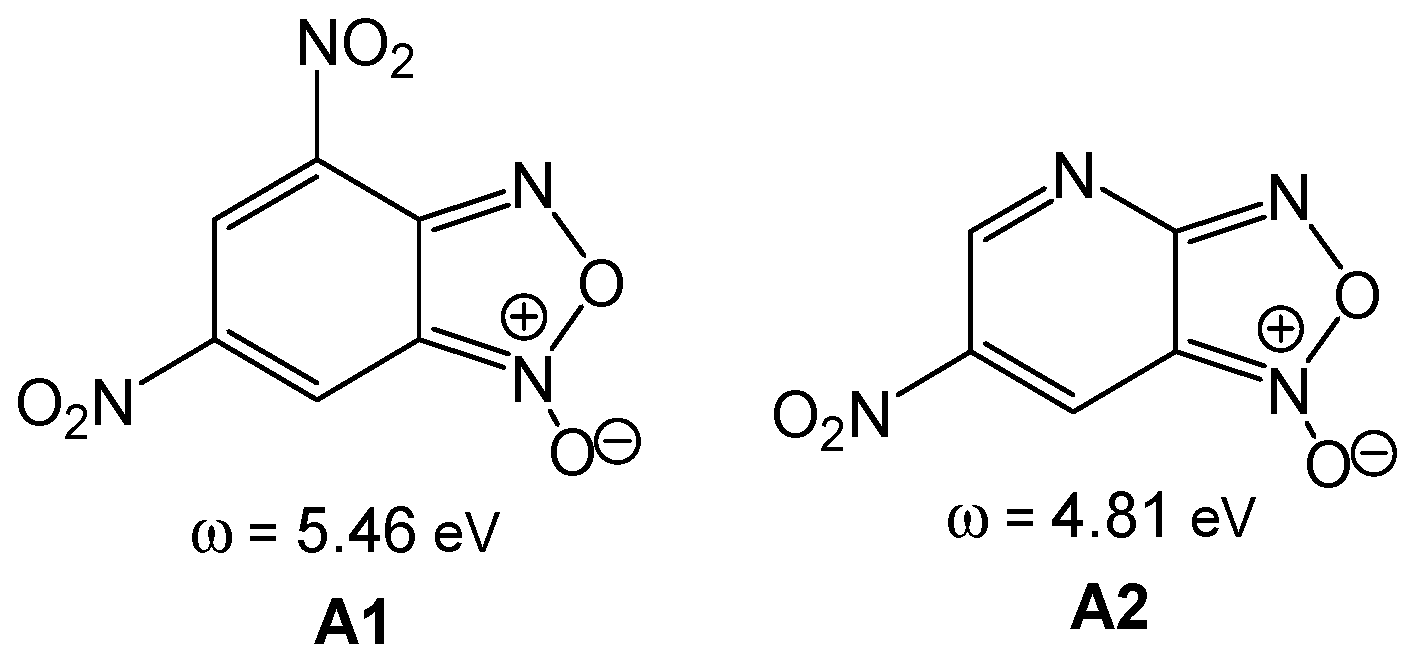
Nitrobenzofuroxans such as 4,6-dinitrobenzofuroxan
A1 and 4-aza-6-nitrobenzofuroxan
A2 (see
Scheme 1) are compounds with a high susceptibility to undergo nucleophilic addition or substitution processes with very weak nucleophiles [
42]. Terrier studied the high reactivity of these species, classifying them as
superelectrophiles [
43]. Thus, neutral species such as 4,6-dinitrobenzofuroxan
A1, ω = 5.56 eV, and 4-aza-6-nitrobenzofuroxan
A2, ω = 4.81 eV, presenting an electrophilicity ω index higher than 4.00 eV, are
superelectrophiles showing high reactivity in polar reactions [
44]. Note that
superelectrophilic species do not react in ionic reactions; the ionic character of an organic reaction is determined by the participation of cationic or anionic species throughout the reaction [
41].
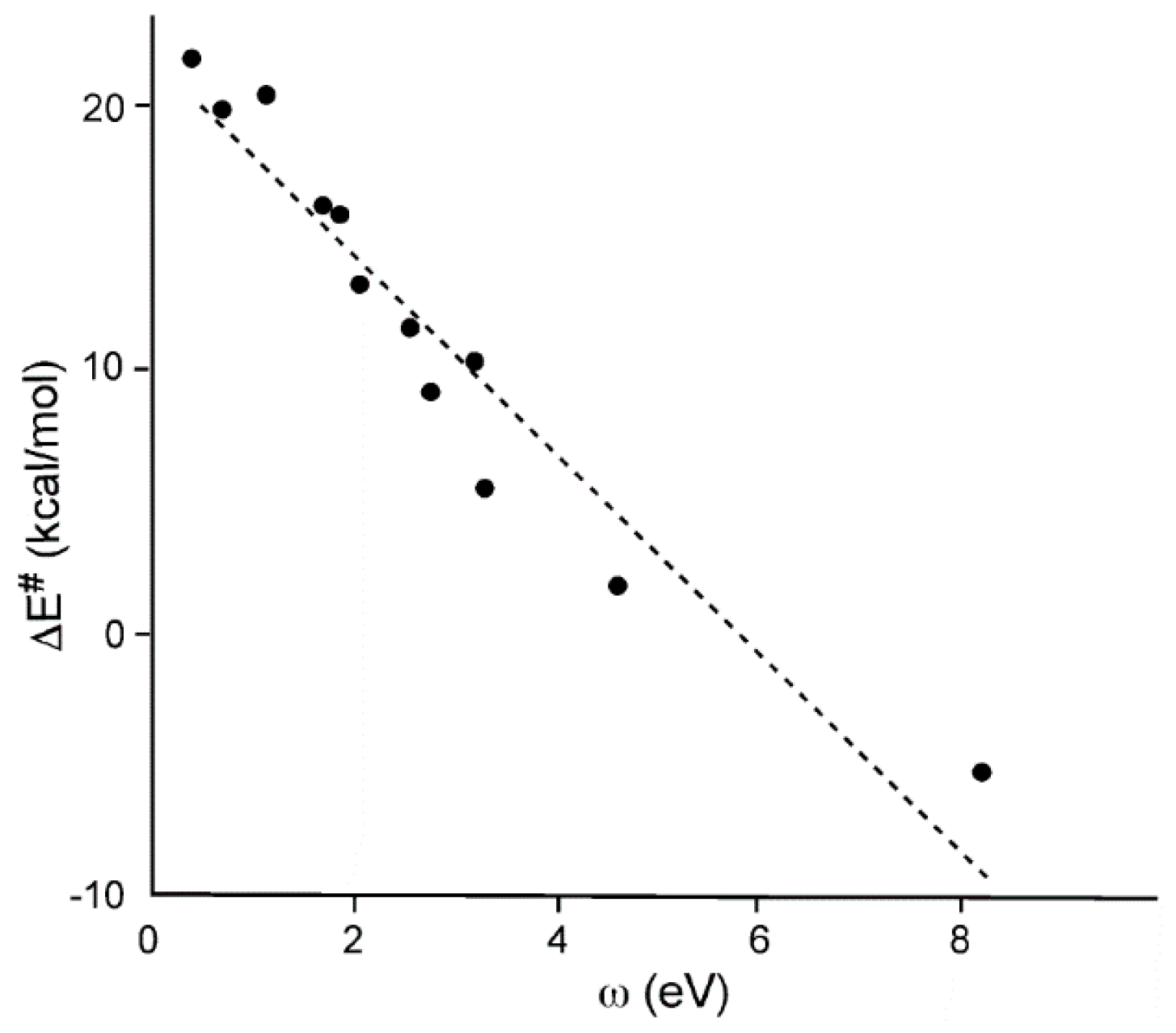
In 2009, Domingo studied the Diels-Alder reactions of cyclopentadiene
2l with twelve ethylenes of increased electrophilicity [
40]. For this short series, a good correlation between the computed activation energies and the electrophilicity ω index of these ethylene derivatives was found (R
2 = 0.92) (see
Figure 2).
As commented, in the short series of simple molecules participating in Diels-Alder reactions given in
Table 2, a good correlation between the inverse of the electrophilicity ω index of the marginal electrophiles and its expected nucleophilicity was found. In a polar reaction, the more electrophilic a reagent is and more nucleophilic the other is, the higher the GEDT that usually takes place. This behaviour accelerates polar reactions through more zwitterionic TSs. In this first study involving simple molecules, a good correlation between the difference of the electrophilicity of the two reagents, Δω, and the polar character of the reaction was established. Thus, it is expected that the Diels-Alder reaction between a pair of reagents located at the extreme positions of
Table 1 and presenting a Δω higher than 3.00 eV takes place easily via a highly polar reaction.
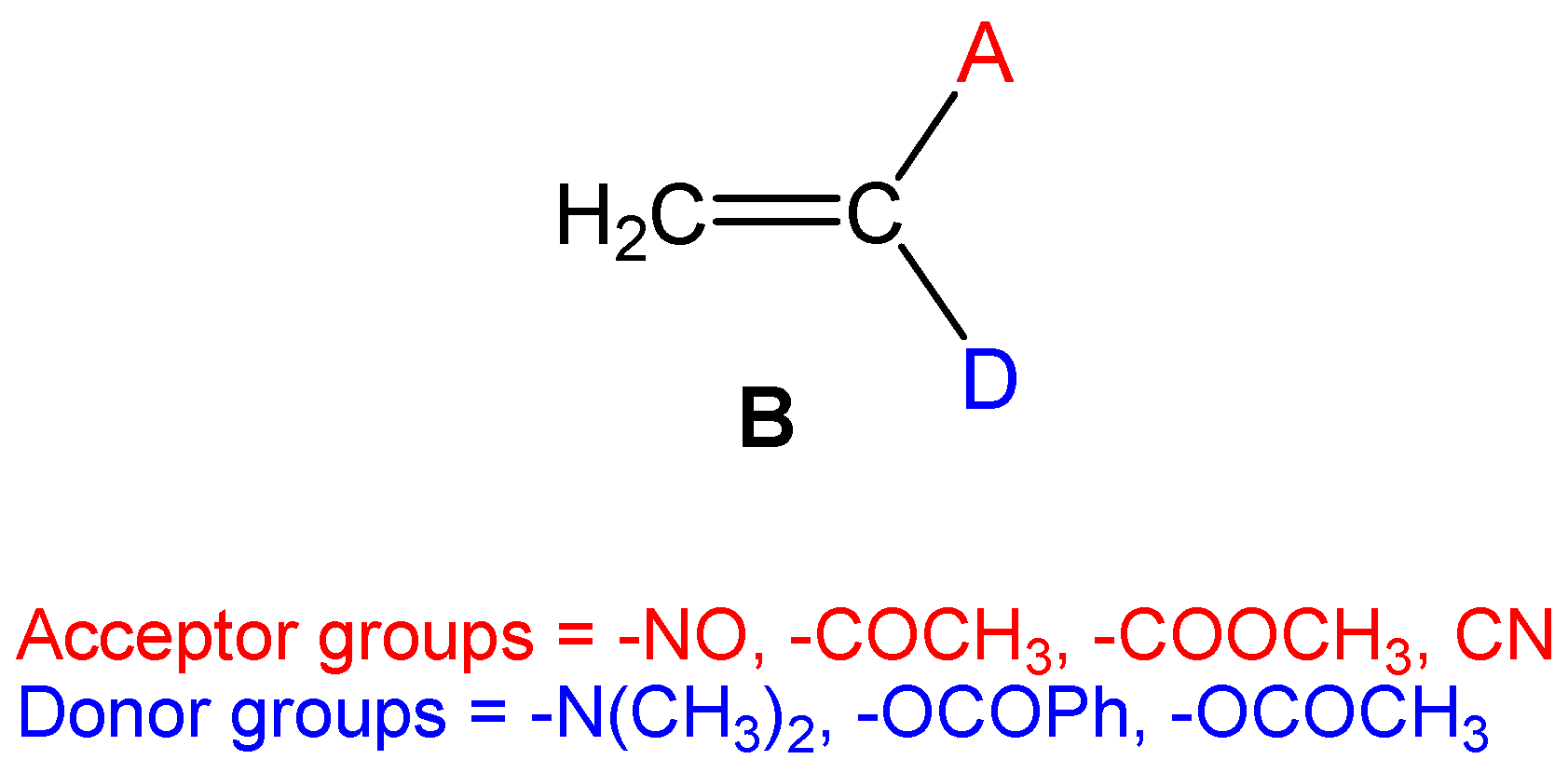
However, this prediction fails for more complex molecules such as captodative ethylenes B (see
Scheme 2), which display concurrently both electrophilic and nucleophilic behaviours (see later). For these species having two or more functional groups of a different electron demand, the value of the electrophilicity ω index does not correlate well with their expected nucleophilicity.
2.5. The Nucleophilicity N Index
While for the electrophilicity ω index only one expression has been established, several approaches for the nucleophilicity index have been gaven. In 1998, Roy [
45] proposed the “relative electrophilicity” (
+/
) and the “relative nucleophilicity” (
/
) descriptors for a k atom, as a way to locate the preferable electrophilic and nucleophilic reactive sites for the study of the intermolecular reactivity. The electrophilic and nucleophilic local softness
and
[
45] are given by the following equations:
where S is the chemical softness and
and
are the electrophilic and nucleophilic Fukui functions.
In 2003, Chattaraj proposed three generalised philicity
(α = +, − or o) indices to identify the most electrophilic, nucleophilic and radical sites in reactivity and regioselectivity studies [
46]. The condensed (or local) electrophilicity
and nucleophilicity
indices were related to the Parr global electrophilicity ω index and the corresponding Fukui functions by Equations (22) and (23).
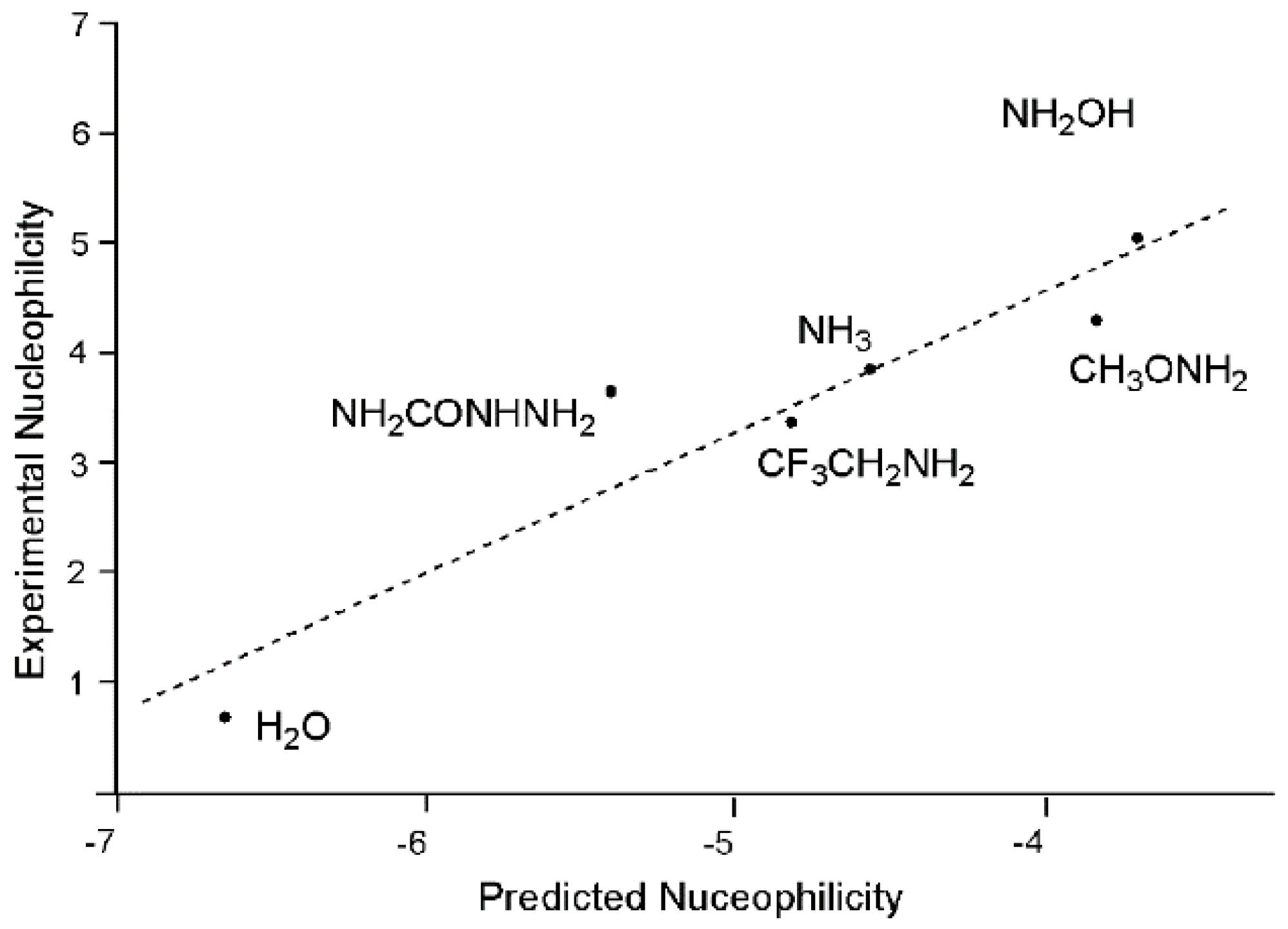
In 2003, Contreras described a method to estimate the relative nucleophilicity for a series of neutral and charged electron-donor species from their solution phase ionisation potential, I
s (Equation (24)) [
47].
A very good correlation between Mayr’s experimental nucleophilicity N
+ [
48] and the predicted solution nucleophilicity obtained by Equation (24) at the IPCM-MP2/6-311G(d,p) level for a series of first-row electron donors was found (see
Figure 3).
In 2007, Gázquez introduced the concepts of the electroaccepting, ω
+, and electrodonating, ω
−, powers as [
49]:
where ω
+ represents a measure of the propensity of a given system to accept electron density, while ω
− represents the propensity of this system to donate electron density.
In 2008, we proposed an empirical (relative) nucleophilicity
N index for closed-shell organic molecules based on the HOMO energies,
, obtained within the Kohn-Sham scheme as an approach to the gas phase I, and defined as [
50]:
The nucleophilicity
N index is referred to tetracyanoethylene (TCE)
2b, which is the most electrophilic neutral species in
Table 2,
i.e., the expected least nucleophilic neutral species. This choice allowed convenient handling of a nucleophilicity scale of positive values.
An analysis of a series of common nucleophilic species participating in polar organic reactions allowed a further classification of organic molecules as strong nucleophiles with
N > 3.0 eV, moderate nucleophiles with 2.0 ≤
N ≤ 3.0 eV and marginal nucleophiles with
N < 2.0 eV (see
Table 3) [
51].
As can be seen, while water,
3z,
N = 1.20 eV, is the poorest nucleophile of this series, hydrazine
3c,
N = 3.65 eV, is one of the best nucleophilic species. Alkyl substitution on the marginal nucleophilic ethylene
3x increases its nucleophilicity
N index from 1.86 eV to 3.35 eV in tetramethylethylene
3f, being classified as a strong nucleophile.
Table 3 also shows that the nucleophilicity
N index predicts correctly the activation/deactivation of benzene with the substitution. Finally, while ethylene
2n is a marginal electrophile, ω = 0.73 eV (see
Table 2), and a marginal nucleophile,
3x,
N = 1.86 eV (see
Table 3), 1,3-butadiene
2j is a moderate electrophile, ω = 1.05 eV (see
Table 2), and on the borderline of strong nucleophiles,
3j,
N = 2.89 eV (see
Table 3). Consequently, while ethylene
3x cannot participate in polar Diels-Alder reactions, butadiene
3j participates as a very good nucleophile. These results completely agree with the experimental observation that while the non-polar Diels-Alder reaction between butadiene and ethylene is one of the most unfavourable ones, only the electrophilic activation of ethylene makes the reaction feasible [
40].
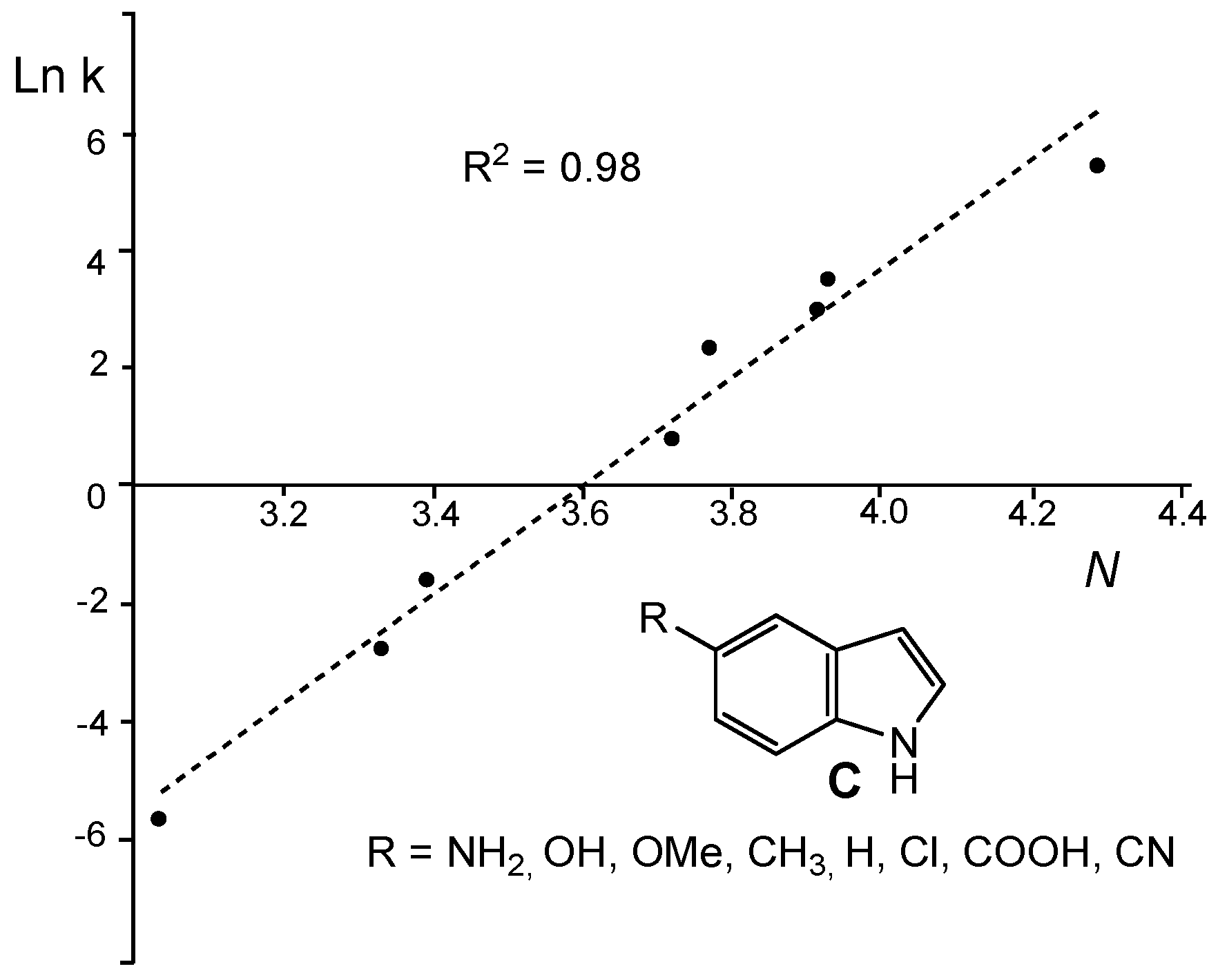
Several theoretical and experimental studies have evidenced the capability of the nucleophilicity
N index to predict the nucleophilic behaviour of organic molecules.
Figure 4 shows a good correlation (R
2 = 0.89) between the experimental Ln k for the reactions of a series of 5-substituted indoles
C with a benzhydryl cation [
52] and the nucleophilicity
N index of the former [
53].
Regarding Chattaraj’s philicity
indices, a greater
value corresponds to a better capability of accepting electron density, whereas a smaller value of
of a system makes it a better donor of electron density. In order to equalise with the general notion that “more is better”, in 2010, Roy proposed the nucleophilicity
N′ index as the inverse of Gázquez’s electrodonating ω
− power [
54]. Moreover, since the nucleophilicity
N′ index obtained as the inverse of ω
− was below 1, the authors later defined the nucleophilicity
N′ index as:
To validate the nucleophilicity
N′ index, the nucleophilicity values of sixty-nine commonly used arenes and heteroarenes were calculated and the corresponding values were compared with the Hammett σ and σ
p values [
55,
56]. In each case, an excellent correlation with the experimental results was obtained. It is interesting to remark that the selected arenes and heteroarenes series are simple molecules having only one functional group, for which, similar to the simple molecules given in
Table 1, the inverse of the Parr electrophilicity ω index also gives good correlations [
54].
In order to test the nucleophilicity
N and
N′ indices, (i) the series of captodative ethylenes
B given in
Scheme 2 and (ii) the series of 2,5-disubstituted bicyclic[2.2.1]hepta-2,5-dienes
D given in
Scheme 3, which display concurrently electrophilic and nucleophilic behaviours, were considered [
53].
While the two indices account well for the nucleophilic behaviour of organic molecules having only one substitution, the nucleophilicity
N index works better for more complex molecules presenting a dual reactivity, but the
N′ index fails. Thus, captodative ethylene
4b, having one of the most electron-withdrawing groups, the nitroso –NO one, and one of the most electron-releasing groups, the amino –N(CH
3)
2 one, presents a high electrophilicity ω index, 2.52 eV, being classified as a strong electrophile, and a high nucleophilicity
N index, 3.29 eV, also being classified as a strong nucleophile, in clear agreement with its expected dual reactivity (see
Table 4). However, the nucleophilicity
N′ index affords a low value,
N′ = 1.89 eV, for this species. A similar behaviour was found for captodative ethylene
4a. For captodative ethylenes
4c–
d, both nucleophilicity
N and
N′ indices yield similar values.
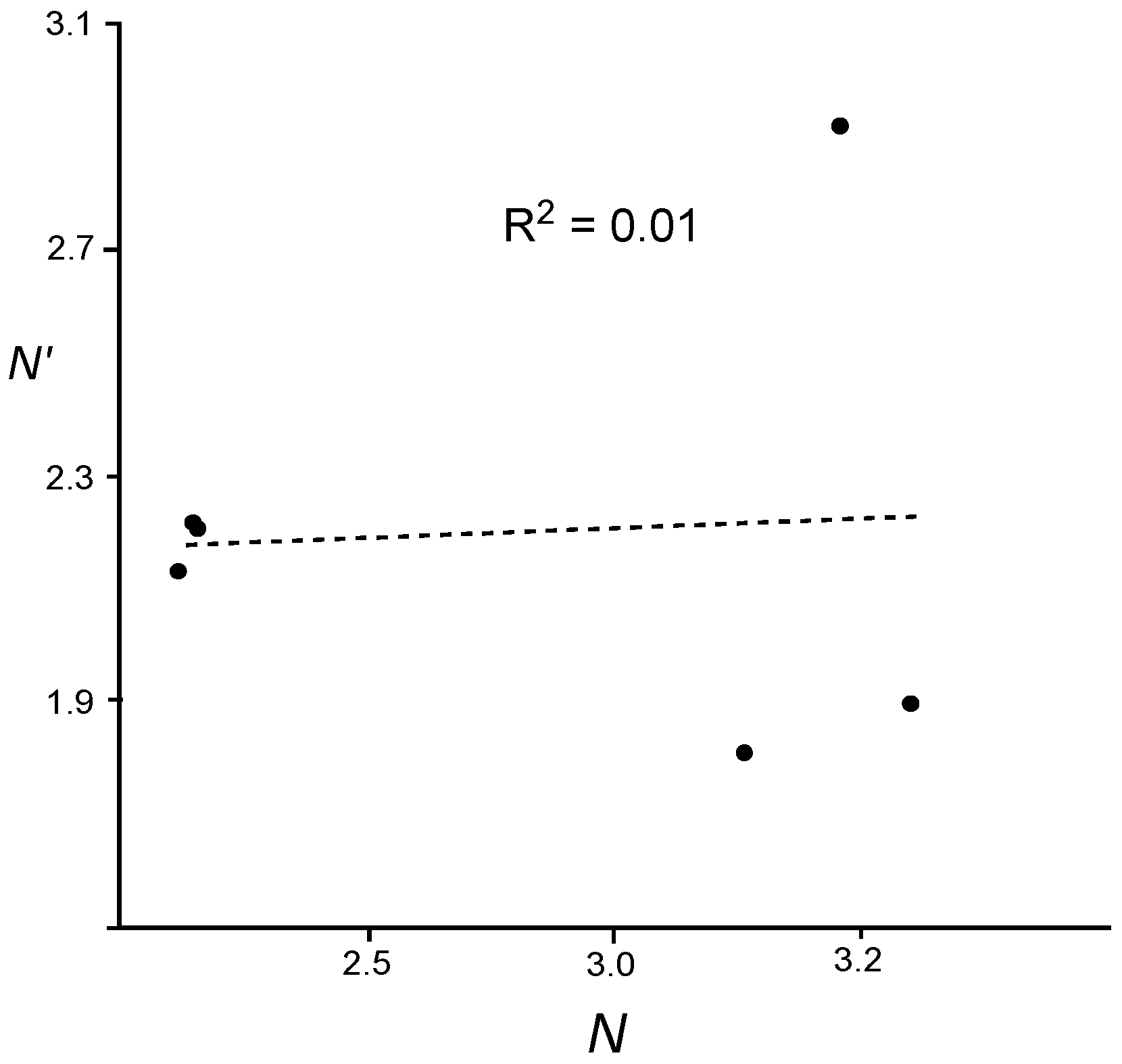
A plot of the nucleophilicity
N′ index
versus our nucleophilicity
N index for the series of captodative ethylenes
4a–
f given in
Table 4 shows that there is no lineal relation, R
2 = 0.01 (see
Figure 5). The nucleophilicity
N′ index fails to predict the nucleophilic character of captodative ethylenes
B presenting both strong electron-drawing and strong electron-donating substituents. In these ambiphilic species, while the electrophilicity ω is well reproduced, the nucleophilicity
N is underestimated by the
N′ index.
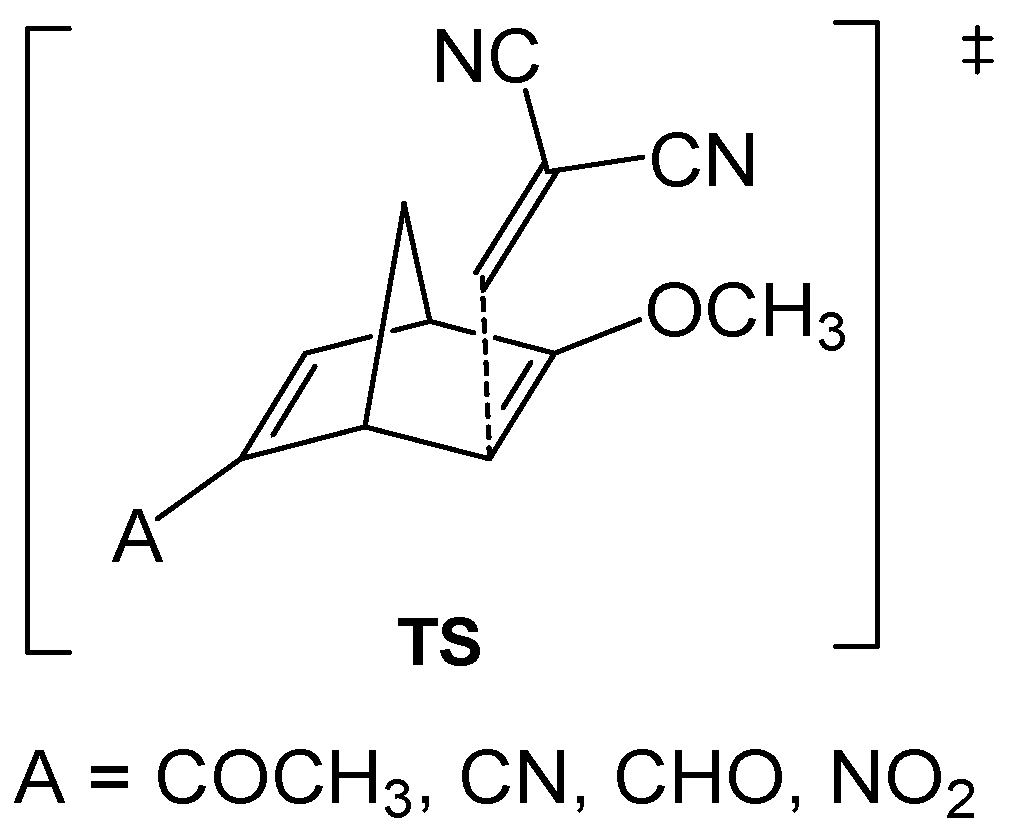
A comparative analysis of the nucleophilicity
N and
N′ indices of twelve 2-substituted-6-methoxy-bicyclic[2.2.1]hepta-2,5-dienes
D versus the computed activation energy associated with the nucleophilic attack on 1,1-dicyanoethylene
E (see
Scheme 3) showed that the nucleophilicity
N index yields a better correlation, R
2 = 0.99, than the
N′ one, R
2 = 0.85 (see
Figure 6) [
53].
These comparative studies asserted that the simple nucleophilicity
N index derived from E
HOMO as an approach of I is an adequate and effective measure of the nucleophilicity of simple and complex organic molecules displaying concurrently electrophilic and nucleophilic behaviours [
53].
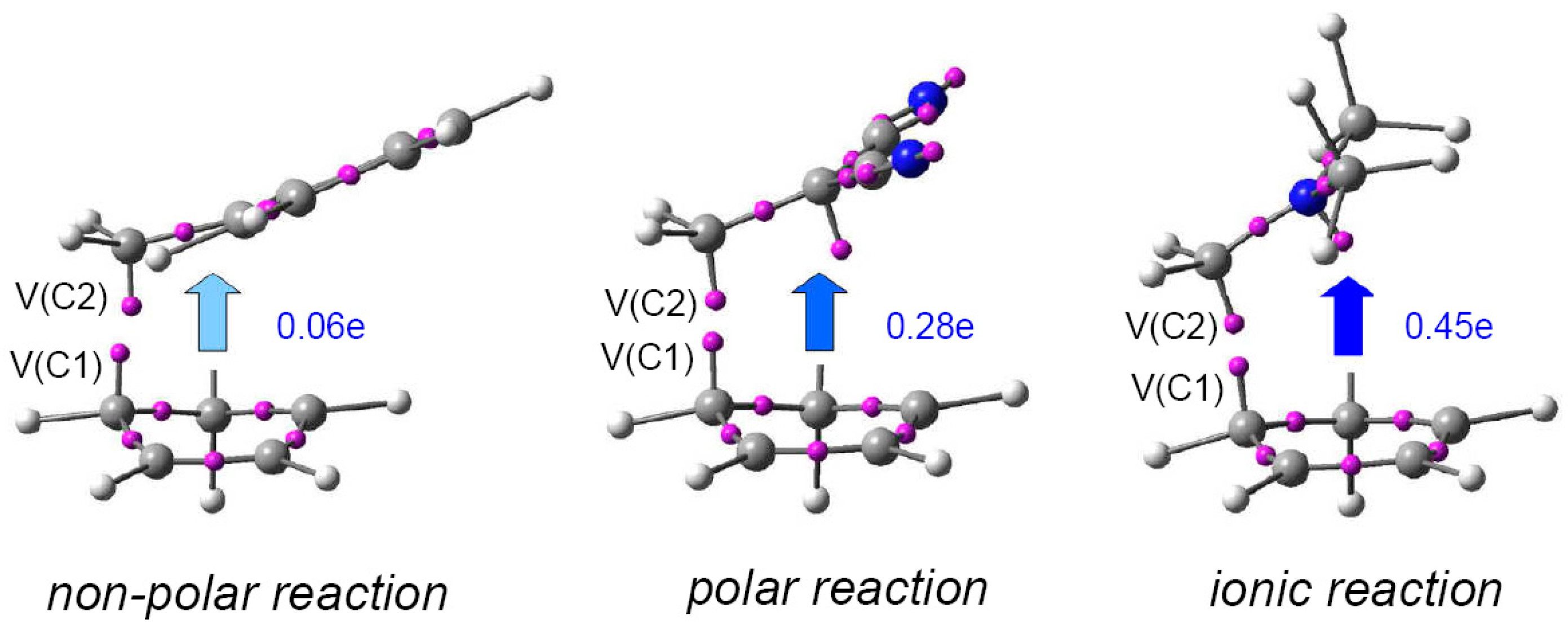
2.7. The Parr Functions P(r)
ELF topological analysis by way of a large number of studies devoted to the characterisation of the molecular mechanism of different organic reactions involving the participation of C–C double bonds made it possible to establish that in non-polar, polar and ionic reactions the formation of C–C single bonds usually takes place within the short range of 2.0–1.9 Å by a C-to-C coupling of two
pseudoradical centers [
14] generated along the reaction (see
Figure 7) [
15]. In polar and ionic reactions, the formation of these
pseudoradical centers is favoured by the GEDT that takes place from the nucleophile to the electrophile. In non-symmetric molecules, a non-symmetric electron density distribution takes place during the GEDT process. Thus, while in the nucleophilic species some atoms lose less electron density, in the electrophilic species some atoms gather more electron density. These relevant atoms correspond to the most nucleophilic and most electrophilic centers of the reactant molecules. Note that the larger the GEDT at the TS, the larger asynchronicity in the C–C single bond formation.
In case an amount equivalent to one electron is transferred, the nucleophile becomes a radical cation, while the electrophile becomes a radical anion. Interestingly, analysis of the atomic spin density (ASD) at the radical cation and the radical anion gives a picture of the distribution of the electron density in the electrophile and the nucleophile when they approach each other along the reaction progress.
Based on these observations, in 2014, Domingo proposed the Parr functions
P(
r) [
59,
60], which are given by the following equations:
and
where
(
r) is the ASD at the
r atom of the radical cation of a considered molecule and
(
r) is the ASD at the
r atom of the radical anion. Each ASD gathered at the different atoms of the radical cation and the radical anion of a molecule provides the local nucleophilic
and electrophilic
Parr functions of the neutral molecule.
Remarkably, prior to the Woodward-Hofmman proposal for the pericyclic mechanism, Woodward proposed in 1942 a mechanism for Diels-Alder reactions in which, before the formation of the two C–C single bonds in the final cycloadducts, an
ion pair complex is formed by one electron transfer process (see
ion pair complex [A·]
+ [B·]
− in
Scheme 4) [
61]. Woodward’s
ion pair complex mechanism can be seen as an earlier assertion of Domingo’s polar Diels-Alder mechanism [
40] in which a zwitterionic transition state is formed resulting from the GEDT. The analysis of the Parr functions can be seen as an analysis of the ASD at the separated [A·]
+ and [B·]
− fragments of Woodward’s
ion pair complex.
In
Table 5, electrophilic
and nucleophilic
Parr functions, and electrophilic
and nucleophilic
Parr and Yang [
34] (PY) and Yang and Mortier [
35] (YM) Fukui functions of four ethylene derivatives are given. A detailed analysis of the three models shows that the YM Fukui functions present some relevant errors. In spite of the fact that the three local functions are normalised, the sum of the YM Fukui functions corresponding to heavy atoms is below 1.0, in discrepancy with the sum of the Parr and the PY Fukui functions, which is closer to 1.0. As can be seen, electrophilic
PY and YM Fukui functions give the N and O atoms of nitroso ethylene
5a as the most electrophilic center, while electrophilic
Parr functions correctly predict the C1 carbon as the most electrophilic center of this electron-deficient ethylene. Similarly, for nitroethylene
5b, electrophilic
PY and YM Fukui functions at the N and O atoms are higher than those at the C1 carbon atom, while again, electrophilic
Parr functions correctly predict the C1 carbon as the most electrophilic center. For the electron-rich vinyl ether
5d, nucleophilic
YM Fukui functions give the O3 oxygen atom slightly more nucleophilic than the carbon C1, while nucleophilic
Parr functions correctly predict the C1 carbon as the most nucleophilic center. For a more comprehensive discussion see reference [
59].
Despite the similar electrophilic and nucleophilic local activations given by Parr and PY Fukui functions for the short series of compounds given in
Table 5, there are two reasons by which the use of the Parr functions, instead of the PY Fukui ones, is recommended: (i) both functions are conceptually different. The electrophilic
PY condensed Fukui functions are obtained from the LUMO electron density (see Equation (14)) as an approximation from the FMO theory. ELF bonding analyses concerning polar reactions involving the participation of C–C double bonds indicate that the C–C single bonds are formed through the C-to-C coupling of two
pseudoradical centers [
15] instead of a HOMO–LUMO donation process as suggested by the FMO theory [
62]. Consequently, the LUMO of electrophiles does not participate in the bond formation process; and (ii) Parr functions, obtained by performing simple unrestricted calculations at the radical anion and the radical cation of a molecule, are easier to obtain than PY condensed Fukui functions, which are obtained from the HOMO and LUMO coefficients and the corresponding overlapping integrals using specific programs [
63].
With the electrophilic
and nucleophilic
Parr functions at hand, the local electrophilicity
and the local nucleophilicity
indices were redefined as follows [
59]:
and
Therefore, a simple analysis of the Parr functions permits characterising the most electrophilic and the most nucleophilic centers in a molecule. These centers are those with the highest electron density developed along the GEDT involved in polar processes.
2.8. Parr Functions and Polar Reactivity
As commented above, electrophilic
or nucleophilic
Parr functions are able to explain regio- and chemoselectivities in most polar reactions. However, the functionality of Parr functions goes further and, interestingly, within Domingo’s C–C single bond formation model involving C–C double bonds, they are also able to explain the reactivity in polar organic reactions. Hence, the reactions of nitrones is an illustrative example of the applicability of Parr functions in this context. Nitrones are TACs participating in
zwitterionic-type (
zw-type) 32CA reactions [
64]. Most of the simplest zwitterionic TACs are characterised by a high nucleophilicity
N index,
N > 3.0 eV, being classified as strong nucleophiles; consequently, they can react with electron-deficient ethylenes [
65]. For these TACs to participate in
zw-type 32CA reactions towards electron-rich ethylenes, an electrophilic activation of the TAC is demanded. Thus, the simplest nitrone
6a, ω = 1.06 eV, is a moderate electrophile, and
C-phenyl-nitrone
6b, ω = 1.57 eV, is on the borderline of strong electrophiles (see
Table 6). Substitution of a hydrogen by a strong electron-withdrawing –NO
2 group in the phenyl substituent of the nitrone considerably increases the electrophilicity of nitrone
6c to 3.13 eV. However, in spite of its very high electrophilic activation, the
zw-type 32CA of
6c with 2-methylene-1,2-dioxolane, a strong nucleophile,
N = 3.53 eV
, presents a high activation energy, 13.2 kcal·mol
−1, which is similar to that associated with the non-polar 32CA reaction between the simplest nitrone
6a and ethylene, 13.1 kcal/mol.
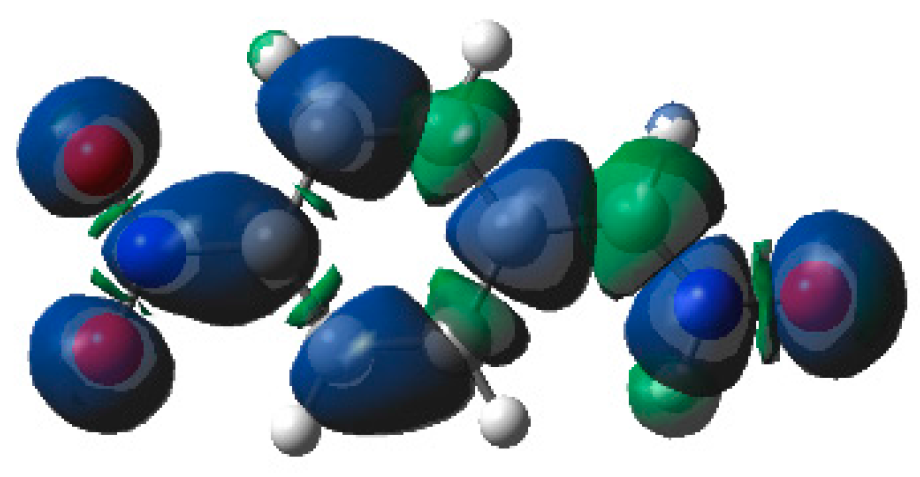
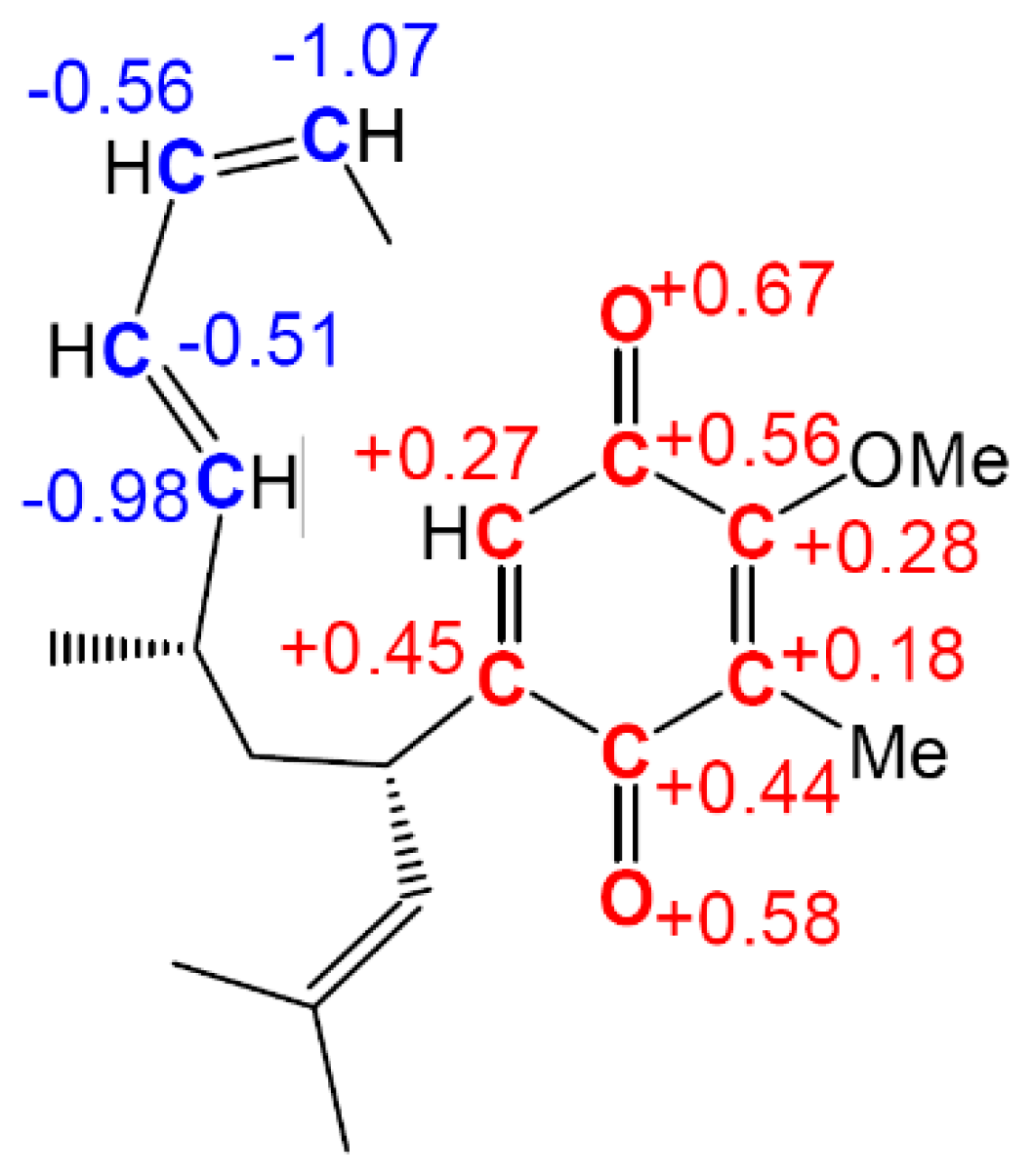
The low reactivity of the electrophilically activated nitrone
6c can be explained by an analysis of the electrophilic
Parr functions. As can be seen in
Figure 8, electrophilic Parr functions of nitrone
6c are mainly located at the
p-nitro-phenyl substituent (see the blue regions of the 3D representations of the ASD of the radical anion of nitrone
6c). While the nitrone oxygen atom presents a low electrophilic
Parr function,
= 0.18, the nitrone carbon atom is electrophilically deactivated,
= −0.07. Consequently, low values of the electrophilic
or nucleophilic
Parr functions at the atoms involved in the formation of C–C single bonds could account for the low reactivity of electrophilically and nucleophilically activated species in polar reactions.
2.10. Electrophilic and Nucleophilic Free Radicals
Non-polar organic reactions involving neutral radical species do not experience any electron density exchange along the reaction. However, the adequate substitution in the radical species may favour the electron density exchange, determining the chemical reactivity of these radical species. Consequently, a set of five DFT reactivity indices, namely, the global electrophilicity
and nucleophilicity
indices, the radical
Parr function and the local electrophilicity
and nucleophilicity
indices, have recently been proposed for the study of the reactivity of free radicals [
68].
Using Parr’s definition, the electrophilicity
index of free radicals at their ground state, namely:
was introduced, where µ° is the global electronic chemical potential and η° is the global chemical hardness of free radicals. Similar to μ and η, these quantities may be approached as:
and
where I° and A° correspond to the ionisation potential and the electron affinity of free radicals. Operational formulations for these quantities can be obtained through:
and
where
and
are the one electron energies of the frontier HOMO and LUMO for the electrons in the α and β spin state of the radicals and the relationships
I° =
and
A° =
were used.
On the other hand, and in analogy to our nucleophilicity
N index, we also introduced the global nucleophilicity
N° index of free radicals as:
where the dicyanomethyl (DCM) radical is taken as a reference to define a positive scale of global nucleophilicity of radicals.
For the local descriptors, the radical Parr function
is defined as:
where
(
r) is the ASD at the r atom of the neutral radical.
Therefore, the local electrophilicity
and nucleophilicity
indices for free radicals can be easily obtained by the following expressions:
where
and
are obtained from Equations (36) and (41), respectively.
The global radical indices were tested for a series of twenty-three free radicals being electrophilically and/or nucleophilically activated (see
Table 7). Thus, analysis of the electrophilicity
and nucleophilicity
indices for the substituted free radicals given in
Table 7 clearly shows that they correctly predict their electrophilic and nucleophilic behaviours. Interestingly, these global indices also account for the ambiphilic behaviour of free radicals, such as
7g and
7h, having concurrently an electron-withdrawing and electron-releasing substitution.
Analysis of the local electrophilicity
and nucleophilicity
indices for free radicals, together with analysis of the local electrophilicity
and nucleophilicity
indices for alkenes, allows explaining the regio- and chemoselectivity in radical additions of free radicals to alkenes. ELF topological analysis for the C–C bond formation along the nucleophilic addition of 2-hydroxyprop-2-yl free radical
7u to electrophilic methyl acrylate evidenced that the new C–C bond is formed through the C-to-C coupling of two radical centers, which were properly characterised through the use of the Parr functions [
68]. Besides, radical Parr functions
have also shown to be an applicable tool to analyse the most electrophilic and nucleophilic centers in cationic and anionic organic molecules participating in ionic reactions [
41,
69].





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
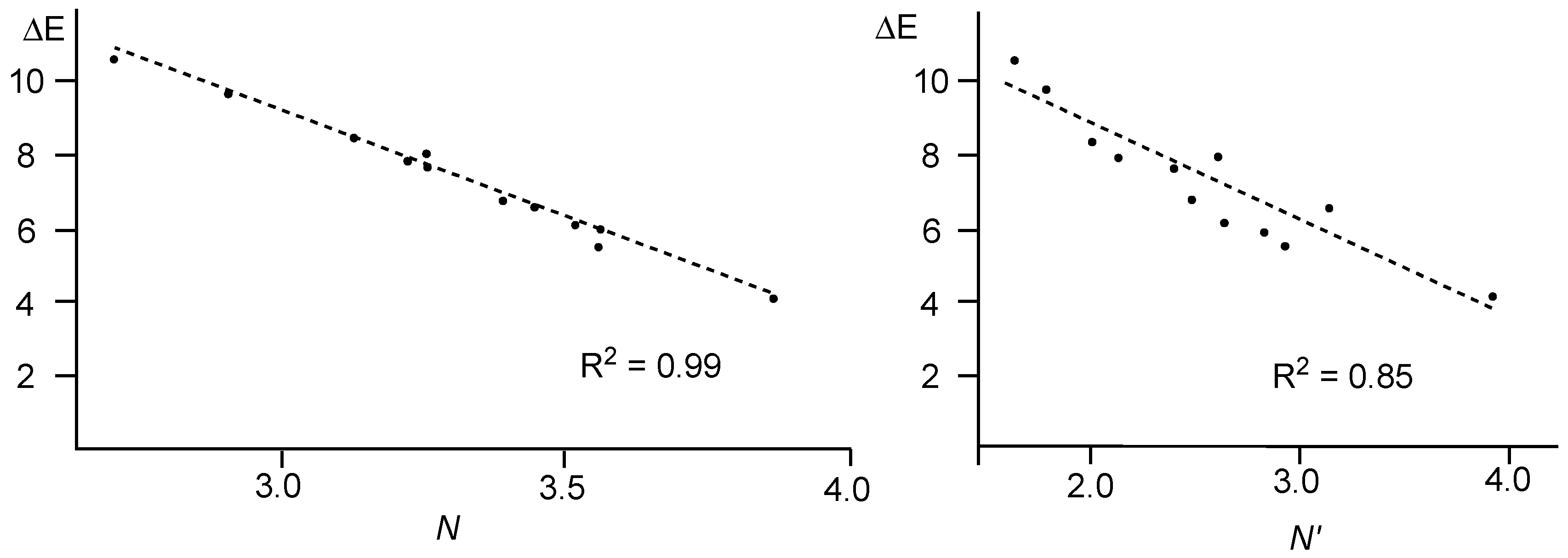{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
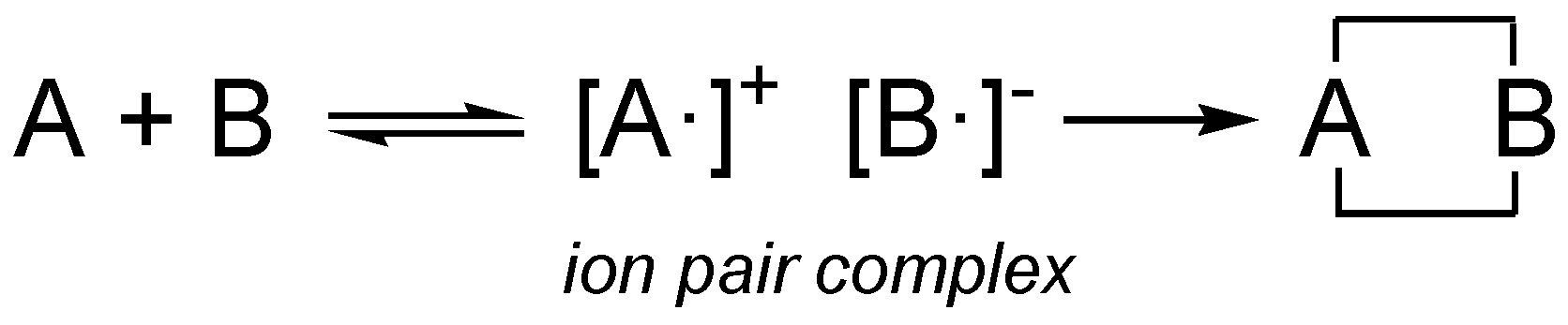{kind=link}