
Synthesis, Biological Evaluation and Molecular Modelling of 2′-Hydroxychalcones as Acetylcholinesterase Inhibitors

Abstract
:1. Introduction
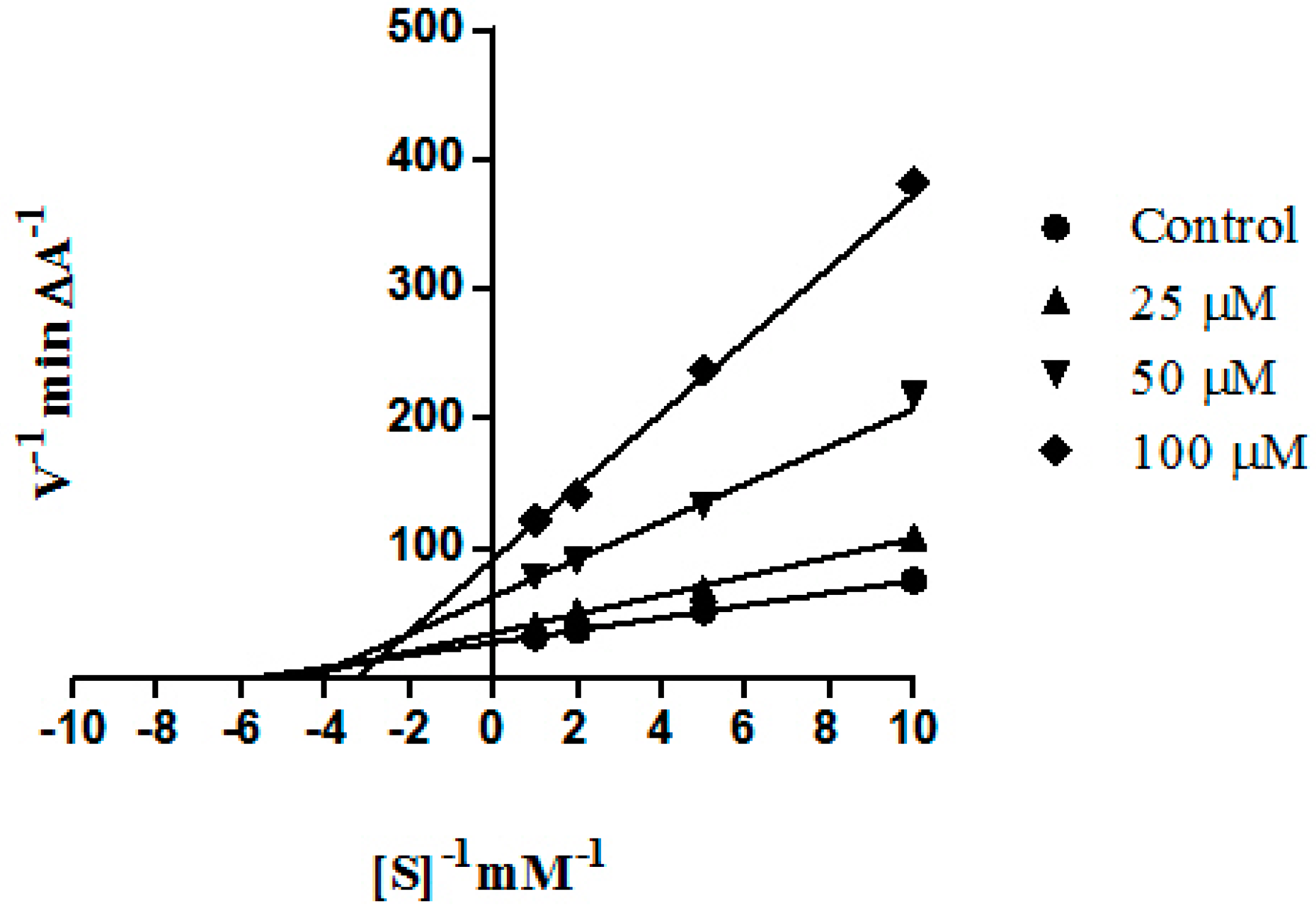
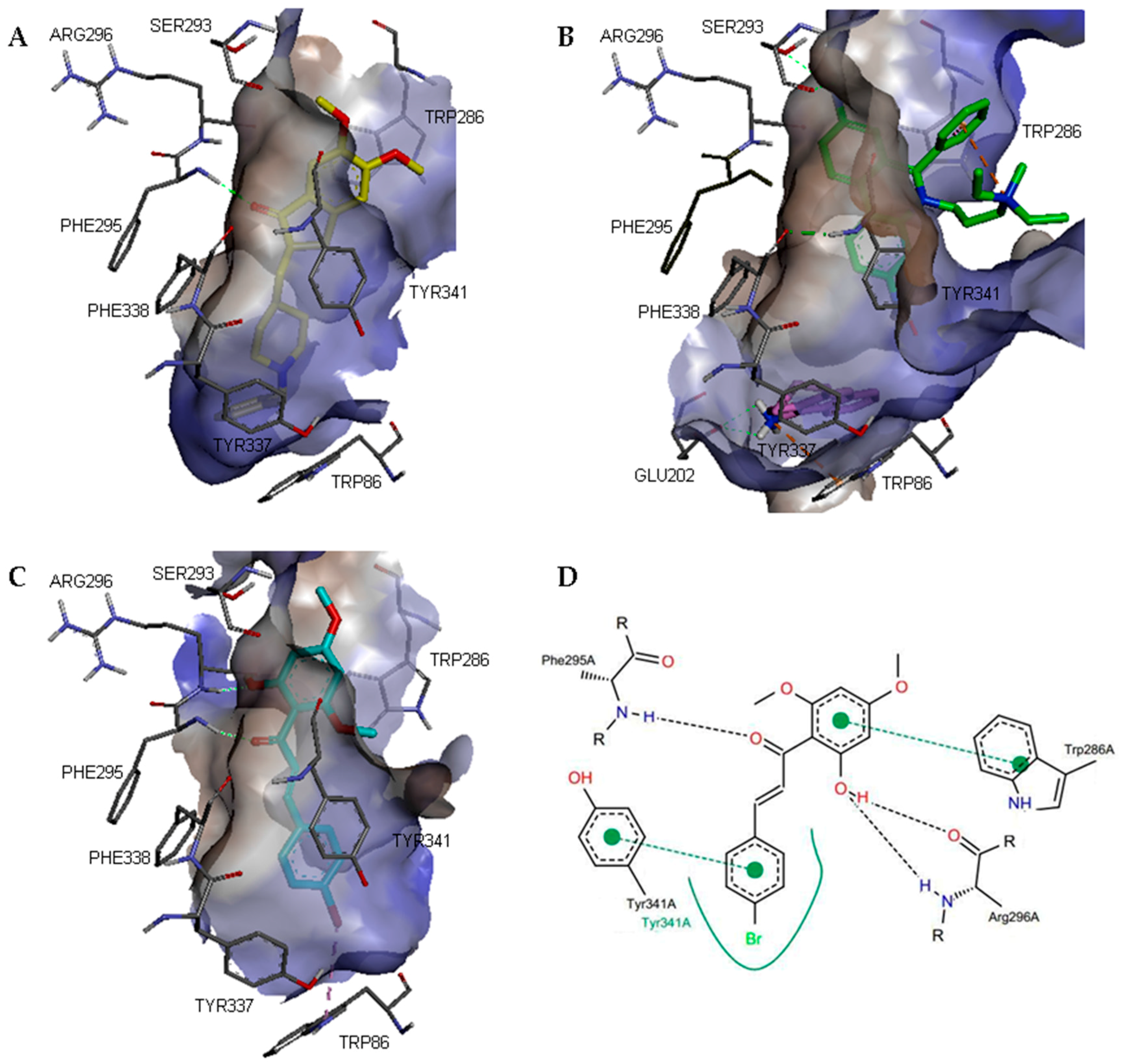
2. Results and Discussion
3. Materials and Methods
3.1. General Information
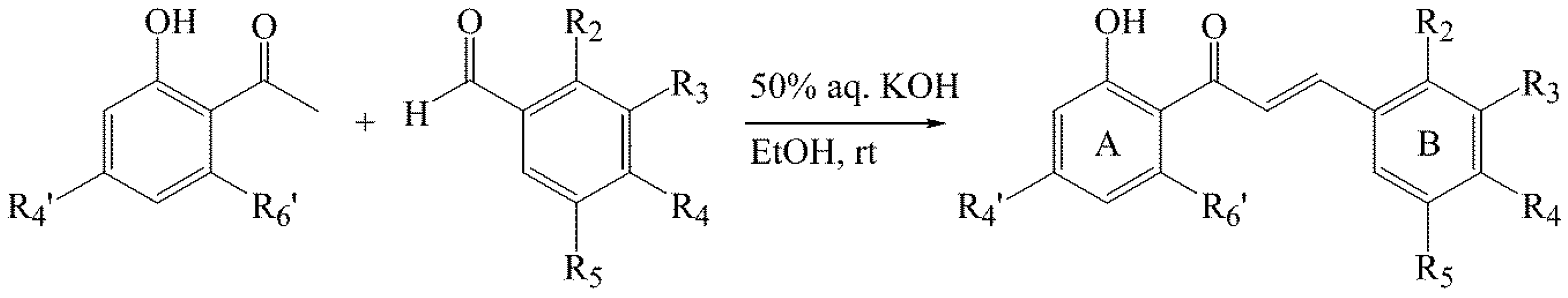
3.2. Synthesis of Chalcones 1–14
3.3. Enzyme Inhibition Studies
3.4. Molecular Modelling Studies
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 2007, 3, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Suh, W.H.; Suslick, K.S.; Suh, Y.-H. Therapeutic agents for Alzheimer’s disease. Curr. Med. Chem. Cent. Nerv. Syst. Agents 2005, 5, 259–269. [Google Scholar] [CrossRef]
- Hansen, R.A.; Gartlehner, G.; Webb, A.P.; Morgan, L.C.; Moore, C.G.; Jonas, D.E. Efficacy and safety of donepezil, galantamine, and rivastigmine for the treatment of Alzheimer’s disease: A systematic review and meta-analysis. Clin. Interv. Aging 2008, 3, 211–225. [Google Scholar] [PubMed]
- Wiesner, J.; Kriz, Z.; Kuca, K.; Jun, D.; Koca, J. Acetylcholinesterases—The structural similarities and differences. J. Enzym. Inhib. Med. Chem. 2007, 22, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Silman, I.; Sussman, J.L. Acetylcholinesterase ‘classical’ and ‘non-classical’ functions and pharmacology. Curr. Opin. Pharmacol. 2005, 5, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.E.; Park, J.Y.; No, K.T.; Shin, J.H.; Lee, S.K.; Eun, J.S.; Yang, J.H.; Shin, T.Y.; Kim, D.K.; Chae, B.S.; et al. Synthesis, in vitro assay, and molecular modeling of new piperidine derivatives having dual inhibitory potency against acetylcholinesterase and A β1–42 aggregation for Alzheimer’s disease therapeutics. Bioorg. Med. Chem. 2007, 15, 6596–6607. [Google Scholar] [CrossRef] [PubMed]
- Andersson, C.D.; Forsgren, N.; Akfur, C.; Allgardsson, A.; Berg, L.; Engdahl, C.; Qian, W.; Ekström, F.; Linusson, A. Divergent structure-activity relationships of structurally similar acetylcholinesterase inhibitors. J. Med. Chem. 2013, 56, 7615–7624. [Google Scholar] [CrossRef] [PubMed]
- Sahu, N.K.; Balbhadra, S.S.; Choudhary, S.S.; Kohli, D.V. Exploring pharmacological significance of chalcone scaffold: A review. Curr. Med. Chem. 2012, 19, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.; Khan, K.M.; Sher, M.; Maharvi, G.M.; Nawaz, S.A.; Choudhary, M.I.; Atta-ur-Rahman; Supuran, C.T. Synthesis and inhibitory potential towards acetylcholinesterase, butyrylcholinesterase and lipoxygenase of some variably substituted chalcones. J. Enzym. Inhib. Med. Chem. 2005, 20, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhang, J.; Sheng, R.; Dong, X.; He, Q.; Yang, B.; Hu, W. Synthesis and biological evaluation of novel flavonoid derivatives as dual binding acetylcholinesterase inhibitors. J. Enzym. Inhib. Med. Chem. 2009, 24, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Sheng, R.; Lin, X.; Zhang, J.; Chol, K.S.; Huang, W.; Yang, B.; He, Q.; Hu, W. Design, synthesis and evaluation of flavonoid derivatives as potent AChE inhibitors. Bioorg. Med. Chem. 2009, 17, 6692–6698. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.E.; Cho, J.K.; Curtis-Long, M.J.; Ryu, H.W.; Kim, J.H.; Kim, H.J.; Yuk, H.J.; Kim, D.W.; Park, K.H. Inhibitory evaluation of sulfonamide chalcones on β-secretase and acylcholinesterase. Molecules 2013, 18, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Bag, A.; Ghosh, S.; Tulsan, R.; Sood, A.; Zhou, W.; Schifone, C.; Foster, M.; LeVine III, H.; Török, B.; Török, M. Design, synthesis and biological activity of multifunctional α, β-unsaturated carbonyl scaffolds. Bioorg. Med. Chem. Lett. 2013, 23, 2614–2618. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.R.; Huang, X.Q.; Lou, D.H.; Liu, X.J.; Liu, W.K.; Wang, Q.A. Synthesis and acetylcholinesterase inhibitory activity of Mannich base derivatives flavokawain B. Bioorg. Med. Chem. Lett. 2014, 24, 4749–4753. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.R.; Liu, X.J.; Fan, H.Q.; Tang, J.J.; Gao, X.H.; Liu, W.K. Design, synthesis and pharmacological evaluation of chalcone derivatives as acetylcholinesterase inhibitors. Bioorg. Med. Chem. 2014, 22, 6124–6133. [Google Scholar] [CrossRef] [PubMed]
- Mai, C.W.; Yaeghoobi, M.; Rahman, N.A.; Kang, Y.B.; Pichika, M.R. Chalcones with electron-withdrawing and electron-donating substituents: Anticancer activity against TRAIL resistant cancer cells, structure-activity relationship analysis and regulation of apoptotic proteins. Eur. J. Med. Chem. 2014, 77, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Shaw, G.; Morse, S.; Ararat, M.; Graham, F.L. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 2002, 16, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, S.A. Blood–brain barrier permeability considerations for CNS-targeted compound library design. Curr. Opin. Chem Biol. 2008, 12, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Sugimoto, H.; Iimura, Y.; Yamanishi, Y.; Yamatsu, K. Synthesis and structure-activity relationships of acetylcholinesterase inhibitors: 1-Benzyl-4-[(5,6-dimethoxy-1-oxoindan-2-yl)methyl]piperidine hydro-chloride and related compounds. J. Med. Chem. 1995, 38, 4821–4829. [Google Scholar] [CrossRef] [PubMed]
- Ucar, G.; Gokhan, N.; Yesilada, A.; Bilgin, A.A. 1-N-Substituted thiocarbamoyl-3-phenyl-5-thienyl-2-pyrazolines: A novel cholinesterase and selective monoamine oxidase B inhibitors. Neurosci. Lett. 2005, 382, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Sawatzky, E.; Wehle, S.; Kling, B.; Wendrich, J.; Bringmann, G.; Sotriffer, C.A.; Heilmann, J.; Decker, M. Discovery of highly selective and nanomolar carbamate-based butyrylcholinesterase inhibitors by rational investigation into their inhibition mode. J. Med. Chem. 2016, 59, 2067–2082. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, A.; Ballard, C.; Bullock, R.; Darreh-Shori, T.; Somogyi, M. A review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer’s disease. Prim. Care Companion CNS Disord. 2013, 15. [Google Scholar] [CrossRef]
- Kryger, G.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with E2020 (Aricept (R)): Implications for the design of new anti-Alzheimer drugs. Structure 1999, 7, 297–307. [Google Scholar] [CrossRef]
- Berg, L.; Andersson, C.D.; Artursson, E.; Hörnberg, A.; Tunemalm, A.K.; Linusson, A.; Ekström, F. Targeting acetylcholinesterase: Identification of chemical leads by high throughput screening, structure determination and molecular modeling. PLoS ONE 2011, 6, e26039. [Google Scholar] [CrossRef] [PubMed]
- Matter, H.; Nazaré, M.; Güssregen, S.; Will, D.W.; Schreuder, H.; Bauer, A.; Urmann, M.; Ritter, K.; Wagner, M.; Wehner, V. Evidence for C-Cl/C-Br···pi interactions as an important contribution to protein-ligand binding affinity. Angew. Chem. Int. Ed. Engl. 2009, 48, 2911–2916. [Google Scholar] [CrossRef] [PubMed]
- Nachon, F.; Carletti, E.; Ronco, C.; Trovaslet, M.; Nicolet, Y.; Jean, L.; Renard, P.Y. Crystal structures of human cholinesterases in complex with huprine W and tacrine: Elements of specificity for anti-Alzheimer’s drugs targeting acetyl- and butyryl-cholinesterase. Biochem. J. 2013, 453, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Imafuku, K.; Honda, H.; McOmie, J.F.W. Cyclodehydrogenation of 2′-hydroxychalcones with 2,3-dichloro-5,6-dicyano-p-benzoquinone: A simple route for flavones and aurones. Synthesis 1987, 2, 199–201. [Google Scholar] [CrossRef]
- Maiti, G.; Karmakar, R.; Bhattacharya, R.N.; Kayal, U. A novel one pot route to flavones under dual catalysis, an organo- and a Lewis acid catalyst. Tetrahedron Lett. 2011, 52, 5610–5612. [Google Scholar] [CrossRef]
- Detsi, A.; Majdalani, M.; Kontogiorgis, C.A.; Hadjipavlou-Litina, D.; Kefalas, P. Natural and synthetic 2′-hydroxy-chalcones and aurones: Synthesis characterization and evaluation of the antioxidant and soybean lipoxygenase inhibitory activity. Bioorg. Med. Chem. 2009, 17, 8073–8085. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.C.; Yang, C.H. Haloflavones. Taiwan Yaoxue Zazhi 1951, 3, 39–41. [Google Scholar]
- Prasad, Y.R.; Prasoona, L.; Rao, A.L.; Lakshmi, K.; Kumar, P.R.; Rao, B.G. Synthesis and antimicrobial activity of some new chalcones. Int. J. Chem. Sci. 2005, 3, 685–689. [Google Scholar]
- Boeck, P.; Leal, P.C.; Yunes, R.A.; Cechinel Filho, V.; Lopez, S.; Sortino, M.; Escalante, A.; Furlan, R.L.E.; Zacchino, S. Antifungal activity and studies on mode of action of novel xanthoxyline-derived chalcones. Arch. der Pharm. 2005, 338, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Aponte, J.C.; Verástegui, M.; Malaga, E.; Zimic, M.; Quiliano, M.; Vaisberg, A.J.; Gilman, R.H.; Hammond, G.B. Synthesis, cytotoxicity, and anti-Trypanosoma cruzi activity of new chalcones. J. Med. Chem. 2008, 51, 6230–6234. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, M.; Simoens, M.; Falchi, G.; Lavaggi, M.L.; Piro, O.E.; Castellano, E.E.; Vidal, A.; Azqueta, A.; Monge, A.; de Ceráin, A.L.; et al. Synthetic chalcones, flavanones, and flavones as antitumoral agents: Biological evaluation and structure–activity relationships. Bioorg. Med. Chem. 2007, 15, 3356–3367. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Stierand, K.; Maaß, P.; Rarey, M. Molecular complexes at a glance: Automated generation of two-dimensional complex diagrams. Bioinformatics 2006, 22, 1710–1716. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 7, 10, 13 and 14 are available from the authors.




{kind=link}
{kind=link}
{kind=link}
| Compound | MW | HBD | HBA | A log P | PSA/Å2 |
|---|---|---|---|---|---|
| 1 | 224.26 | 1 | 2 | 3.46 | 38.12 |
| 2 | 238.28 | 1 | 2 | 3.95 | 38.12 |
| 3 | 303.15 | 1 | 2 | 4.21 | 38.12 |
| 4 | 258.70 | 1 | 2 | 4.12 | 38.12 |
| 5 | 267.32 | 1 | 3 | 3.62 | 41.49 |
| 6 | 254.28 | 1 | 3 | 3.44 | 47.05 |
| 7 | 303.15 | 1 | 2 | 4.21 | 38.12 |
| 8 | 309.14 | 2 | 3 | 4.55 | 58.93 |
| 9 | 298.33 | 1 | 4 | 3.91 | 55.98 |
| 10 | 363.20 | 1 | 4 | 4.18 | 55.98 |
| 11 | 327.37 | 1 | 5 | 3.59 | 59.33 |
| 12 | 314.33 | 1 | 5 | 3.41 | 64.91 |
| 13 | 363.20 | 1 | 4 | 4.18 | 55.98 |
| 14 | 369.20 | 2 | 5 | 4.51 | 76.79 |
| Donepezil | 379.49 | 0 | 4 | 4.57 | 38.51 |
| Compound | R4´ | R6´ | R2 | R3 | R4 | R5 | % Inhibition at 10 µM a | IC50/µM b |
|---|---|---|---|---|---|---|---|---|
| 1 | H | H | H | H | H | H | 11 | nd c |
| 2 | H | H | H | H | Me | H | na d | nd c |
| 3 | H | H | H | H | Br | H | na d | nd c |
| 4 | H | H | H | H | Cl | H | na d | nd c |
| 5 | H | H | H | H | NMe2 | H | 28 | 266 ± 48 |
| 6 | H | H | H | H | OMe | H | 6 | nd c |
| 7 | H | H | Br | H | H | H | 38 | 55 ± 12 |
| 8 | H | H | OH | Cl | H | Cl | na d | nd c |
| 9 | OMe | OMe | H | H | Me | H | 23 | 354 ± 33 |
| 10 | OMe | OMe | H | H | Br | H | 30 | 41 ± 13 |
| 11 | OMe | OMe | H | H | NMe2 | H | 5 | nd c |
| 12 | OMe | OMe | H | H | OMe | H | 7 | nd c |
| 13 | OMe | OMe | Br | H | H | H | 23 | 85 ± 16 |
| 14 | OMe | OMe | OH | Cl | H | Cl | 51 | 73 ± 22 |
| Propidium e | 50 | 11 ± 3 | ||||||
| Tacrine f | 91 | 0.19 ± 0.04 |
| Compound | Human AChE | Equine BChE | Selectivity | |
|---|---|---|---|---|
| Ki /µM | Ki′ /µM | IC50 /µM a | for AchE b | |
| 7 | 40 | 59 | 108 ± 18 | 2.0 |
| 10 | 31 | 47 | 59 ± 12 | 1.4 |
| 13 | 113 | 165 | 71 ± 14 | 0.8 |
| 14 | 56 | 92 | 81 ± 18 | 1.1 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sukumaran, S.D.; Chee, C.F.; Viswanathan, G.; Buckle, M.J.C.; Othman, R.; Abd. Rahman, N.; Chung, L.Y. Synthesis, Biological Evaluation and Molecular Modelling of 2′-Hydroxychalcones as Acetylcholinesterase Inhibitors. Molecules 2016, 21, 955. https://doi.org/10.3390/molecules21070955
Sukumaran SD, Chee CF, Viswanathan G, Buckle MJC, Othman R, Abd. Rahman N, Chung LY. Synthesis, Biological Evaluation and Molecular Modelling of 2′-Hydroxychalcones as Acetylcholinesterase Inhibitors. Molecules. 2016; 21(7):955. https://doi.org/10.3390/molecules21070955
Chicago/Turabian StyleSukumaran, Sri Devi, Chin Fei Chee, Geetha Viswanathan, Michael J. C. Buckle, Rozana Othman, Noorsaadah Abd. Rahman, and Lip Yong Chung. 2016. "Synthesis, Biological Evaluation and Molecular Modelling of 2′-Hydroxychalcones as Acetylcholinesterase Inhibitors" Molecules 21, no. 7: 955. https://doi.org/10.3390/molecules21070955
APA StyleSukumaran, S. D., Chee, C. F., Viswanathan, G., Buckle, M. J. C., Othman, R., Abd. Rahman, N., & Chung, L. Y. (2016). Synthesis, Biological Evaluation and Molecular Modelling of 2′-Hydroxychalcones as Acetylcholinesterase Inhibitors. Molecules, 21(7), 955. https://doi.org/10.3390/molecules21070955