
A PDMS-Based Microfluidic Hanging Drop Chip for Embryoid Body Formation



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
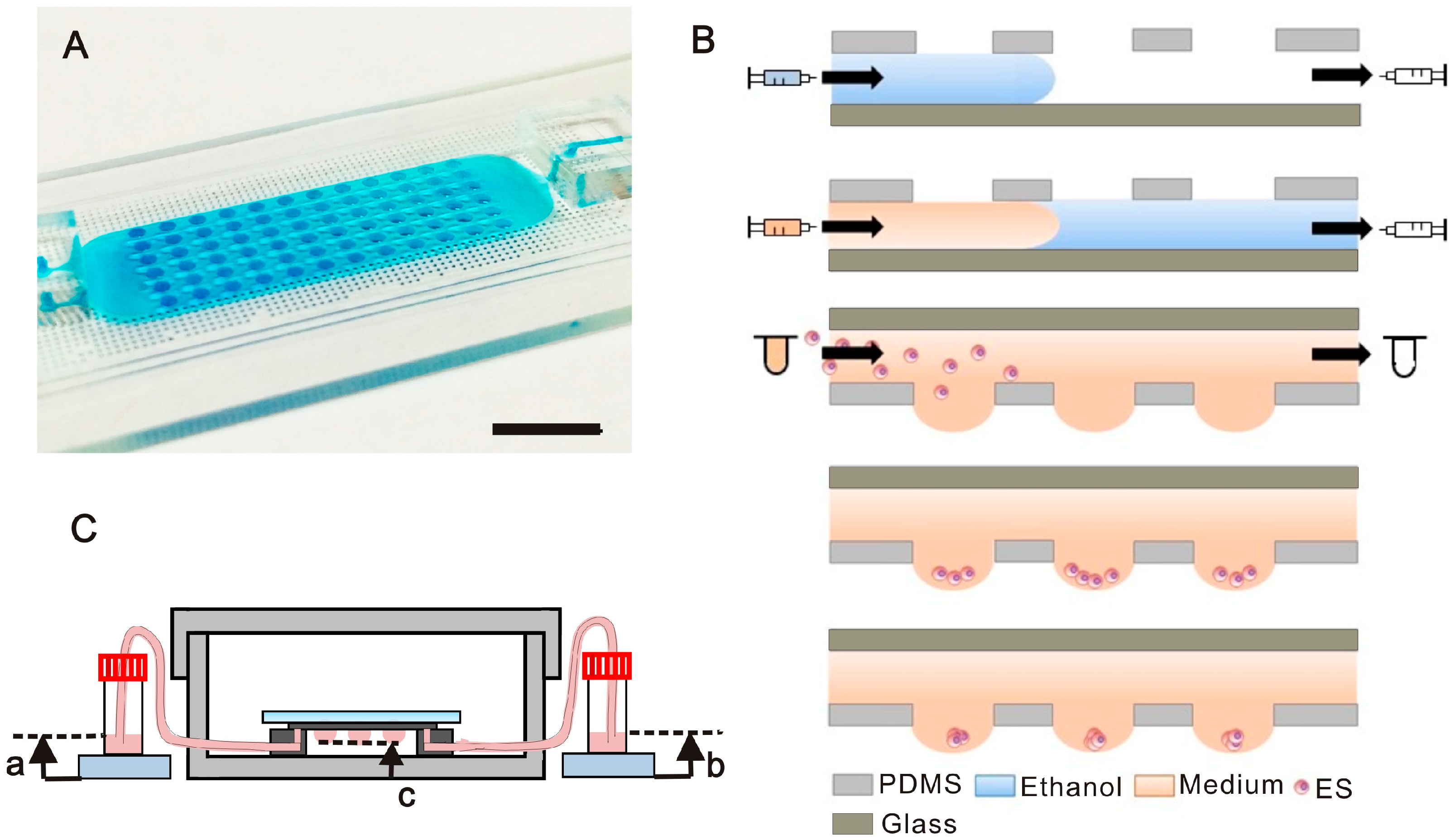
2.1. Chip Design and Experimental Process and Setup
2.2. Characteristics of the Microfluidic Device
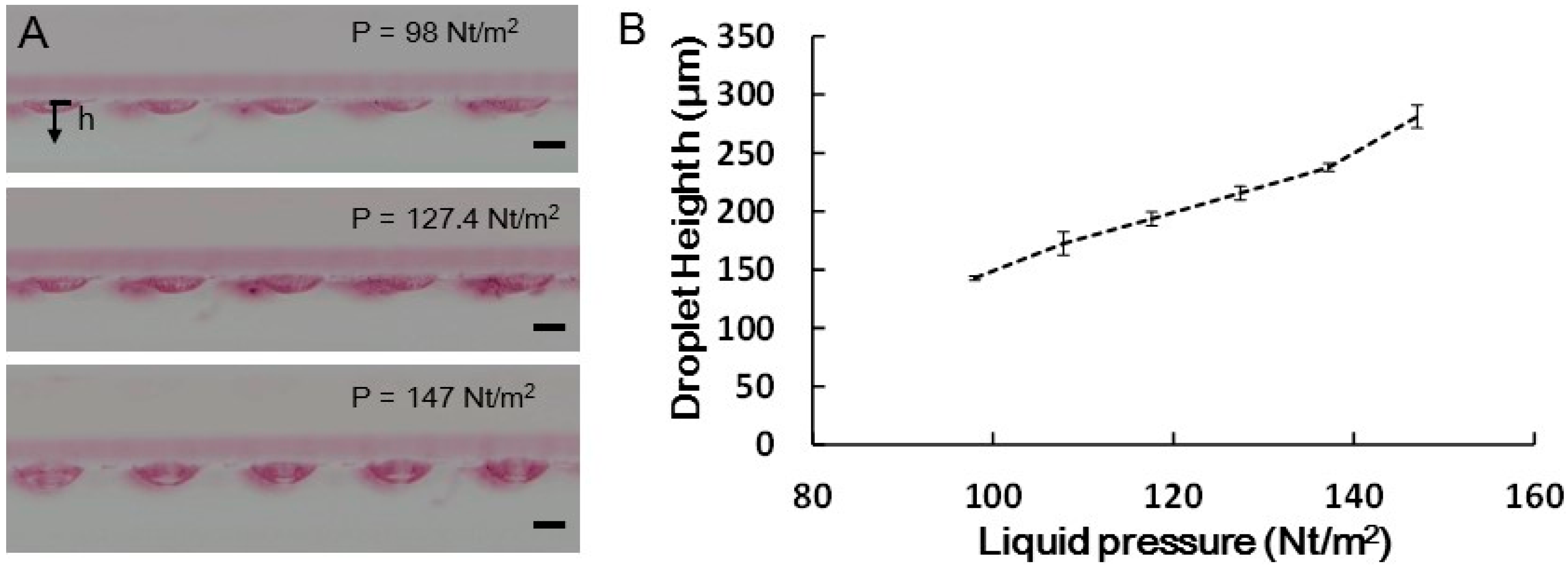
2.2.1. Hanging Drop Formation
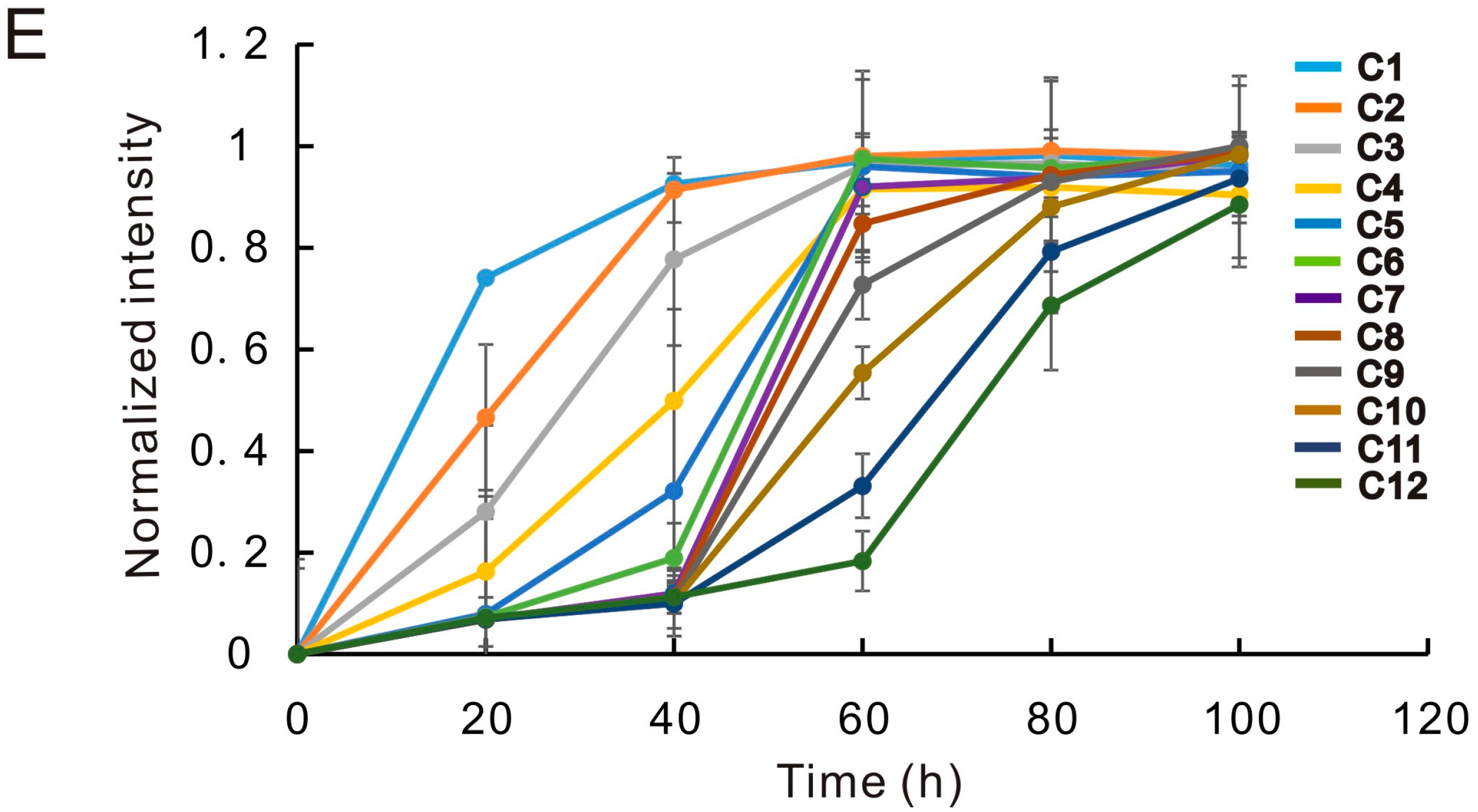
2.2.2. Demonstration of Solution Exchange in μHD Chip
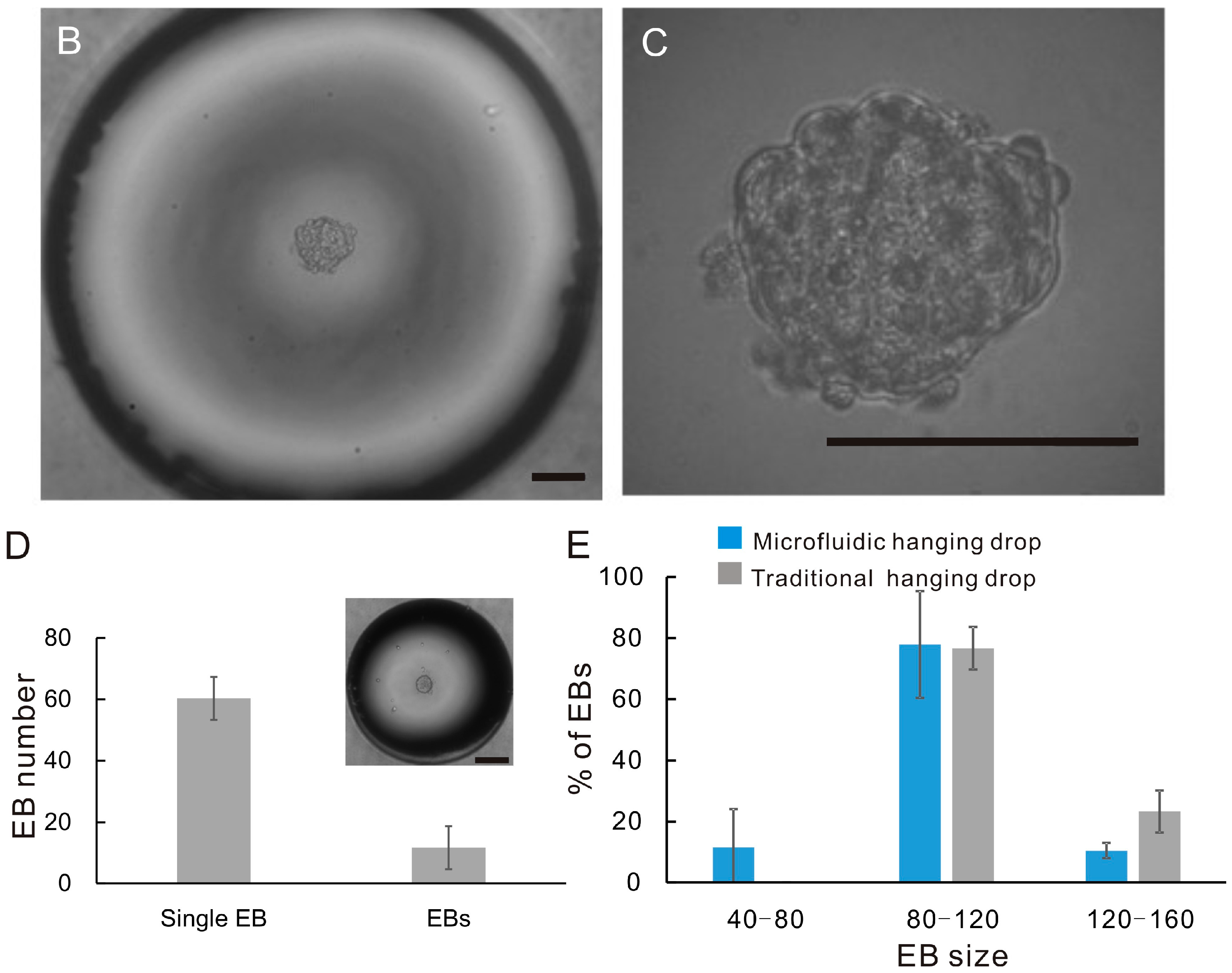
2.3. EB Formation
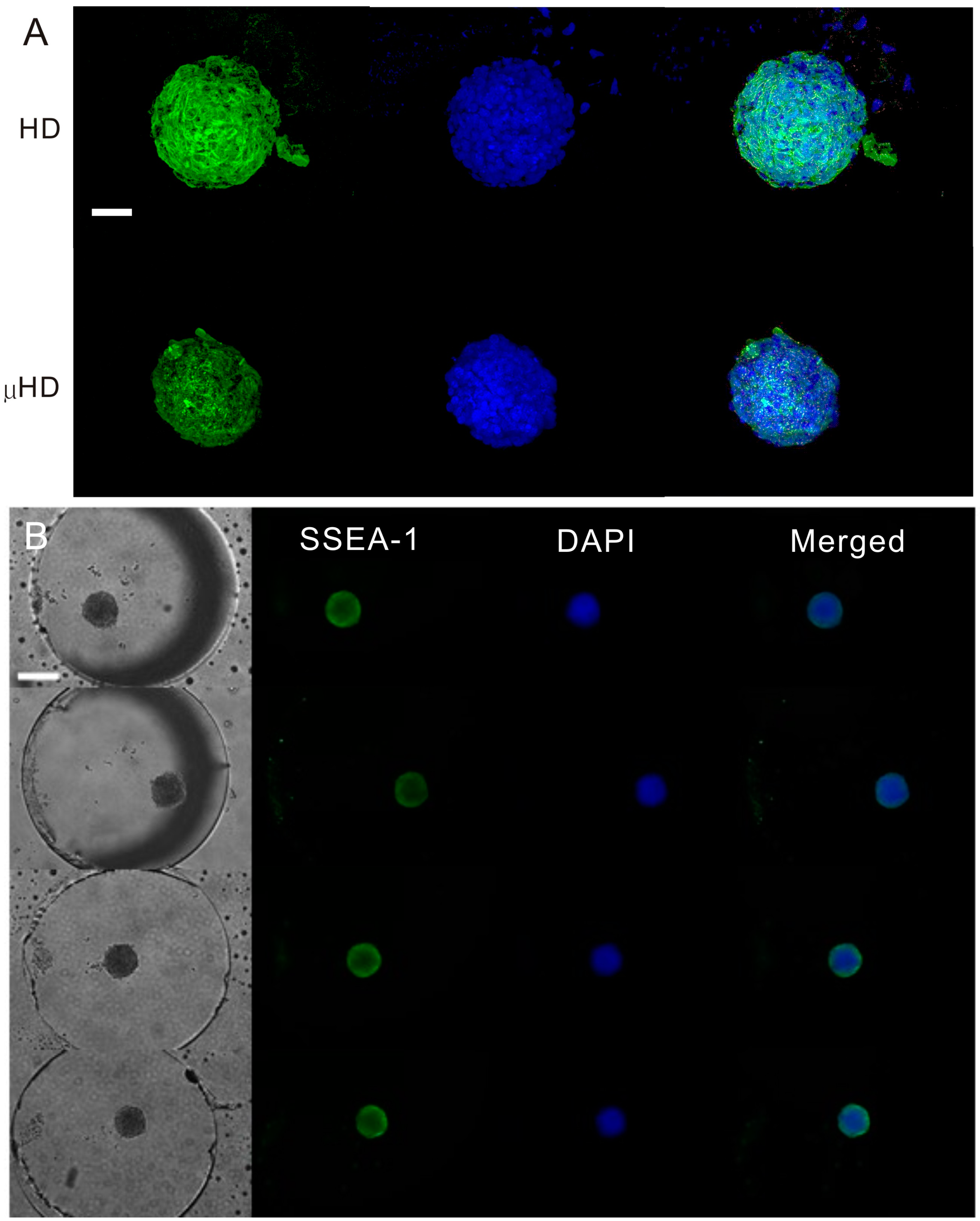
2.4. Immunostaining of EBs
3. Materials and Methods
3.1. Fabrication Process
3.2. Droplet and EB Measurement
3.3. Medium Exchange Experiment
3.4. mES Cell Culture, Cell Line: D3
3.5. Immunochemistry Staining
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ESCs | Embryonic stem cells |
| EB | Embryoid body |
| BSA | Bovine serum albumin |
| CV | Coefficient of variance |
| DAPI | 4′,6-Diamidino-2-phenylindole |
| DMEM | Dulbecco’s modified eagle’s medium |
| EC | Embryonic carcinoma |
| FITC | Fluorescin isothiocyanate |
| MEF | Mouse embryonic fibroblast |
| MPC | 2-Methacryloyloxyethyl phosphorylcholine |
| mESCs | Mouse embryonic stem cells |
| NEAA | Non-essential amino acids |
| PBS | Phosphate buffered saline |
| PE | Polyester |
| PDMS | Polydimethylsiloxane |
| SSEA-1 | Stage-specific embryonic antigen-1 |
| LIF | Leukemia inhibitory factor |
| μHD | Microfluidic hanging drop |
References
- Lewis, G.G.; DiTucci, M.J.; Baker, M.S.; Phillips, S.T. High throughput method for prototyping three-dimensional, paper-based microfluidic devices. Lab Chip 2012, 12, 2630–2633. [Google Scholar] [CrossRef] [PubMed]
- Desbaillets, I.; Ziegler, U.; Groscurth, P.; Gassmann, M. Embryoid bodies: An in vitro model of mouse embryogenesis. Exp. Physiol. 2000, 85, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Itskovitz-Eldor, J.; Schuldiner, M.; Karsenti, D.; Eden, A.; Yanuka, O.; Amit, M.; Soreq, H.; Benvenisty, N. Differentiation of human embryonic stem cells into embryoid bodies comprising the three embryonic germ layers. Mol. Med. 2000, 6, 88–95. [Google Scholar] [PubMed]
- Shen, M.M.; Leder, P. Leukemia inhibitory factor is expressed by the preimplantation uterus and selectively blocks primitive ectoderm formation invitro. Proc. Natl. Acad. Sci. USA 1992, 89, 8240–8244. [Google Scholar] [CrossRef] [PubMed]
- Odorico, J.S.; Kaufman, D.S.; Thomson, J.A. Multilineage differentiation from human embryonic stem cell lines. Stem Cells 2001, 19, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Keller, G. Embryonic stem cell differentiation: Emergence of a new era in biology and medicine. Genes Dev. 2005, 19, 1129–1155. [Google Scholar] [CrossRef] [PubMed]
- Plachta, N.; Bibel, M.; Tucker, K.L.; Barde, Y.A. Developmental potential of defined neural progenitors derived from mouse embryonic stem cells. Development 2004, 131, 5449–5456. [Google Scholar] [CrossRef] [PubMed]
- Klug, M.G.; Soonpaa, M.H.; Koh, G.Y.; Field, L.J. Genetically selected cardiomyocytes from differentiating embryonic stem cells form stable intracardiac grafts. J. Clin. Investig. 1996, 98, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.A.; Tosh, D.; Wilson, D.I.; Lindsay, S.; Forrester, L.M. Hepatic differentiation of murine embryonic stem cells. Exp. Cell Res. 2002, 272, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Toyooka, Y.; Tsunekawa, N.; Akasu, R.; Noce, T. Embryonic stem cells can form germ cells in vitro. Proc. Natl. Acad. Sci. USA 2003, 100, 11457–11462. [Google Scholar] [CrossRef] [PubMed]
- Potocnik, A.J.; Nielsen, P.J.; Eichmann, K. In vitro generation of lymphoid precursors from embryonic stem-cells. EMBO J. 1994, 13, 5274–5283. [Google Scholar] [PubMed]
- Shinji, T.; Koide, N.; Tsuji, T. Glycosaminoglycans partially substitute for proteoglycans in spheroid formation of adult-rat hepatocytes in primary culture. Cell Struct. Funct. 1988, 13, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Dang, S.M.; Kyba, M.; Perlingeiro, R.; Daley, G.Q.; Zandstra, P.W. Efficiency of embryoid body formation and hematopoietic development from embryonic stem cells in different culture systems. Biotechnol. Bioeng. 2002, 78, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Koike, M.; Kurosawa, H.; Amano, Y. A round-bottom 96-well polystyrene plate coated with 2-methacryloyloxyethyl phosphorylcholine as an effective tool for embryoid body formation. Cytotechnology 2005, 47, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, M.; Niebruegge, S.; Werner, A.; Willbold, E.; Burg, M.; Ruediger, M.; Field, L.J.; Lehmann, J.; Zweigerdt, R. Differentiation and lineage selection of mouse embryonic stem cells in a stirred bench scale bioreactor with automated process control. Biotechnol. Bioeng. 2005, 92, 920–933. [Google Scholar] [CrossRef] [PubMed]
- Gerecht-Nir, S.; Cohen, S.; Itskovitz-Eldor, J. Bioreactor cultivation enhances the efficiency of human embryoid body (hEB) formation and differentiation. Biotechnol. Bioeng. 2004, 86, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kniss, D.A.; Lasky, L.C.; Yang, S.T. Culturing and differentiation of murine embryonic stem cells in a three-dimensional fibrous matrix. Cytotechnology 2003, 41, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Torisawa, Y.-S.; Chueh, B.-H.; Huh, D.; Ramamurthy, P.; Roth, T.M.; Barald, K.F.; Takayama, S. Efficient formation of uniform-sized embryoid bodies using a compartmentalized microchannel device. Lab Chip 2007, 7, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Ahadian, S.; Yamada, S.; Ramón-Azcón, J.; Ino, K.; Shiku, H.; Khademhosseini, A.; Matsue, T. Rapid and high-throughput formation of 3D embryoid bodies in hydrogels using the dielectrophoresis technique. Lab Chip 2014, 14, 3690–3694. [Google Scholar] [CrossRef] [PubMed]
- Vrij, E.J.; Espinoza, S.; Heilig, M.; Kolew, A.; Schneider, M.; van Blitterswijk, C.; Truckenmüller, R.; Rivron, N.C. 3D high throughput screening and profiling of embryoid bodies in thermoformed microwell plates. Lab Chip 2016, 16, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Mohr, J.C.; de Pablo, J.J.; Palecek, S.P. 3-D microwell culture of human embryonic stem cells. Biomaterials 2006, 27, 6032–6042. [Google Scholar] [CrossRef] [PubMed]
- Karp, J.M.; Yeh, J.; Eng, G.; Fukuda, J.; Blumling, J.; Suh, K.-Y.; Cheng, J.; Mahdavi, A.; Borenstein, J.; Langer, R.; et al. Controlling size, shape and homogeneity of embryoid bodies using poly(ethylene glycol) microwells. Lab Chip 2007, 7, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.Y.; Chung, B.G.; Lee, D.H.; Khademhosseini, A.; Kim, J.-H.; Lee, S.-H. Controlled-size embryoid body formation in concave microwell arrays. Biomaterials 2010, 31, 4296–4303. [Google Scholar] [CrossRef] [PubMed]
- Hescheler, J.; Fleischmann, B.K.; Lentini, S.; Maltsev, V.A.; Rohwedel, J.; Wobus, A.M.; Addicks, K. Embryonic stem cells: A model to study structural and functional properties in cardiomyogenesis. Cardiovasc. Res. 1997, 36, 149–162. [Google Scholar] [CrossRef]
- Lee, W.G.; Ortmann, D.; Hancock, M.J.; Bae, H.; Khademhosseini, A. A Hollow Sphere Soft Lithography Approach for Long-Term Hanging Drop Methods. Tissue Eng. Part C Methods 2010, 16, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Tung, Y.-C.; Hsiao, A.Y.; Allen, S.G.; Torisawa, Y.-S.; Ho, M.; Takayama, S. High-throughput 3D spheroid culture and drug testing using a 384 hanging drop array. Analyst 2011, 136, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H.; Chen, C.C.; Folch, A. “Microcanals” for micropipette access to single cells in microfluidic environments. Lab Chip 2004, 4, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.








© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, H.-W.; Hsiao, Y.-H.; Chen, C.-C.; Yet, S.-F.; Hsu, C.-H. A PDMS-Based Microfluidic Hanging Drop Chip for Embryoid Body Formation. Molecules 2016, 21, 882. https://doi.org/10.3390/molecules21070882
Wu H-W, Hsiao Y-H, Chen C-C, Yet S-F, Hsu C-H. A PDMS-Based Microfluidic Hanging Drop Chip for Embryoid Body Formation. Molecules. 2016; 21(7):882. https://doi.org/10.3390/molecules21070882
Chicago/Turabian StyleWu, Huei-Wen, Yi-Hsing Hsiao, Chih-Chen Chen, Shaw-Fang Yet, and Chia-Hsien Hsu. 2016. "A PDMS-Based Microfluidic Hanging Drop Chip for Embryoid Body Formation" Molecules 21, no. 7: 882. https://doi.org/10.3390/molecules21070882
APA StyleWu, H. -W., Hsiao, Y. -H., Chen, C. -C., Yet, S. -F., & Hsu, C. -H. (2016). A PDMS-Based Microfluidic Hanging Drop Chip for Embryoid Body Formation. Molecules, 21(7), 882. https://doi.org/10.3390/molecules21070882