
Glutamine Synthetase Drugability beyond Its Active Site: Exploring Oligomerization Interfaces and Pockets


Abstract
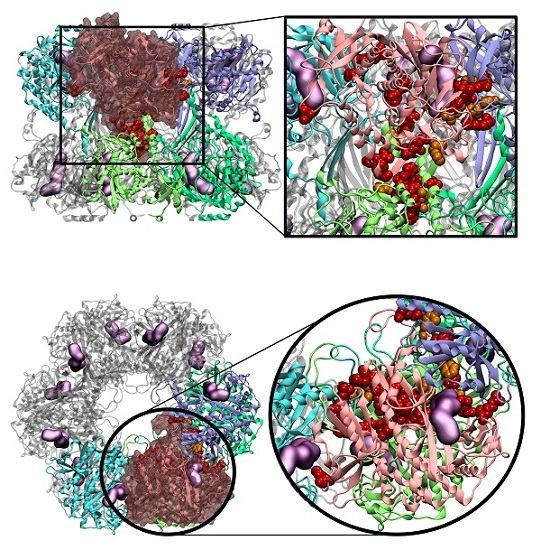
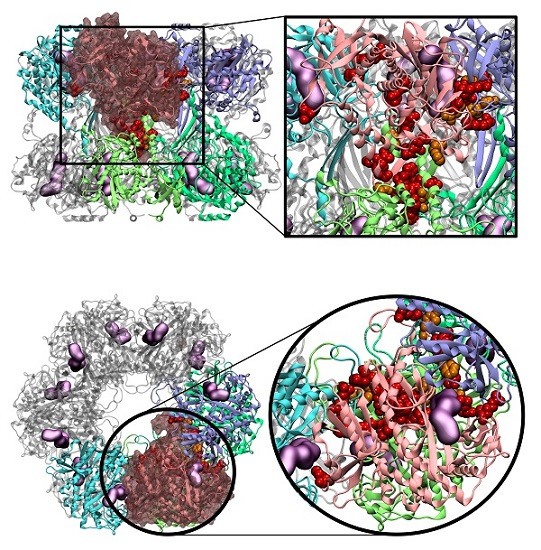
:
1. Introduction
2. Results
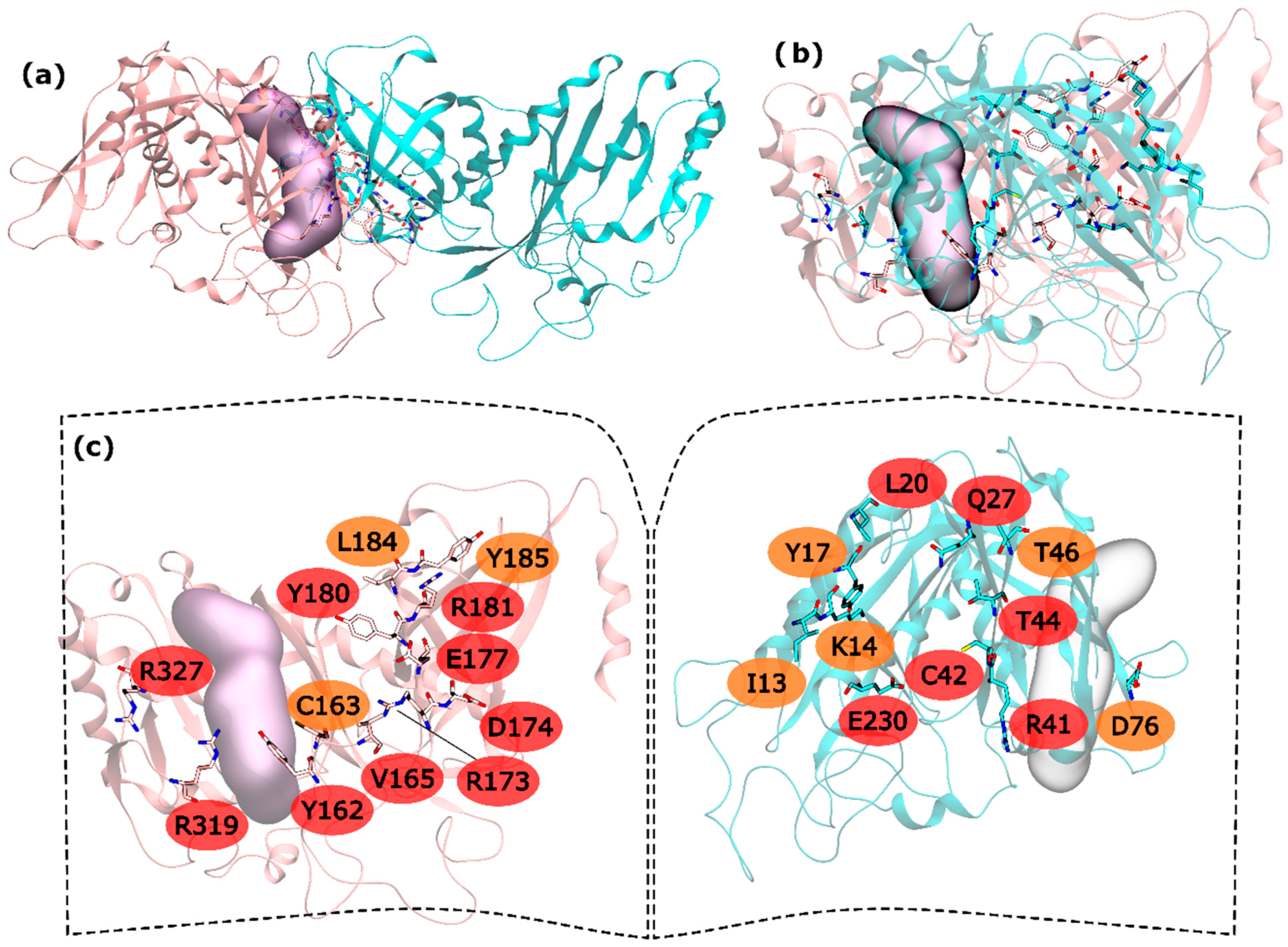
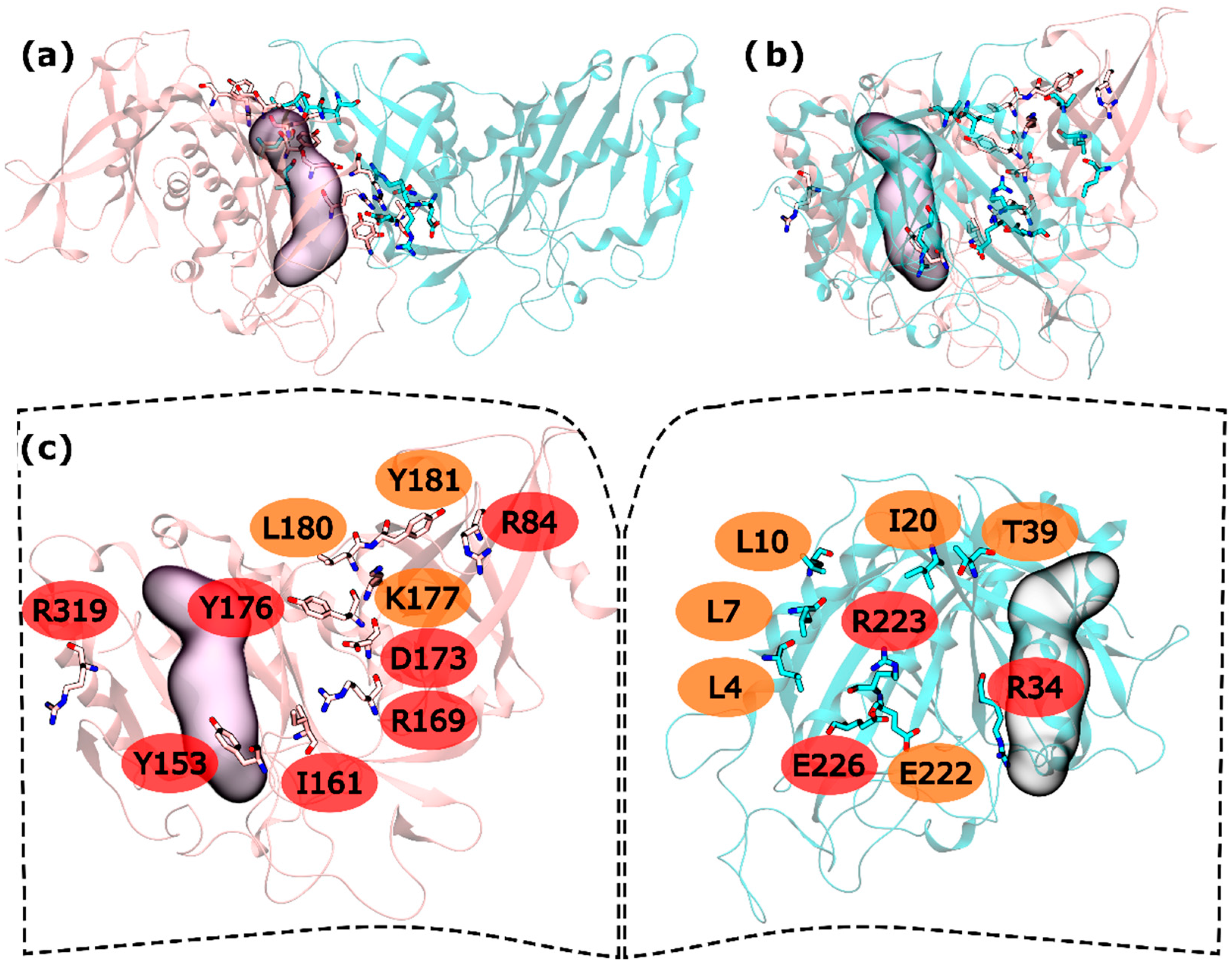
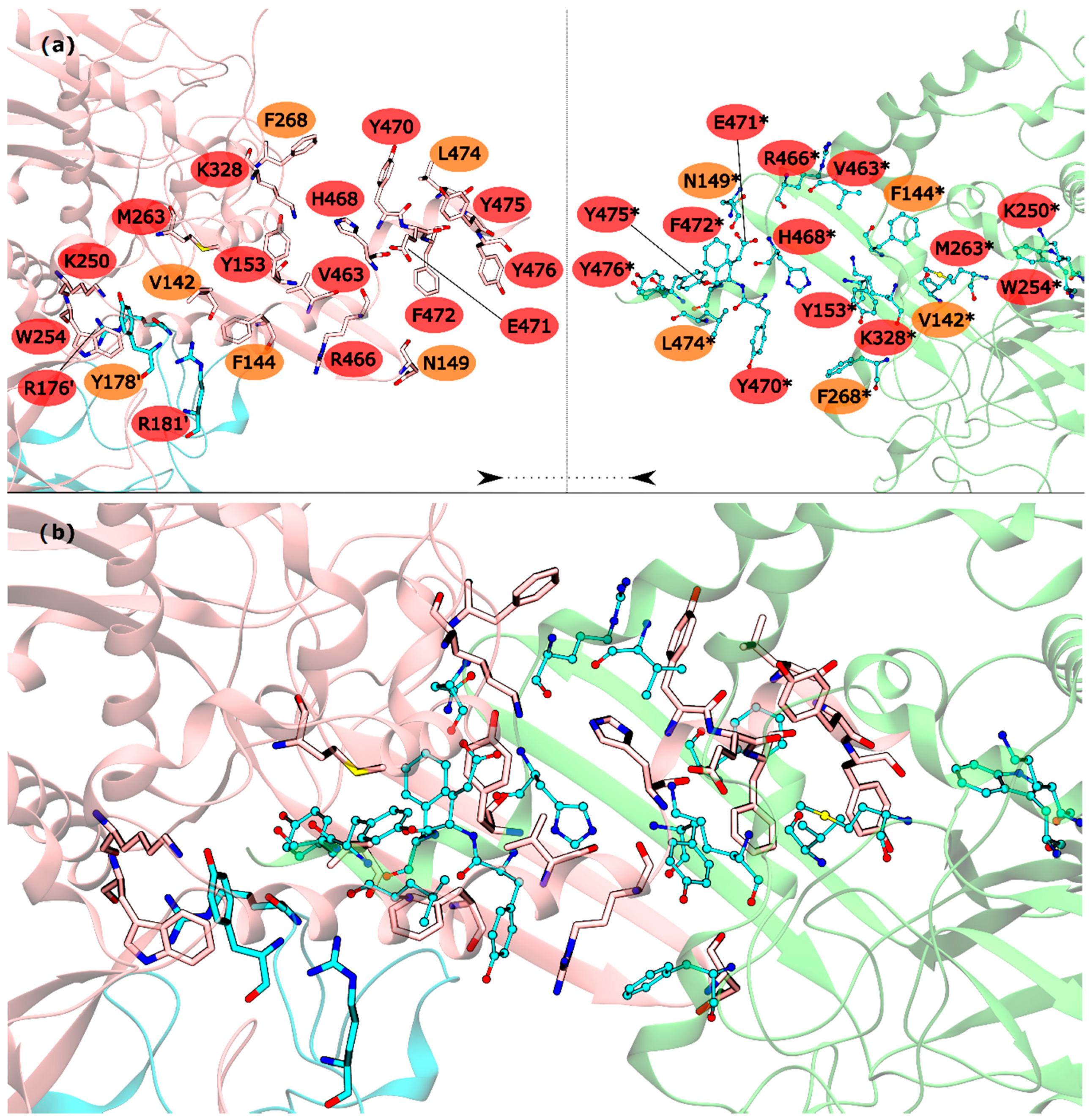
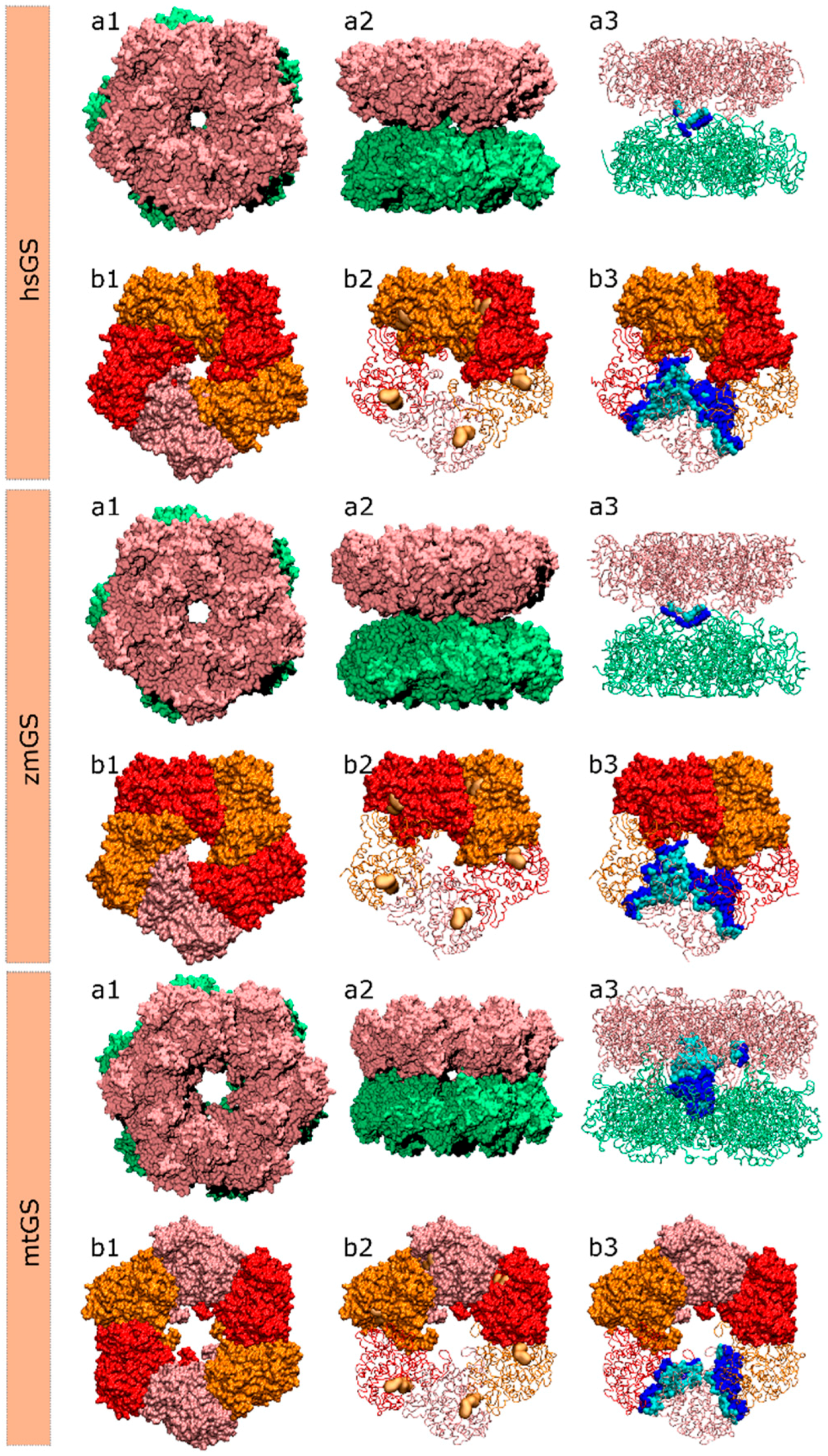
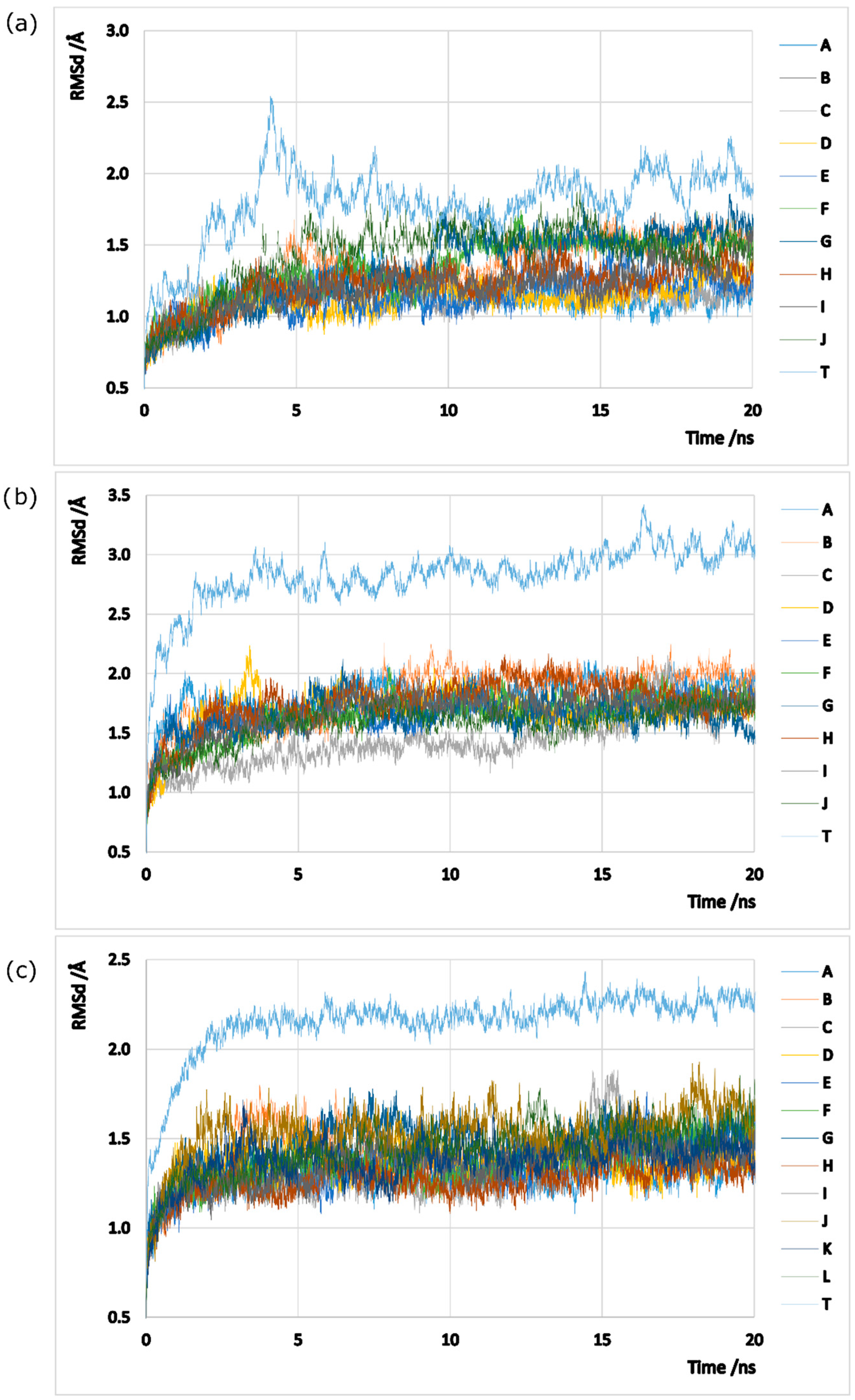
2.1. hsGS PPI
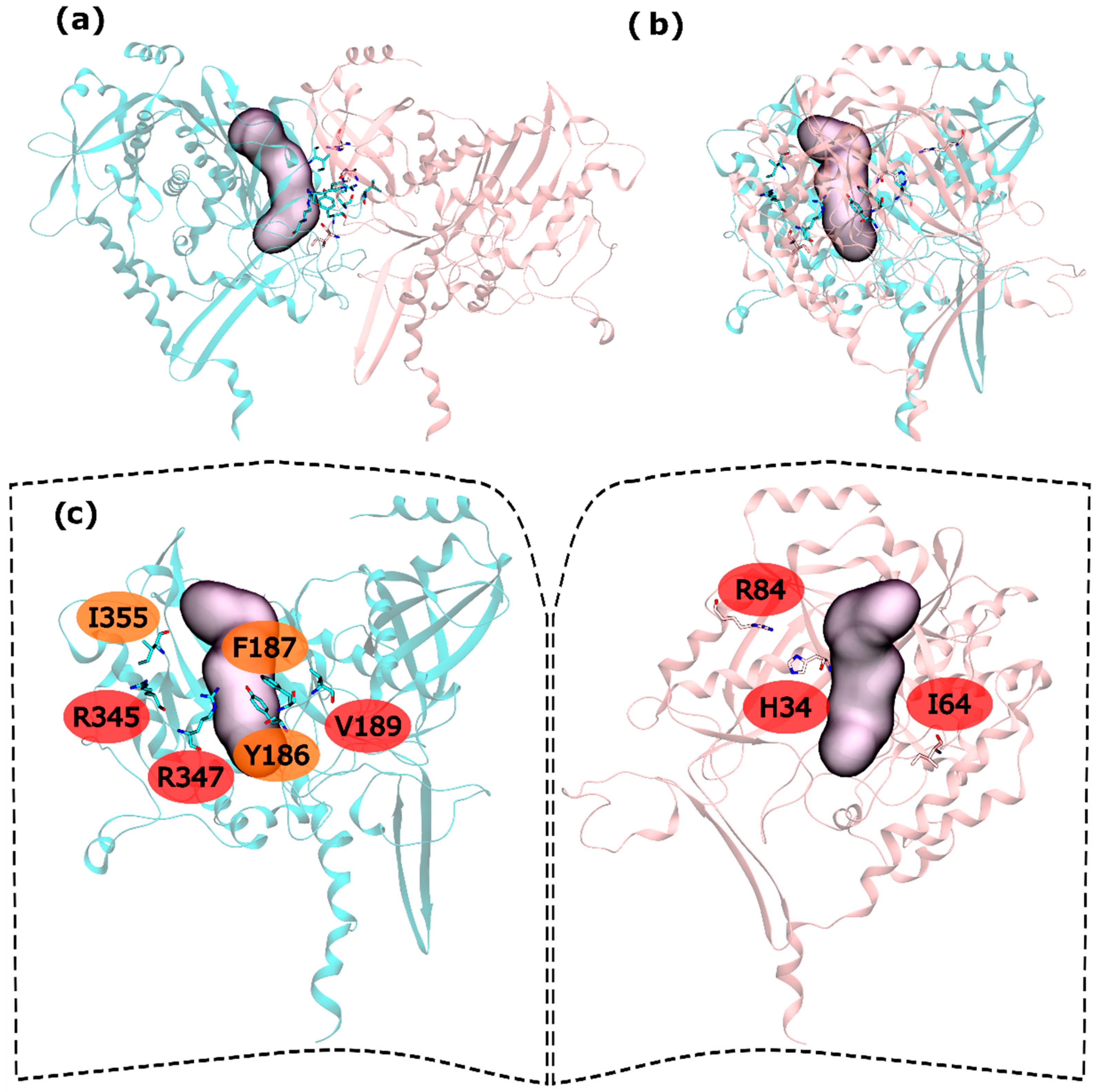
2.2. zmGS PPI
2.3. mtGS PPI
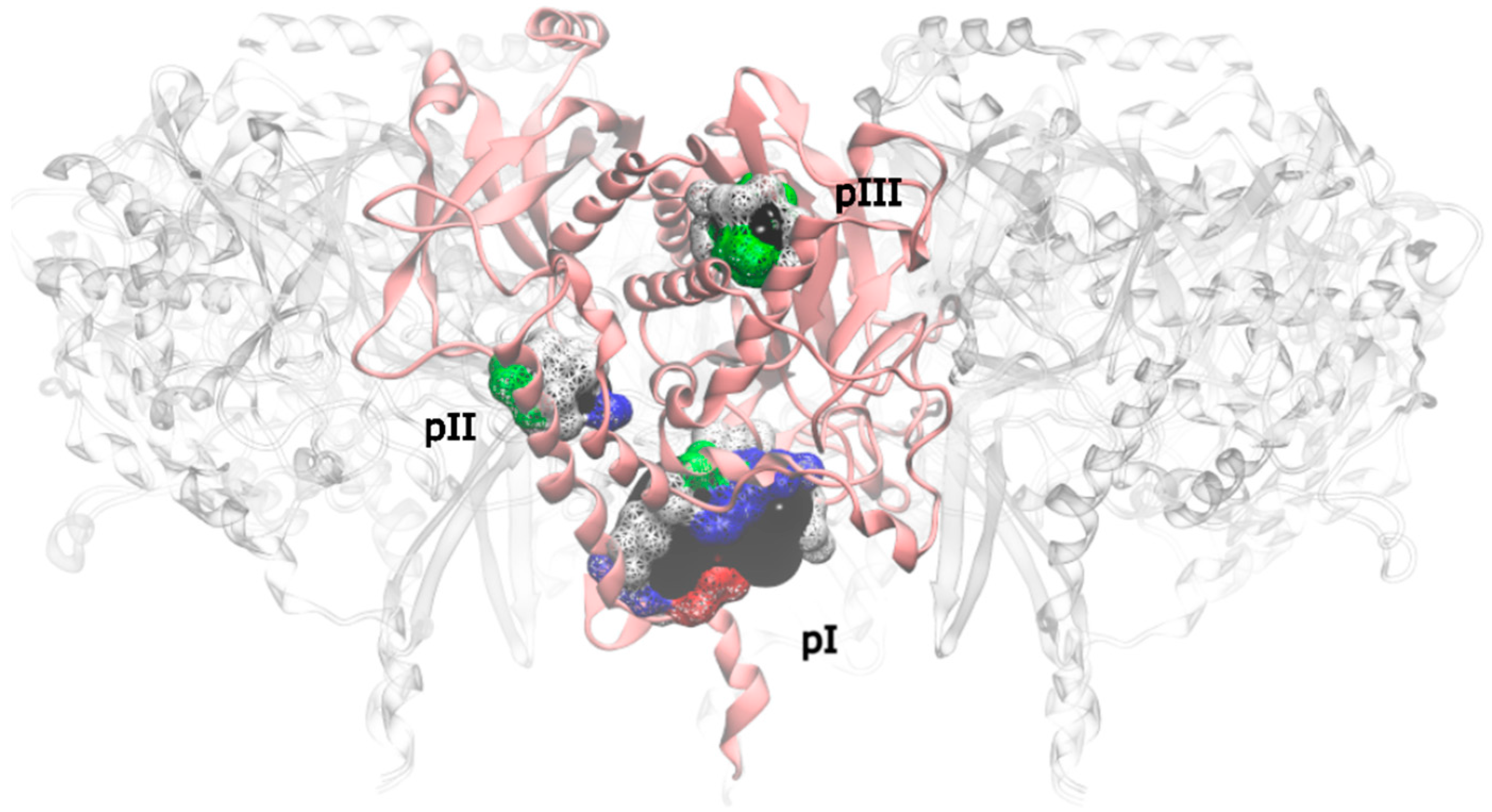
2.4. Novel Drugable Pockets
3. Discussion
4. Materials and Methods
4.1. Selection of Crystallographic Structures
4.2. Study of the Oligomerization Interface by cASM
4.3. Search for Drugable Pockets
4.4. Docking of Molecules on the Drugable Pockets
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Resid | A | B | C | D | E | F | G | H | I | J | Tested | Classif |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 | x | x | x | x | x | x | no | |||||
| 11 | x | x | x | x | x | x | no | |||||
| 13 | x | x | x | x | x | x | x | x | x | yes | WS | |
| 14 | x | x | x | x | x | x | x | yes | WS | |||
| 15 | x | x | x | x | x | no | ||||||
| 16 | x | x | x | x | x | x | no | |||||
| 17 | x | x | x | x | x | x | x | x | x | x | yes | WS |
| 18 | x | x | x | x | x | x | x | x | yes | NS | ||
| 20 | x | x | x | x | x | x | x | x | x | x | yes | HS |
| 22 | x | x | x | x | x | x | x | x | x | x | yes | NS |
| 25 | x | no | ||||||||||
| 27 | x | x | x | x | x | x | x | x | x | yes | HS | |
| 41 | x | x | x | x | x | x | x | x | x | x | yes | HS |
| 42 | x | x | x | x | x | x | x | x | x | yes | HS | |
| 43 | x | x | x | x | x | x | x | x | yes | NS | ||
| 44 | x | x | x | x | x | x | x | x | yes | HS | ||
| 46 | x | x | x | x | x | x | x | x | x | x | yes | WS |
| 63 | x | x | x | x | no | |||||||
| 66 | x | x | x | x | x | x | x | x | yes | NS | ||
| 73 | x | x | x | x | x | no | ||||||
| 74 | x | no | ||||||||||
| 76 | x | x | x | x | x | x | x | x | x | x | yes | WS |
| 78 | x | x | x | x | no | |||||||
| 90 | x | x | x | x | no | |||||||
| 91 | x | x | no | |||||||||
| 104 | x | no | ||||||||||
| 142 | x | x | x | x | x | x | no | |||||
| 143 | x | x | no | |||||||||
| 145 | x | no | ||||||||||
| 152 | x | x | x | x | x | x | x | x | x | yes | NS | |
| 154 | x | x | x | x | x | x | x | x | yes | NS | ||
| 162 | x | x | x | x | x | x | x | x | x | x | yes | HS |
| 163 | x | x | x | x | x | x | x | x | x | x | yes | WS |
| 165 | x | x | x | x | x | x | x | x | x | x | yes | HS |
| 168 | x | x | x | x | no | |||||||
| 173 | x | x | x | x | x | x | x | x | yes | HS | ||
| 174 | x | x | x | x | x | x | x | x | x | x | yes | HS |
| 177 | x | x | x | x | x | x | x | x | x | x | yes | HS |
| 180 | x | x | x | x | x | x | x | x | x | yes | HS | |
| 181 | x | x | x | x | x | x | x | x | x | x | yes | HS |
| 184 | x | x | x | x | x | x | x | x | x | x | yes | WS |
| 185 | x | x | x | x | x | x | x | x | x | x | yes | WS |
| 193 | x | x | x | x | no | |||||||
| 226 | x | x | x | x | no | |||||||
| 227 | x | x | no | |||||||||
| 230 | x | x | x | x | x | x | x | x | x | x | yes | HS |
| 231 | x | x | x | x | x | no | ||||||
| 232 | x | x | x | x | x | x | x | x | x | x | yes | NS |
| 235 | x | x | x | x | x | no | ||||||
| 318 | x | no | ||||||||||
| 319 | x | x | x | x | x | x | x | x | x | x | yes | HS |
| 324 | x | no | ||||||||||
| 327 | x | x | x | x | x | x | x | x | x | x | yes | HS |
| 328 | x | no |
| Resid | A | B | C | D | E | F | G | H | I | J | Tested | Classif |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | x | x | x | x | x | x | x | x | x | yes | NS | |
| 4 | x | x | x | x | x | x | x | x | x | x | yes | WS |
| 5 | x | x | x | x | x | x | no | |||||
| 6 | x | x | x | x | x | x | x | x | yes | NS | ||
| 7 | x | x | x | x | x | x | x | x | x | x | yes | |
| 8 | x | x | x | x | x | x | x | x | x | x | yes | NS |
| 9 | x | no | ||||||||||
| 10 | x | x | x | x | x | x | x | x | x | x | yes | WS |
| 12 | x | x | x | x | x | x | x | x | yes | NS | ||
| 15 | x | x | x | x | x | x | x | yes | NS | |||
| 16 | x | no | ||||||||||
| 18 | x | x | x | x | no | |||||||
| 20 | x | x | x | x | x | x | x | x | x | yes | WS | |
| 32 | x | x | no | |||||||||
| 33 | x | x | no | |||||||||
| 34 | x | x | x | x | x | x | x | x | x | yes | HS | |
| 35 | x | x | x | x | x | x | no | |||||
| 36 | x | x | x | x | x | x | no | |||||
| 38 | x | no | ||||||||||
| 39 | x | x | x | x | x | x | x | x | x | x | yes | WS |
| 56 | x | x | x | x | x | x | x | x | yes | NS | ||
| 59 | x | x | x | x | x | x | x | x | x | x | yes | NS |
| 66 | x | x | x | x | x | no | ||||||
| 67 | x | x | x | x | no | |||||||
| 68 | x | no | ||||||||||
| 69 | x | x | x | x | x | x | x | x | x | yes | NS | |
| 79 | x | x | no | |||||||||
| 81 | x | no | ||||||||||
| 82 | x | x | no | |||||||||
| 83 | x | x | x | x | x | x | no | |||||
| 84 | x | x | x | x | x | x | x | x | yes | HS | ||
| 137 | x | x | x | x | x | no | ||||||
| 138 | x | x | x | x | x | no | ||||||
| 139 | x | no | ||||||||||
| 140 | x | x | no | |||||||||
| 141 | x | no | ||||||||||
| 147 | x | x | x | x | x | x | no | |||||
| 148 | x | no | ||||||||||
| 150 | x | x | x | x | x | x | x | x | yes | NS | ||
| 158 | x | x | x | x | x | x | x | x | x | x | yes | HS |
| 159 | x | x | x | x | x | x | x | x | yes | NS | ||
| 161 | x | x | x | x | x | x | x | x | x | x | yes | HS |
| 164 | x | x | x | x | x | x | x | x | x | x | yes | NS |
| 165 | x | x | x | x | x | x | no | |||||
| 169 | x | x | x | x | x | x | x | x | x | yes | HS | |
| 170 | x | x | x | x | x | x | x | x | x | x | yes | NS |
| 173 | x | x | x | x | x | x | x | yes | HS | |||
| 176 | x | x | x | x | x | x | x | x | x | yes | HS | |
| 177 | x | x | x | x | x | x | x | x | x | x | yes | WS |
| 180 | x | x | x | x | x | x | x | x | x | x | yes | WS |
| 181 | x | x | x | x | x | x | x | x | x | x | yes | WS |
| 187 | x | x | x | no | ||||||||
| 189 | x | x | x | x | x | x | no | |||||
| 190 | x | no | ||||||||||
| 193 | x | no | ||||||||||
| 201 | x | x | x | no | ||||||||
| 222 | x | x | x | x | x | x | x | x | yes | WS | ||
| 223 | x | x | x | x | x | x | x | yes | NS | |||
| 224 | x | no | ||||||||||
| 226 | x | x | x | x | x | x | x | x | x | x | yes | HS |
| 227 | x | x | x | x | x | x | x | x | x | x | yes | NS |
| 229 | x | x | x | x | x | no | ||||||
| 231 | x | x | x | x | no | |||||||
| 310 | x | x | x | x | x | x | x | yes | NS | |||
| 311 | x | x | x | x | x | x | no | |||||
| 316 | x | no | ||||||||||
| 319 | x | x | x | x | x | x | x | x | x | x | yes | HS |
| Resid | A | B | C | D | E | F | G | H | I | J | K | L | Tested | Classif |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 20 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | NS |
| 31 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | NS |
| 32 | x | x | x | No | ||||||||||
| 33 | x | x | No | |||||||||||
| 34 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | HS |
| 35 | x | x | x | x | x | x | x | x | x | x | x | Yes | NS | |
| 36 | x | x | x | x | x | x | x | x | x | x | No | |||
| 40 | x | x | x | No | ||||||||||
| 54 | x | x | x | x | x | x | x | x | x | No | ||||
| 59 | x | x | x | x | x | No | ||||||||
| 64 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | HS |
| 65 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | NS |
| 66 | x | x | No | |||||||||||
| 68 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | NS |
| 84 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | HS |
| 99 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | NS |
| 141 | x | x | x | x | x | No | ||||||||
| 142 | x | x | x | x | x | x | x | x | x | x | x | Yes | WS | |
| 143 | x | x | x | x | x | No | ||||||||
| 144 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | WS |
| 146 | x | No | ||||||||||||
| 147 | x | x | x | x | x | x | No | |||||||
| 149 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | WS |
| 151 | x | x | x | x | x | x | x | x | x | x | x | Yes | NS | |
| 152 | x | x | x | x | x | x | x | x | x | No | ||||
| 153 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | HS |
| 154 | x | x | x | x | x | x | x | x | x | x | x | Yes | NS | |
| 161 | x | x | x | x | x | x | x | No | ||||||
| 162 | x | x | No | |||||||||||
| 164 | x | x | x | x | x | x | x | x | x | x | No | |||
| 175 | x | x | x | x | x | x | x | No | ||||||
| 176 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | HS |
| 178 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | WS |
| 179 | x | x | x | x | x | No | ||||||||
| 181 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | HS |
| 186 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | WS |
| 187 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | WS |
| 189 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | WS |
| 194 | x | No | ||||||||||||
| 196 | x | x | x | x | x | x | x | x | x | x | x | Yes | NS | |
| 197 | x | No | ||||||||||||
| 199 | x | x | x | x | No | |||||||||
| 200 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | NS |
| 203 | x | x | x | x | x | x | x | x | x | No | ||||
| 207 | x | x | No | |||||||||||
| 212 | x | x | No | |||||||||||
| 213 | x | x | x | No | ||||||||||
| 214 | x | x | x | x | x | No | ||||||||
| 215 | x | x | x | x | x | x | x | x | x | x | No | |||
| 247 | x | x | x | x | x | No | ||||||||
| 250 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | HS |
| 251 | x | x | x | x | x | x | No | |||||||
| 254 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | HS |
| T261 | x | x | x | x | x | No | ||||||||
| M263 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | HS |
| P265 | x | x | No | |||||||||||
| F268 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | WS |
| N325 | x | x | x | No | ||||||||||
| Y327 | x | No | ||||||||||||
| K328 | x | x | x | x | x | x | x | x | x | x | x | Yes | HS | |
| V331 | x | x | x | No | ||||||||||
| R345 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | HS |
| N346 | x | No | ||||||||||||
| R347 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | HS |
| R352 | x | x | No | |||||||||||
| I355 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | WS |
| T356 | x | No | ||||||||||||
| D404 | x | x | x | x | x | x | x | x | No | |||||
| T421 | x | x | x | x | No | |||||||||
| Q422 | x | x | x | x | x | x | x | x | x | x | No | |||
| S424 | x | No | ||||||||||||
| E457 | x | No | ||||||||||||
| E459 | x | x | x | No | ||||||||||
| V463 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | HS |
| N464 | x | x | x | x | x | x | x | x | x | x | No | |||
| I465 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | NS |
| R466 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | HS |
| H468 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | HS |
| Y470 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | WS |
| E471 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | HS |
| F472 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | HS |
| L474 | x | x | x | x | x | x | x | x | x | x | x | Yes | WS | |
| Y475 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | HS |
| Y476 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | HS |
| D477 | x | x | x | x | x | x | x | x | x | x | x | x | Yes | NS |
| V478 | x | x | x | x | x | x | x | x | x | x | x | x | No |
References
- Rhee, S.G.; Chock, P.B. Mechanistic studies of glutamine synthetase from Escherichia coli: Kinetic evidence for two reaction intermediates in biosynthetic reaction. Proc. Natl. Acad. Sci. USA 1976, 73, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, K.; Kayumov, A.; Woyda, K.; Ilinskaja, O.; Forchhammer, K. Transcription factor TnrA inhibits the biosynthetic activity of glutamine synthetase in Bacillus subtilis. FEBS Lett. 2013, 587, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Almassy, R.J.; Janson, C.A.; Hamlin, R.; Xuong, N.H.; Eisenberg, D. Novel subunit[mdash]subunit interactions in the structure of glutamine synthetase. Nature 1986, 323, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Moreira, C.; Ramos, M.J.; Fernandes, P.A. Reaction Mechanism of Mycobacterium Tuberculosis Glutamine Synthetase Using Quantum Mechanics/Molecular Mechanics Calculations. Chem Eur. J. 2016, 22, 9218–9225. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R. The Story of Glutamine Synthetase Regulation. J. Biol. Chem. 2001, 276, 44357–44364. [Google Scholar] [CrossRef] [PubMed]
- He, Y.-X.; Gui, L.; Liu, Y.-Z.; Du, Y.; Zhou, Y.; Li, P.; Zhou, C.-Z. Crystal structure of Saccharomyces cerevisiae glutamine synthetase Gln1 suggests a nanotube-like supramolecular assembly. Proteins Struct. Funct. Bioinform. 2009, 76, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Ghoshroy, S.; Robertson, D.L. Molecular evolution of glutamine synthetase II and III in the chromalveolates1. J. Phycol. 2012, 48, 768–783. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, S.; Isu, S.; Kaneko, G.; Yamada, H.; Hara, T.; Itoh, Y.; Watabe, S. The occurrence of eukaryotic type III glutamine synthetase in the marine diatom Chaetoceros compressum. Mar. Genom. 2009, 2, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Van Rooyen, J.M.; Abratt, V.R.; Belrhali, H.; Sewell, T. Crystal Structure of Type III Glutamine Synthetase: Surprising Reversal of the Inter-Ring Interface. Structure 2011, 19, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Brookes, G.; Barfoot, P. Economic impact of GM crops. GM Crops Food 2014, 5, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.-C.; Hung, D.-Z.; Wu, M.-L.; Tsai, W.-J.; Wang, L.-M.; Ger, J.; Deng, J.-F.; Yang, C.-C. Acute human Glufosinate-containing herbicide poisoning. Clin. Toxicol. 2012, 50, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Onodera, M.; Fujita, Y.; Fujino, Y.; Kikuchi, S.; Endo, S. Factors associated with severe effects following acute glufosinate poisoning. Clin. Toxicol. 2013, 51, 846–849. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Tanaka, T.; Fujisawa, M.; Shimada, K. Toxicokinetics of DL-Glufosinate Enantiomer in Human BASTA Poisoning. Biol. Pharm. Bull. 2003, 26, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Occhipinti, A.; Berlicki, Ł.; Giberti, S.; Dziȩdzioła, G.; Kafarski, P.; Forlani, G. Effectiveness and mode of action of phosphonate inhibitors of plant glutamine synthetase. Pest Manag. Sci. 2010, 66, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Evstigneeva, Z.G.; Solov’eva, N.A.; Sidel’nikova, L.I. Methionine Sulfoximine and Phosphinothrycin: A Review of Their Herbicidal Activity and Effects on Glutamine Synthetase. Appl. Biochem. Microbiol. 2003, 39, 539–543. [Google Scholar] [CrossRef]
- Calas, A.-G.; Richard, O.; Même, S.; Beloeil, J.-C.; Doan, B.-T.; Gefflaut, T.; Même, W.; Crusio, W.E.; Pichon, J.; Montécot, C. Chronic exposure to glufosinate-ammonium induces spatial memory impairments, hippocampal MRI modifications and glutamine synthetase activation in mice. NeuroToxicology 2008, 29, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Mowbray, S.; Kathiravan, M.; Pandey, A.; Odell, L. Inhibition of Glutamine Synthetase: A Potential Drug Target in Mycobacterium tuberculosis. Molecules 2014, 19, 13161–13176. [Google Scholar] [CrossRef] [PubMed]
- Lukasz, B. Inhibitors of Glutamine Synthetase and their Potential Application in Medicine. Mini-Rev. Med. Chem. 2008, 8, 869–878. [Google Scholar]
- Harth, G.; Clemens, D.L.; Horwitz, M.A. Glutamine synthetase of Mycobacterium tuberculosis: Extracellular release and characterization of its enzymatic activity. Proc. Natl Acad. Sci. USA 1994, 91, 9342–9346. [Google Scholar] [CrossRef] [PubMed]
- Harth, G.; Horwitz, M.A. Inhibition of Mycobacterium tuberculosis Glutamine Synthetase as a Novel Antibiotic Strategy against Tuberculosis: Demonstration of Efficacy In Vivo. Infect. Immun. 2003, 71, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Couturier, C.; Silve, S.; Morales, R.; Pessegue, B.; Llopart, S.; Nair, A.; Bauer, A.; Scheiper, B.; Pöverlein, C.; Ganzhorn, A.; et al. Nanomolar inhibitors of Mycobacterium tuberculosis glutamine synthetase 1: Synthesis, biological evaluation and X-ray crystallographic studies. Bioorg. Med. Chem. Lett. 2015, 25, 1455–1459. [Google Scholar] [CrossRef] [PubMed]
- Gising, J.; Nilsson, M.T.; Odell, L.R.; Yahiaoui, S.; Lindh, M.; Iyer, H.; Sinha, A.M.; Srinivasa, B.R.; Larhed, M.; Mowbray, S.L.; et al. Trisubstituted Imidazoles as Mycobacterium tuberculosis Glutamine Synthetase Inhibitors. J. Med. Chem. 2012, 55, 2894–2898. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.T.; Krajewski, W.W.; Yellagunda, S.; Prabhumurthy, S.; Chamarahally, G.N.; Siddamadappa, C.; Srinivasa, B.R.; Yahiaoui, S.; Larhed, M.; Karlén, A.; et al. Structural Basis for the Inhibition of Mycobacterium tuberculosis Glutamine Synthetase by Novel ATP-Competitive Inhibitors. J. Mol. Biol. 2009, 393, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Berlicki, Ł.; Kafarski, P. Computer-aided analysis of the interactions of glutamine synthetase with its inhibitors. Bioorg. Med. Chem. 2006, 14, 4578–4585. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, W.W.; Jones, T.A.; Mowbray, S.L. Structure of Mycobacterium tuberculosis glutamine synthetase in complex with a transition-state mimic provides functional insights. Proc. Natl. Acad. Sci. USA 2005, 102, 10499–10504. [Google Scholar] [CrossRef] [PubMed]
- Gill, H.S.; Eisenberg, D. The Crystal Structure of Phosphinothricin in the Active Site of Glutamine Synthetase Illuminates the Mechanism of Enzymatic Inhibition. Biochemistry 2001, 40, 1903–1912. [Google Scholar] [CrossRef] [PubMed]
- Adeva, M.M.; Souto, G.; Blanco, N.; Donapetry, C. Ammonium metabolism in humans. Metabolism 2012, 61, 1495–1511. [Google Scholar] [CrossRef] [PubMed]
- Frieg, B.; Görg, B.; Homeyer, N.; Keitel, V.; Häussinger, D.; Gohlke, H. Molecular Mechanisms of Glutamine Synthetase Mutations that Lead to Clinically Relevant Pathologies. PLoS Comput. Biol. 2016, 12, e1004693. [Google Scholar] [CrossRef] [PubMed]
- Häberle, J.; Shahbeck, N.; Ibrahim, K.; Hoffmann, G.F.; Ben-Omran, T. Natural course of glutamine synthetase deficiency in a 3 year old patient. Mol. Genet. Metab. 2011, 103, 89–91. [Google Scholar] [CrossRef] [PubMed]
- Betti, M.; García-Calderón, M.; Pérez-Delgado, C.M.; Credali, A.; Estivill, G.; Galván, F.; Vega, J.M.; Márquez, A.J. Glutamine Synthetase in Legumes: Recent Advances in Enzyme Structure and Functional Genomics. Int. J. Mol. Sci. 2012, 13, 7994–8024. [Google Scholar] [CrossRef] [PubMed]
- Bernard, S.M.; Habash, D.Z. The importance of cytosolic glutamine synthetase in nitrogen assimilation and recycling. New Phytol. 2009, 182, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Brusilow, S.W.; Koehler, R.C.; Traystman, R.J.; Cooper, A.J.L. Astrocyte Glutamine Synthetase: Importance in Hyperammonemic Syndromes and Potential Target for Therapy. Neurotherapeutics 2010, 7, 452–470. [Google Scholar] [CrossRef] [PubMed]
- Suárez, I.; Bodega, G.; Fernández, B. Glutamine synthetase in brain: Effect of ammonia. Neurochem. Int. 2002, 41, 123–142. [Google Scholar] [CrossRef]
- Gebhardt, R.; Gaunitz, F.; Mecke, D. Heterogeneous (positional) expression of hepatic glutamine synthetase: Features, regulation and implications for hepatocarcinogenesis. Adv. Enzyme Regul. 1994, 34, 27–56. [Google Scholar] [CrossRef]
- Gebhardt, R.; Baldysiak-Figiel, A.; Krügel, V.; Ueberham, E.; Gaunitz, F. Hepatocellular expression of glutamine synthetase: An indicator of morphogen actions as master regulators of zonation in adult liver. Prog. Histochem. Cytochem. 2007, 41, 201–266. [Google Scholar] [CrossRef] [PubMed]
- Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. Hot spots—A review of the protein–protein interface determinant amino-acid residues. Proteins Struct. Funct. Bioinform. 2007, 68, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. Computational Alanine Scanning Mutagenesis-an Improved Methodological Approach. J. Comput. Chem. 2007, 28, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Wisniewski, J.A.; Ji, H. Hot spot-based design of small-molecule inhibitors for protein–protein interactions. Bioorganic Med. Chem. Lett. 2014, 24, 2546–2554. [Google Scholar] [CrossRef] [PubMed]
- Lise, S.; Archambeau, C.; Pontil, M.; Jones, D.T. Prediction of hot spot residues at protein-protein interfaces by combining machine learning and energy-based methods. BMC Bioinform. 2009, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.; Dong, G.; Miao, Z.; Zhang, W.; Wang, W. State-of-the-art strategies for targeting protein-protein interactions by small-molecule inhibitors. Chem. Soc. Rev. 2015, 44, 8238–8259. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Tang, Y.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing towards the reality. Chem. Biol. 2014, 21, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- González-Ruiz, D.; Gohlke, H. Targeting Protein-protein Interactions with Small Molecules: Challenges and Perspectives for Computational Binding Epitope Detection and Ligand Finding. Curr. Med. Chem. 2006, 13, 2607–2625. [Google Scholar] [CrossRef] [PubMed]
- Schames, J.R.; Henchman, R.H.; Siegel, J.S.; Sotriffer, C.A.; Ni, H.; McCammon, J.A. Discovery of a Novel Binding Trench in HIV Integrase. J. Med. Chem. 2004, 47, 1879–1881. [Google Scholar] [CrossRef] [PubMed]
- Summa, V.; Petrocchi, A.; Bonelli, F.; Crescenzi, B.; Donghi, M.; Ferrara, M.; Fiore, F.; Gardelli, C.; Gonzalez Paz, O.; Hazuda, D.J.; et al. Discovery of Raltegravir, a Potent, Selective Orally Bioavailable HIV-Integrase Inhibitor for the Treatment of HIV-AIDS Infection. J. Med. Chem. 2008, 51, 5843–5855. [Google Scholar] [CrossRef] [PubMed]
- Wassman, C.D.; Baronio, R.; Demir, Ö.; Wallentine, B.D.; Chen, C.-K.; Hall, L.V.; Salehi, F.; Lin, D.-W.; Chung, B.P.; Wesley Hatfield, G.; et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013, 4, 1407. [Google Scholar] [CrossRef] [PubMed]
- Pietrucci, F.; Vargiu, A.V.; Kranjc, A. HIV-1 Protease Dimerization Dynamics Reveals a Transient Druggable Binding Pocket at the Interface. Sci. Rep. 2015, 5, 18555. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, J.V.; Cerqueira, N.M.F.S.A.; Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. CompASM: An Amber-VMD alanine scanning mutagenesis plug-in. Theor. Chem. Acc. 2012, 131, 1–7. [Google Scholar] [CrossRef]
- Kay, B.K.; Williamson, M.P.; Sudol, M. The importance of being proline: The interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000, 14, 231–241. [Google Scholar] [PubMed]
- Zarrinpar, A.; Bhattacharyya, R.P.; Lim, W.A. The Structure and Function of Proline Recognition Domains. Sci. Signal. 2003, 2003. [Google Scholar] [CrossRef] [PubMed]
- Zondlo, N.J. Aromatic–Proline Interactions: Electronically Tunable CH/π Interactions. Acc. Chem. Res. 2013, 46, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Masalkar, P.D.; Roberts, D.M. Glutamine synthetase isoforms in nitrogen-fixing soybean nodules: Distinct oligomeric structures and thiol-based regulation. FEBS Lett. 2015, 589, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, P.; Barril, X. Understanding and Predicting Druggability. A High-Throughput Method for Detection of Drug Binding Sites. J. Med. Chem. 2010, 53, 5858–5867. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties that Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.W.; Mutkus, L.A.; Aschner, M. Mercuric chloride, but not methylmercury, inhibits glutamine synthetase activity in primary cultures of cortical astrocytes. Brain Res. 2001, 891, 148–157. [Google Scholar] [CrossRef]
- Singh, U.; Panchanadikar, V.; Sarkar, D. Development of a Simple Assay Protocol for High-Throughput Screening of Mycobacterium tuberculosis Glutamine Synthetase for the Identification of Novel Inhibitors. J. Biomol. Screen. 2005, 10, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Olof, L.; Luke, R.O.; Sherry, L.M.; Mikael, T.N.; Wojciech, W.K.; Anneli, N.; Anders, K.; Mats, L. Microwave-Enhanced alpha-Arylation of a Protected Glycine in Water:Evaluation of 3-Phenylglycine Derivatives as Inhibitors of the Tuberculosis Enzyme, Glutamine Synthetase. Comb. Chem. High Throughput Screen. 2007, 10, 783–789. [Google Scholar]
- Gxoyiya, B.S.B.; Kaye, P.T.; Kenyon, C. Benzimidazole-Derived ATP Analogues as Potential Glutamine Synthetase Inhibitors. Synth. Commun. 2010, 40, 2578–2587. [Google Scholar] [CrossRef]
- Salisu, S.; Kenyon, C.; Kaye, P.T. Studies Towards the Synthesis of ATP Analogs as Potential Glutamine Synthetase Inhibitors. Synth. Commun. 2011, 41, 2216–2225. [Google Scholar] [CrossRef]
- Odell, L.R.; Nilsson, M.T.; Gising, J.; Lagerlund, O.; Muthas, D.; Nordqvist, A.; Karlén, A.; Larhed, M. Functionalized 3-amino-imidazo[1,2-a]pyridines: A novel class of drug-like Mycobacterium tuberculosis glutamine synthetase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 4790–4793. [Google Scholar] [CrossRef] [PubMed]
- Reddy Chichili, V.P.; Kumar, V.; Sivaraman, J. Linkers in the structural biology of protein–protein interactions. Protein Sci. Publ. Protein Soc. 2013, 22, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of Molecular Recognition Features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Tompa, P.; Simon, I.; Dosztányi, Z. Molecular Principles of the Interactions of Disordered Proteins. J. Mol. Biol. 2007, 372, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K. Another Window into Disordered Protein Function. Structure 2007, 15, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Stec, B.; Godzik, A. Between Order and Disorder in Protein Structures: Analysis of “Dual Personality” Fragments in Proteins. Structure 2007, 15, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [PubMed]
- Turoverov, K.K.; Kuznetsova, I.M.; Uversky, V.N. The protein kingdom extended: Ordered and intrinsically disordered proteins, their folding, supramolecular complex formation, and aggregation. Prog. Biophys. Mol. Biol. 2010, 102, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Harth, G.; Masleša-Galić, S.; Tullius, M.V.; Horwitz, M.A. All four Mycobacterium tuberculosis glnA genes encode glutamine synthetase activities but only GlnA1 is abundantly expressed and essential for bacterial homeostasis. Mol. Microbiol. 2005, 58, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Unno, H.; Uchida, T.; Sugawara, H.; Kurisu, G.; Sugiyama, T.; Yamaya, T.; Sakakibara, H.; Hase, T.; Kusunoki, M. Atomic Structure of Plant Glutamine Synthetase: A key enzyme for plant productivity. J. Biol. Chem. 2006, 281, 29287–29296. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, W.W.; Collins, R.; Holmberg-Schiavone, L.; Jones, T.A.; Karlberg, T.; Mowbray, S.L. Crystal Structures of Mammalian Glutamine Synthetases Illustrate Substrate-Induced Conformational Changes and Provide Opportunities for Drug and Herbicide Design. J. Mol. Biol. 2008, 375, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Maurizi, M.R.; Ginsburg, A. Active site ligand stabilization of quaternary structures of glutamine synthetase from Escherichia coli. J. Biol. Chem. 1982, 257, 7246–7251. [Google Scholar] [PubMed]
- Murray, D.S.; Chinnam, N.; Tonthat, N.K.; Whitfill, T.; Wray, L.V.; Fisher, S.H.; Schumacher, M.A. Structures of the Bacillus subtilis Glutamine Synthetase Dodecamer Reveal Large Intersubunit Catalytic Conformational Changes Linked to a Unique Feedback Inhibition Mechanism. J. Biol. Chem. 2013, 288, 35801–35811. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AMBER 12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Sun, H.; Li, Y.; Wang, J.; Hou, T. Assessing the Performance of MM/PBSA and MM/GBSA Methods. 3. The Impact of Force Fields and Ligand Charge Models. J. Phys. Chem. B 2013, 117, 8408–8421. [Google Scholar] [CrossRef] [PubMed]
- Martins, S.A.; Perez, M.A.S.; Moreira, I.S.; Sousa, S.F.; Ramos, M.J.; Fernandes, P.A. Computational Alanine Scanning Mutagenesis: MM-PBSA vs. TI. J. Chem. Theory Comput. 2013, 9, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not Available.






| Resid | ΔΔGbind | SEM | Classif |
|---|---|---|---|
| I13 | 3.82 | 0.40 | WS |
| K14 | 2.67 | 0.29 | WS |
| Y17 | 2.69 | 0.16 | WS |
| M18 | 0.59 | 0.22 | NS |
| L20 | 4.57 | 0.16 | HS |
| Q22 | 1.38 | 0.04 | NS |
| Q27 | 5.84 | 0.30 | HS |
| R41 | 6.12 | 0.26 | HS |
| C42 | 10.63 | 1.07 | HS |
| K43 | 1.23 | 0.89 | NS |
| T44 | 11.21 | 1.23 | HS |
| T46 | 2.07 | 0.10 | WS |
| S66 | 0.09 | 0.09 | NS |
| D76 | 2.86 | 0.25 | WS |
| N152 | 0.94 | 0.43 | NS |
| F154 | 0.42 | 0.23 | NS |
| Y162 | 4.37 | 0.18 | HS |
| C163 | 2.22 | 0.11 | WS |
| V165 | 5.29 | 0.15 | HS |
| R173 | 4.22 | 0.32 | HS |
| D174 | 5.15 | 0.41 | HS |
| E177 | 9.52 | 0.53 | HS |
| Y180 | 4.85 | 0.38 | HS |
| R181 | 7.90 | 0.57 | HS |
| L184 | 3.32 | 0.10 | WS |
| Y185 | 3.54 | 0.07 | WS |
| E230 | 12.47 | 0.58 | HS |
| F232 | −0.02 | 0.01 | NS |
| R319 | 5.14 | 0.71 | HS |
| R327 | 5.40 | 0.38 | HS |
| Resid | ΔΔGbind | SEM | Classif |
|---|---|---|---|
| C3 | 0.75 | 0.15 | NS |
| L4 | 3.14 | 0.27 | WS |
| D6 | 1.84 | 0.21 | NS |
| L7 | 3.62 | 0.26 | WS |
| V8 | 0.35 | 0.22 | NS |
| L10 | 3.78 | 0.30 | WS |
| L12 | −4.30 | 0.45 | NS |
| T15 | −1.99 | 0.43 | NS |
| I20 | 3.67 | 0.33 | WS |
| R34 | 7.21 | 0.24 | HS |
| T39 | 2.21 | 0.07 | WS |
| D56 | −0.10 | 0.50 | NS |
| S59 | 0.59 | 0.19 | NS |
| E69 | 2.42 | 0.78 | NS |
| R84 | 4.74 | 0.23 | HS |
| F150 | 1.26 | 0.29 | NS |
| Y158 | 3.88 | 0.32 | HS |
| C159 | 1.64 | 0.11 | NS |
| I161 | 7.08 | 0.20 | HS |
| E164 | −1.68 | 0.44 | NS |
| R169 | 8.28 | 0.25 | HS |
| D170 | −1.46 | 0.06 | NS |
| D173 | 5.44 | 0.69 | HS |
| Y176 | 4.07 | 0.20 | HS |
| K177 | 3.39 | 0.40 | WS |
| L180 | 3.91 | 0.23 | WS |
| Y181 | 2.90 | 0.36 | WS |
| E222 | 1.62 | 0.53 | WS |
| R223 | −4.57 | 0.90 | NS |
| E226 | 6.27 | 0.53 | HS |
| I227 | 0.57 | 0.21 | NS |
| N310 | −0.59 | 0.41 | NS |
| R319 | 3.94 | 0.32 | HS |
| Resid | ΔΔGbind | SEM | Classif |
|---|---|---|---|
| Y20 | 0.41 | 0.20 | NS |
| I31 | 1.15 | 0.32 | NS |
| H34 | 4.12 | 0.31 | HS |
| F35 | 1.88 | 0.13 | NS |
| I64 | 4.48 | 0.40 | HS |
| H65 | 1.20 | 0.30 | NS |
| D68 | −0.12 | 0.52 | NS |
| R84 | 7.06 | 0.74 | HS |
| F99 | 1.53 | 0.43 | NS |
| V142 | 2.92 | 0.26 | WS |
| F144 | 2.60 | 0.21 | WS |
| N149 | 3.48 | 0.13 | WS |
| S151 | −0.09 | 0.07 | NS |
| Y153 | 7.18 | 0.17 | HS |
| E154 | −0.33 | 0.27 | NS |
| R176 | 6.96 | 1.20 | HS |
| Y178 | 3.42 | 0.22 | WS |
| R181 | 9.57 | 1.12 | HS |
| Y186 | 3.79 | 0.26 | WS |
| F187 | 3.84 | 0.29 | WS |
| V189 | 3.71 | 0.21 | WS |
| V196 | 0.85 | 0.24 | NS |
| D200 | −1.28 | 0.44 | NS |
| K250 | 14.54 | 0.28 | HS |
| W254 | 4.46 | 0.28 | HS |
| M263 | 6.94 | 0.17 | HS |
| F268 | 3.60 | 0.13 | WS |
| K328 | 13.60 | 0.17 | HS |
| R345 | 16.09 | 0.36 | HS |
| R347 | 12.97 | 0.49 | HS |
| I355 | 3.17 | 0.15 | WS |
| V463 | 4.40 | 0.24 | HS |
| I465 | 0.87 | 0.17 | NS |
| R466 | 8.82 | 0.14 | HS |
| H468 | 4.50 | 0.08 | HS |
| Y470 | 3.89 | 0.06 | WS |
| E471 | 9.67 | 0.29 | HS |
| F472 | 5.54 | 0.09 | HS |
| L474 | 2.06 | 0.14 | WS |
| Y475 | 5.49 | 0.10 | HS |
| Y476 | 5.74 | 0.29 | HS |
| D477 | −2.78 | 0.28 | NS |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreira, C.; Ramos, M.J.; Fernandes, P.A. Glutamine Synthetase Drugability beyond Its Active Site: Exploring Oligomerization Interfaces and Pockets. Molecules 2016, 21, 1028. https://doi.org/10.3390/molecules21081028
Moreira C, Ramos MJ, Fernandes PA. Glutamine Synthetase Drugability beyond Its Active Site: Exploring Oligomerization Interfaces and Pockets. Molecules. 2016; 21(8):1028. https://doi.org/10.3390/molecules21081028
Chicago/Turabian StyleMoreira, Cátia, Maria J. Ramos, and Pedro A. Fernandes. 2016. "Glutamine Synthetase Drugability beyond Its Active Site: Exploring Oligomerization Interfaces and Pockets" Molecules 21, no. 8: 1028. https://doi.org/10.3390/molecules21081028
APA StyleMoreira, C., Ramos, M. J., & Fernandes, P. A. (2016). Glutamine Synthetase Drugability beyond Its Active Site: Exploring Oligomerization Interfaces and Pockets. Molecules, 21(8), 1028. https://doi.org/10.3390/molecules21081028